

Abstract
Miltefosine is an alkyl phosphocholine with good oral bioavailability and in vitro activity against Cryptococcus species that has gained interest as an additional agent for cryptococcal infections. Our objective was to further evaluate the in vivo efficacy of miltefosine in experimental in vivo models of cryptococcal meningoencephalitis and disseminated cryptococcosis. Mice were infected intracranially or intravenously with either C. neoformans USC1597 or H99. Miltefosine treatment (1.8 to 45 mg/kg of body weight orally once daily) began at either 1 h or 1 day postinoculation. Fluconazole (10 mg/kg orally twice daily) or amphotericin B deoxycholate (3 mg/kg intraperitoneally once daily) served as positive controls. In our standard models, miltefosine did not result in significant improvements in survival or reductions in fungal burden against either C. neoformans isolate. There was a trend toward improved survival with miltefosine at 7.2 mg/kg against disseminated cryptococcosis with the H99 strain but only at a low infecting inoculum. In contrast, both fluconazole and amphotericin B significantly improved survival in mice with cryptococcal meningoencephalitis and disseminated cryptococcosis due to USC1597. Amphotericin B also improved survival against both cryptococcal infections caused by H99. Combination therapy with miltefosine demonstrated neither synergy nor antagonism in both models. These results demonstrate limited efficacy of miltefosine and suggest caution with the potential use of this agent for the treatment of C. neoformans infections.
INTRODUCTION
Cryptococcal meningoencephalitis remains a substantial infectious disease challenge. Populations at risk for infections caused by Cryptococcus neoformans include HIV/AIDS patients, recipients of solid organ or hematopoietic stem cells transplants, as well as those receiving high-dose chemotherapy or those having some other immunocompromised condition (1, 2). The Centers for Disease Control and Prevention have estimated that in HIV/AIDS patients the global burden of cryptococcosis is 1 million cases annually (3), while other studies have reported the 3-month mortality for the treatment of acute cryptococcal meningoencephalitis to be approximately 20% (4, 5). Effective treatment is available in the United States and other developed countries and includes amphotericin B and flucytosine as induction therapy followed by fluconazole (2). However, intravenous medications as well as oral flucytosine may not be available in less-developed areas, and the use of high-dose oral fluconazole is not as effective as induction therapy with amphotericin B (6, 7). Thus, there is a clinical need for new or alternative strategies, including agents that may be administered orally, for the treatment of cryptococcal meningoencephalitis.
Miltefosine (hexadecylphosphocholine) is an alkyl phosphocholine that has been used in the treatment of leishmaniasis, including New World cutaneous, mucocutaneous, and visceral disease (8–15). Miltefosine can be administered by mouth, and good oral bioavailability (82 to 94%) has been reported in rats and dogs (16). Recently, this agent has gained interest as a potent therapeutic against invasive mycoses, including cryptococcal infections as in vitro studies have demonstrated potent activity against C. neoformans and dermatophytes (17, 18). In one case report, miltefosine in combination with voriconazole and terbinafine was successfully used to treat a child with Scedosporium prolificans osteomyelitis (19). Improvements in survival and reductions in tissue fungal burden with the oral administration of miltefosine were also reported in a murine model of disseminated cryptococcosis (18). Based on the preliminary in vitro data that demonstrated activity against C. neoformans and in vivo efficacy against disseminated cryptococcosis, we sought to further evaluate the effectiveness of miltefosine in our murine model of cryptococcal meningoencephalitis, both as monotherapy and in combination with other antifungals. Studies were also conducted to further characterize the efficacy of this agent against disseminated cryptococcosis.
MATERIALS AND METHODS
Isolates and antifungal agents.
Cryptococcus neoformans clinical isolates USC1597 and H99 were used throughout this study. Each isolate was subcultured at least twice on Sabouraud dextrose agar (SDA) (Remel, Inc., Lenexa, KS) before each in vitro and in vivo experiment. Prior to inoculation, the isolates were grown in brain heart infusion broth overnight at 37°C with shaking. The cells were then collected by centrifugation and were washed three times in sterile saline. The inoculum concentrations were determined with a hemocytometer, and viability was confirmed by plating serial dilutions and enumeration of CFU. The clinically available intravenous formulations of fluconazole (Pfizer, Inc., New York, NY) and amphotericin B deoxycholate (X-Gen Pharmaceuticals, Big Flats, NY) were used in all studies and were prepared according to the package inserts. Miltefosine powder was obtained from Cayman Chemical Company (Ann Arbor, MI) and was dissolved in physiologic saline prior to each experiment.
Susceptibility testing.
Susceptibility testing was done by broth microdilution in accordance with the CLSI M27-A3 methodology in RPMI 1640 growth medium buffered with 0.165 M morpholinepropanesulfonic acid (MOPS; pH 7.0) (20). Stock solutions were prepared in water (fluconazole and amphotericin B deoxycholate) or RPMI 1640 medium (miltefosine). Aliquots of each agent (0.1 ml) at 2× concentrations were dispensed into 96-well microdilution trays. C. neoformans cells in RPMI medium were added (final inoculum, 0.5 × 103 to 2.5 × 103 cells/ml), and the trays were incubated at 35°C. Final antifungal concentrations ranged from 0.125 to 64 μg/ml for miltefosine and fluconazole and from 0.03 to 16 μg/ml for amphotericin B. The MICs for fluconazole were read at 50% reduction in turbidity compared to growth of the control at 72 h. For amphotericin B and miltefosine, MICs were determined as 100% inhibition relative to growth of the controls. In later experiments, 5% (50 mg/ml) bovine serum albumin (BSA; Equitech Bio, Inc. Kerrville, TX) was added to the RPMI medium to assess the effects of protein binding on the in vitro potency of miltefosine.
Animal model.
Previously described murine models of cryptococcal meningoencephalitis and disseminated cryptococcosis were utilized for all studies (18, 21–23). Immunocompetent outbred ICR mice (Harlan) weighing approximately 25 g were housed five mice per cage and had access to food and water ad libitum. To establish cryptococcal meningoencephalitis, mice were anesthetized using isoflurane, and C. neoformans at 1,500 to 2,600 cells/mouse was delivered in a 0.06-ml volume through a 27-gauge needle fastened to a tuberculin syringe with a cuff to prevent penetration of more than 1 mm. A midline puncture through the cranial vault approximately 6 mm posterior to the orbit was made, and the inoculum was injected, after which the mice were allowed to recover. To establish disseminated cryptococcosis, mice were inoculated intravenously via the lateral tail vein with C. neoformans over a range of inocula in different experiments (4.0 × 103 to 6.1 × 105 cells/mouse) delivered in a 0.2-ml volume of sterile saline. This study was approved by the Institutional Animal Care and Use Committee at the University of Texas (UT) Health Science Center at San Antonio, and all animals were maintained in accordance with the American Association for Accreditation of Laboratory Animal Care (24).
Antifungal treatment.
Antifungal therapy with miltefosine was initiated either 1 h or 1 day postinoculation at doses ranging between 1.8 and 60 mg/kg of body weight once daily by oral gavage. Amphotericin B was initiated at either 1 h or 1 day postinoculation at a dose of 3 mg/kg once daily by intraperitoneal injection, while fluconazole was initiated at 1 day postinoculation in all experiments at a dose of 10 mg/kg once daily by oral gavage. Between 8 and 20 mice were included per dosage group of each antifungal in each experiment. The duration of therapy for each agent in each study is specified in Results.
Outcome measures.
Survival and brain tissue fungal burden were used to measure the in vivo efficacy of antifungal therapy. In the survival experiments, mice were followed off therapy to either day 21 or day 30 postinoculation, and any animal that appeared moribund was humanely euthanized. In tissue burden experiments, brain tissues were aseptically collected 1 day after treatment stopped in order to measure fungal burden. Brains were weighed and then homogenized in sterile saline with a tissue homogenizer. Serial dilutions were prepared in sterile saline and plated on SDA. After 48 to 72 h of incubation, colonies were counted, and the number of CFU per gram of tissue for each animal was calculated.
Statistics.
Survival was plotted by Kaplan-Meier analysis, and log rank and Fisher's exact tests were used to assess for significant differences in median survival and percent survival, respectively. Differences in tissue fungal burden were assessed using analysis of variance (ANOVA) with Tukey's posttest for multiple comparisons. A P value of ≤0.05 was considered statistically significant.
RESULTS
Susceptibility.
Miltefosine demonstrated in vitro activity against C. neoformans USC1597 and H99 (MICs of 1 μg/ml and 0.5 μg/ml, respectively) in RPMI medium. Fluconazole was active against each C. neoformans isolate (MIC against USC1597, 1 μg/ml; MIC against H99, 2 μg/ml), and amphotericin B also demonstrated potent activity against both isolates (MIC of 0.25 μg/ml). In contrast to the activity observed in RPMI medium for miltefosine, the potency of this agent was significantly reduced with the addition of 5% BSA (MIC of >64 μg/ml).
Cryptococcal meningoencephalitis caused by C. neoformans USC1597.
The objective of this study was to evaluate the in vivo efficacy of miltefosine against cryptococcal meningoencephalitis, and the initial experiments utilized a fully susceptible C. neoformans isolate (USC1597) to establish infection. Monotherapy with miltefosine at doses ranging from 5 to 60 mg/kg/day, amphotericin B at 3 mg/kg/day, or fluconazole at 10 mg/kg twice daily was initiated at 1 day postchallenge, and each agent was continued for 10 days in the survival arm and for 7 days in the fungal burden arm. Miltefosine did not lead to a survival advantage at doses as high as 45 mg/kg as the median survival (range, 13.5 to 23.5 days) and percent survival (0 to 10%) at each dose did not differ significantly from rates for untreated controls (19 days and 10.5%, respectively) (Fig. 1A and B). In contrast, median survival was significantly prolonged with fluconazole and amphotericin B (29 and >30 days, respectively) compared to controls and each miltefosine dose (P < 0.05 for all comparisons). In addition, amphotericin B significantly improved the percentage of animals surviving to day 30 (95%; P < 0.05) (Fig. 1B). Although therapy with miltefosine at 60 mg/kg was initially tried, mice were unable to tolerate a dose this high as several died following a single administration; therefore, this dose was discontinued. Combination therapy with miltefosine was also ineffective. Neither a survival advantage nor disadvantage was observed with miltefosine at 45 mg/kg plus amphotericin B or fluconazole as the median and percent survival values for these combinations were similar to those observed with monotherapy with these antifungals (Fig. 1C).
Fig 1.

Survival curves in mice inoculated intracranially with C. neoformans USC1597 at 1,900 to 2,800 CFU/animal. Antifungal monotherapy therapy with miltefosine (MTF), fluconazole (FLC), or amphotericin B (AMB) or combination therapy began at 1 day postchallenge (A, B, and C) and continued for 10 days (n = 19 to 20 mice per group for untreated controls, fluconazole, and amphotericin B and 8 to 10 mice per miltefosine dose). The early initiation of miltefosine and amphotericin B or combination therapy at 1 h postchallenge was also evaluated (D). Daily miltefosine therapy continued for 10 days, and amphotericin B was administered for only two doses (n = 9 to 10 mice per group). *, P < 0.05 for median survival compared to untreated controls; §, P < 0.05 for percent survival at study endpoint compared to untreated controls.
Since the marked survival advantage observed with daily administration of amphotericin B may have masked any benefit of combination therapy, we also evaluated daily high-dose miltefosine with a short course of this polyene. In this experiment, both miltefosine at 45 mg/kg and amphotericin B were initiated at 1 h postchallenge, and amphotericin B was discontinued after two doses. Even when therapy was initiated shortly after intracranial challenge, no improvement in survival was observed with miltefosine monotherapy as the median and percent survival values (17.5 days and 0%, respectively) were similar to those of untreated controls (15.5 days and 0%, respectively) (Fig. 1D). In contrast, monotherapy with a short course of amphotericin B still resulted in longer survival than that of controls and miltefosine treatment (24 days; P < 0.01). Similar to the previous results, miltefosine combination therapy did not lead to a significant benefit compared to amphotericin B alone (median survival, 21.5 days). The lack of efficacy of miltefosine was also evident in the brain tissue fungal burden results (Table 1). No reductions in fungal burden were observed in any miltefosine monotherapy group compared to burden in untreated controls. In contrast, significant reductions were observed in mice that received fluconazole or amphotericin B as well as in those that received combination therapy. Furthermore, the addition of miltefosine did not result in further reductions in fungal burden compared to treatment with fluconazole or amphotericin B alone.
Table 1.
Fungal burden in the brains or lungs of mice with cryptococcal meningoencephalitis or disseminated cryptococcosis following intracranial or intravenous inoculation with C. neoformans USC1597
| Inoculation method and tissue |
C. neoformans USC1597 fungal burden (log10 CFU/gram) by treatmenta |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | MTF (mg/kg) |
FLC | AMB | MTF (45 mg/kg) + FLC | MTF (45 mg/kg) + AMB | ||||
| 5 | 10 | 15 | 45 | ||||||
| Intracranialb | |||||||||
| Brain | 6.04 ± 0.51 | 5.57 ± 0.35 | 5.98 ± 0.51 | 5.61 ± 0.54 | 6.03 ± 0.42 | 2.64 ± 1.05* | 2.20 ± 0.92* | 3.32 ± 0.90* | 2.83 ± 1.32* |
| Intravenousc | |||||||||
| Lung | 7.22 ± 0.62 | 7.46 ± 0.28 | 5.77 ± 1.99** | 5.67 ± 1.61** | |||||
| Brain | 6.23 ± 0.23 | 6.34 ± 0.15 | 4.35 ± 1.20* | 4.04 ± 1.02* | |||||
MTF, miltefosine; FLC, fluconazole; AMB, amphotericin B. Values are means ± standard deviations. Fluconazole was administered at 10 mg/kg, and amphotericin B was administered at 3 mg/kg. * P < 0.001; **, P < 0.05 (versus untreated controls and all-miltefosine monotherapy).
In the cryptococcal meningoencephalitis model, antifungal therapy with miltefosine, fluconazole, or amphotericin B was continued through day 7, and brains were collected on day 8 for fungal burden quantification (n = 19 to 20 mice per group for untreated controls, fluconazole, and amphotericin B and 8 to 10 mice per miltefosine dose).
In the disseminated cryptococcosis model, miltefosine therapy was continued through day 7 while amphotericin B was administered for two doses. The lungs and brains were collected on day 8 for fungal burden quantification (n = 10 mice per group).
Cryptococcal meningoencephalitis caused by C. neoformans H99.
Since the previous study that reported in vivo efficacy of miltefosine against disseminated cryptococcosis used a different C. neoformans strain (H99), we also evaluated the effectiveness of this agent against cryptococcal meningoencephalitis caused by this isolate. As shown in Fig. 2, miltefosine was also ineffective against central nervous system (CNS) infection caused by H99. No improvements in survival were observed when miltefosine was initiated 1 h after inoculation at lower doses (1.8 to 7.2 mg/kg/day through day 4) (Fig. 2A) or when higher doses with a longer period of administration were used (15 and 45 mg/kg/day through day 10) (Fig. 2B). In contrast, treatment with both amphotericin B and fluconazole resulted in significant improvements in survival (median survival, 21 and >30 days, respectively) (Fig. 2C) compared to survival of untreated controls (18 days; P < 0.05) and each miltefosine dose (range, 11.5 to 16.5 days; P < 0.0001). The percentage of animals surviving to day 30 was also significantly higher in animals treated with amphotericin B than for all other groups (70% versus 0%; P < 0.001).
Fig 2.
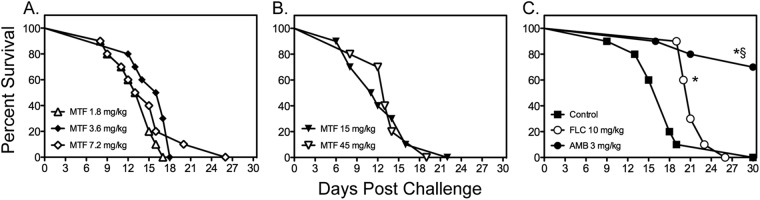
Survival curves of mice inoculated intracranially with C. neoformans H99 at 1,335 CFU CFU/animal. Antifungal therapy with miltefosine (MTF) began at 1 h following infection, while therapy with fluconazole (FLC) and amphotericin B (AMB) was initiated at 1 day postchallenge. Therapy with miltefosine at doses of 1.8, 3.6, and 7.2 mg/kg (A) continued through day 4, and therapy with miltefosine at 15 and 45 mg/kg (B) or with fluconazole and amphotericin B (C) continued through day 10 (n = 10 mice per group). *, P < 0.05 for median survival compared to untreated controls; §, P < 0.05 for percent survival at study endpoint compared to untreated controls.
Disseminated cryptococcosis caused by C. neoformans USC1597 and H99.
We also evaluated the efficacy of miltefosine for the treatment of disseminated cryptococcosis following intravenous challenge. In these experiments, miltefosine (1.8 to 45 mg/kg/day) was initiated 1 h after inoculation, while fluconazole and amphotericin B were started 1 day after infection. As seen against cryptococcal meningoencephalitis, miltefosine did not improve survival at any dose tested (median survival range, 10 to 14 days) compared to rates for untreated controls (12 days) (Fig. 3A and B). Survival was significantly improved with fluconazole and amphotericin B (>21 days for both agents; P < 0.0001). Combination therapy also did not significantly improve survival compared to treatment with amphotericin B alone (Fig. 3C). Furthermore, no significant reductions in fungal burden were observed within the lungs or brain tissue with high-dose miltefosine (45 mg/kg) as monotherapy (Table 1), nor did combination therapy result in further reductions in fungal burden compared to amphotericin B monotherapy.
Fig 3.
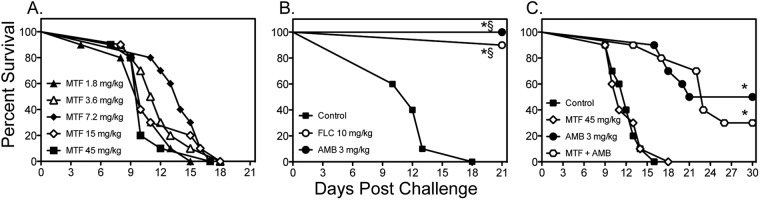
Survival curves in mice inoculated intravenously with C. neoformans USC1597 at 0.54 × 105 to 1.6 × 105 CFU/animal. In the monotherapy experiments (A and B), miltefosine (MTF) began at 1 h postchallenge, while fluconazole (FLC) and amphotericin B (AMB) were initiated at 1 day postchallenge. Therapy with miltefosine at doses of 1.8, 3.6, and 7.2 mg/kg continued through day 4, and therapy with miltefosine at 15 and 45 mg/kg, fluconazole, and amphotericin B continued through day 10. For the combination therapy experiment (C), miltefosine at 45 mg/kg was combined with amphotericin B (n = 10 mice per group). *, P < 0.05 for median survival compared to untreated controls; §, P < 0.05 for percent survival at study endpoint compared to untreated controls.
We also tested the effectiveness of miltefosine against disseminated cryptococcosis caused by C. neoformans H99. As shown in Fig. 4, miltefosine and fluconazole did not improve survival compared to survival of untreated controls, while amphotericin B significantly improved survival versus all other groups. As these results were in contrast to what was previously reported in a similar model of disseminated cryptococcosis, we also evaluated the effects of different infecting inoculum levels on the survival of untreated controls. As expected, the higher inocula (4.7 × 104 and 5.9 × 105 cells/mouse) resulted in 100% mortality early during the course of infection (Fig. 5A). As the survival curve for the inoculum of 4.8 × 103 cells/mouse was similar to that observed in untreated controls in the previous study that reported in vivo efficacy for miltefosine (18), we also tested the effectiveness of this agent at this lower infecting inoculum. Interestingly, a trend toward improved survival was observed with miltefosine at 7.2 mg/kg/day versus survival of untreated controls (17.5 versus 20.5 days, respectively; P = 0.09), which was in agreement with the results of the previous study. This suggests that the in vivo efficacy of this agent may be inoculum dependent.
Fig 4.
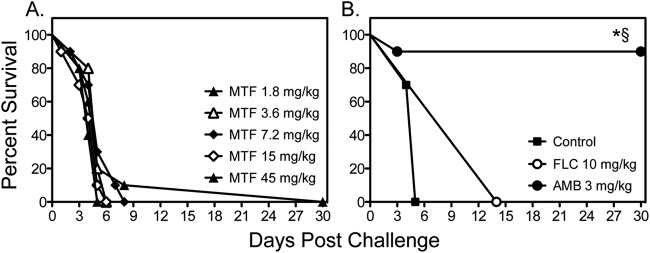
Survival curves in mice inoculated intravenously with C. neoformans H99 at 0.54 × 105 to 6.1 × 105 CFU/animal. Treatment with miltefosine (MTF) began at 1 h postchallenge, while fluconazole (FLC) and amphotericin B (AMB) were started 1 day after infection. Therapy with miltefosine at doses of 1.8, 3.6, and 7.2 mg/kg continued through day 4, and therapy with miltefosine at 15 and 45 mg/kg, fluconazole, and amphotericin B continued through day 10 (n = 10 mice per group). *, P < 0.05 for median survival compared to untreated controls; §, P < 0.05 for percent survival at study endpoint compared to untreated controls.
Fig 5.
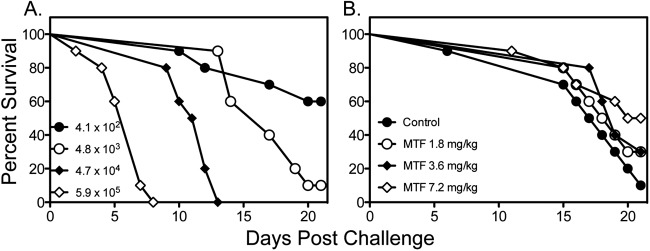
Survival curves in untreated mice challenged intravenously with C. neoformans H99 at different inoculum levels (A) or with a single inoculum (4.0 × 103 CFU/animal) and treated with miltefosine (MTF) (B). Miltefosine was initiated at 1 h postchallenge and continued through day 4 (n = 10 mice per group).
DISCUSSION
Miltefosine, an alkyl phosphocholine, has been used as therapy for various forms of leishmaniasis and has recently garnered attention for its potential use against invasive fungal infections, including cryptococcosis. Current interest in this agent is due to the in vitro activity against Cryptococcus species and dermatophytes and the clinical success that was reported in one patient treated with miltefosine in combination with other antifungal agents for Scedosporium prolificans osteomyelitis (17, 19). Additionally, studies have also reported in vivo efficacy of miltefosine monotherapy in a murine model of disseminated cryptococcosis as well as meningitis due to Naegleria fowleri (18, 25). The mechanism by which miltefosine inhibits fungi has also been elucidated. Following incorporation into Saccharomyces cerevisiae, miltefosine rapidly penetrates the mitochondrial inner membrane, where it inhibits cytochrome c oxidase and disrupts membrane potential, leading to apoptosis-like cell death (26).
Cryptococcal meningoencephalitis is a common manifestation of disseminated disease caused by C. neoformans, and there is a clear need to develop new antifungal agents as well as to evaluate available drugs for the treatment of this invasive mycosis; as such, we sought to evaluate the efficacy of miltefosine in our established murine model of this CNS infection. Based on the in vitro activity that we observed and that reported by others, as well as previous reports of in vivo efficacy, we expected to see benefit with the use of this agent. However, the results were negative in the majority of our experiments as miltefosine monotherapy did not improve survival in our animal models of cryptococcal meningoencephalitis or disseminated cryptococcosis caused by two different C. neoformans isolates. In contrast, efficacy was observed with the clinically available antifungals fluconazole and amphotericin B. In addition, miltefosine failed to reduce fungal burden within the brains or lungs of these animals and did not improve the effectiveness of therapy when used in combination. The exact reason for this lack of in vivo efficacy in our models is unclear as in vitro activity was observed using the CLSI methodology for antifungal susceptibility testing against yeasts. Furthermore, the MIC values we observed against the two C. neoformans isolates were in agreement with those reported by others (18). However, the in vitro potency of this agent was significantly reduced when albumin was added to the growth medium. Miltefosine is highly protein bound (∼95%) (27, 28), which could partially explain the lack of in vivo activity we observed as there may be only a small fraction of unbound drug available to inhibit the organism at the site of infection. In addition, the effectiveness against disseminated cryptococcosis appeared to be dependent on the size of the infecting inoculum as a trend toward improved survival was observed when a lower inoculum was used to establish infection. The clinical relevance of this finding is unknown, but it is noteworthy that miltefosine lacked activity when mice were challenged with our standard inocula in the cryptococcal meningoencephalitis and disseminated cryptococcosis models, while the clinically available agents fluconazole and amphotericin B remained effective.
A limitation of this study is that we did not evaluate the pharmacokinetics of miltefosine in these animal models, nor did we evaluate different dosing strategies in an effort to potentially improve efficacy. Thus, it is unknown whether effective concentrations were achieved either within the bloodstream or at the sites of infection, including the CNS. However, other investigators have reported favorable pharmacokinetics for miltefosine in mice and other animals, including good oral bioavailability (82% to 95%), a long terminal half-life (range, 73 to 104 h), with extensive tissue distribution, including the brain (16, 29, 30). In addition, peak miltefosine serum concentrations above the MICs for the two C. neoformans isolates used in our study and high values for the area under the concentration-time curve (AUC) have been reported in mice that received doses similar to those we tested (29). Furthermore, in our initial experiment with miltefosine, acute toxicity was observed in several mice following a single dose of 60 mg/kg. Thus, it is unlikely that the concentrations that we achieved were suboptimal.
Although the results of this study do not eliminate the potential utility of miltefosine for the treatment of invasive fungal infections, including those caused by Cryptococcus species, caution is warranted. Additional concerns that may limit the clinical use of this agent include dose-limiting side effects (i.e., nausea, vomiting, and loss of appetite) reported in humans as well as the embryotoxic and teratogenic effects that have been observed in animals (16). Further work is also needed in order to determine the potential role of miltefosine in the treatment of invasive mycoses prior to the widespread clinical use of this agent.
ACKNOWLEDGMENTS
We thank Marcos Olivo for his assistance with the animal models.
This work was supported by a contract from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (N01-AI-25475) to T.F.P. T.C.S. is a Sydney Medical School Foundation Fellow.
N.P.W. has received research support from Pfizer, Schering-Plough, Merck, Basilea, and Astellas and has served on advisory boards for Merck, Astellas, Toyama, and Viamet. T.F.P. has received research grants to UT Health Science Center San Antonio from Astellas, Basilea, Merck, and Pfizer and has been a consultant for Astellas, Basilea, Merck, Pfizer, and Toyama. We have no other conflicts of interest.
Footnotes
Published ahead of print 19 November 2012
REFERENCES
- 1. Chayakulkeeree M, Perfect JR. 2006. Cryptococcosis. Infect. Dis. Clin. North Am. 20:507–544, v-vi [DOI] [PubMed] [Google Scholar]
- 2. Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, Harrison TS, Larsen RA, Lortholary O, Nguyen MH, Pappas PG, Powderly WG, Singh N, Sobel JD, Sorrell TC. 2010. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 50:291–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530 [DOI] [PubMed] [Google Scholar]
- 4. Dromer F, Mathoulin-Pelissier S, Launay O, Lortholary O. 2007. Determinants of disease presentation and outcome during cryptococcosis: the CryptoA/D study. PLoS Med. 4:e21 doi:10.1371/journal.pmed.0040021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lortholary O, Poizat G, Zeller V, Neuville S, Boibieux A, Alvarez M, Dellamonica P, Botterel F, Dromer F, Chene G. 2006. Long-term outcome of AIDS-associated cryptococcosis in the era of combination antiretroviral therapy. AIDS 20:2183–2191 [DOI] [PubMed] [Google Scholar]
- 6. Larsen RA, Leal MA, Chan LS. 1990. Fluconazole compared with amphotericin B plus flucytosine for cryptococcal meningitis in AIDS. A randomized trial. Ann. Intern. Med. 113:183–187 [DOI] [PubMed] [Google Scholar]
- 7. Saag MS, Powderly WG, Cloud GA, Robinson P, Grieco MH, Sharkey PK, Thompson SE, Sugar AM, Tuazon CU, Fisher JF, et al. 1992. Comparison of amphotericin B with fluconazole in the treatment of acute AIDS-associated cryptococcal meningitis. The NIAID Mycoses Study Group and the AIDS Clinical Trials Group. N. Engl. J. Med. 326:83–89 [DOI] [PubMed] [Google Scholar]
- 8. Bhattacharya SK, Jha TK, Sundar S, Thakur CP, Engel J, Sindermann H, Junge K, Karbwang J, Bryceson AD, Berman JD. 2004. Efficacy and tolerability of miltefosine for childhood visceral leishmaniasis in India. Clin. Infect. Dis. 38:217–221 [DOI] [PubMed] [Google Scholar]
- 9. Bhattacharya SK, Sinha PK, Sundar S, Thakur CP, Jha TK, Pandey K, Das VR, Kumar N, Lal C, Verma N, Singh VP, Ranjan A, Verma RB, Anders G, Sindermann H, Ganguly NK. 2007. Phase 4 trial of miltefosine for the treatment of Indian visceral leishmaniasis. J. Infect. Dis. 196:591–598 [DOI] [PubMed] [Google Scholar]
- 10. Jha TK, Sundar S, Thakur CP, Bachmann P, Karbwang J, Fischer C, Voss A, Berman J. 1999. Miltefosine, an oral agent, for the treatment of Indian visceral leishmaniasis. N. Engl. J. Med. 341:1795–1800 [DOI] [PubMed] [Google Scholar]
- 11. Soto J, Arana BA, Toledo J, Rizzo N, Vega JC, Diaz A, Luz M, Gutierrez P, Arboleda M, Berman JD, Junge K, Engel J, Sindermann H. 2004. Miltefosine for new world cutaneous leishmaniasis. Clin. Infect. Dis. 38:1266–1272 [DOI] [PubMed] [Google Scholar]
- 12. Soto J, Toledo J, Gutierrez P, Nicholls RS, Padilla J, Engel J, Fischer C, Voss A, Berman J. 2001. Treatment of American cutaneous leishmaniasis with miltefosine, an oral agent. Clin. Infect. Dis. 33:E57–61 [DOI] [PubMed] [Google Scholar]
- 13. Soto J, Toledo J, Valda L, Balderrama M, Rea I, Parra R, Ardiles J, Soto P, Gomez A, Molleda F, Fuentelsaz C, Anders G, Sindermann H, Engel J, Berman J. 2007. Treatment of Bolivian mucosal leishmaniasis with miltefosine. Clin. Infect. Dis. 44:350–356 [DOI] [PubMed] [Google Scholar]
- 14. Sundar S, Jha TK, Thakur CP, Engel J, Sindermann H, Fischer C, Junge K, Bryceson A, Berman J. 2002. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 347:1739–1746 [DOI] [PubMed] [Google Scholar]
- 15. Sundar S, Rosenkaimer F, Makharia MK, Goyal AK, Mandal AK, Voss A, Hilgard P, Murray HW. 1998. Trial of oral miltefosine for visceral leishmaniasis. Lancet 352:1821–1823 [DOI] [PubMed] [Google Scholar]
- 16. Sindermann H, Engel J. 2006. Development of miltefosine as an oral treatment for leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 100(Suppl 1):S17–S20 [DOI] [PubMed] [Google Scholar]
- 17. Tong Z, Widmer F, Sorrell TC, Guse Z, Jolliffe KA, Halliday C, Lee OC, Kong F, Wright LC, Chen SC. 2007. In vitro activities of miltefosine and two novel antifungal biscationic salts against a panel of 77 dermatophytes. Antimicrob. Agents Chemother. 51:2219–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Widmer F, Wright LC, Obando D, Handke R, Ganendren R, Ellis DH, Sorrell TC. 2006. Hexadecylphosphocholine (miltefosine) has broad-spectrum fungicidal activity and is efficacious in a mouse model of cryptococcosis. Antimicrob. Agents Chemother. 50:414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kesson AM, Bellemore MC, O'Mara TJ, Ellis DH, Sorrell TC. 2009. Scedosporium prolificans osteomyelitis in an immunocompetent child treated with a novel agent, hexadecylphospocholine (miltefosine), in combination with terbinafine and voriconazole: a case report. Clin. Infect. Dis. 48:1257–1261 [DOI] [PubMed] [Google Scholar]
- 20. CLSI 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A3, 3rd ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 21. Diamond DM, Bauer M, Daniel BE, Leal MA, Johnson D, Williams BK, Thomas AM, Ding JC, Najvar L, Graybill JR, Larsen RA. 1998. Amphotericin B colloidal dispersion combined with flucytosine with or without fluconazole for treatment of murine cryptococcal meningitis. Antimicrob. Agents Chemother. 42:528–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ding JC, Bauer M, Diamond DM, Leal MA, Johnson D, Williams BK, Thomas AM, Najvar L, Graybill JR, Larsen RA. 1997. Effect of severity of meningitis on fungicidal activity of flucytosine combined with fluconazole in a murine model of cryptococcal meningitis. Antimicrob. Agents Chemother. 41:1589–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson GR, III, Wiederhold NP, Najvar LK, Bocanegra R, Kirkpatrick WR, Graybill JR, Patterson TF. 2012. A murine model of Cryptococcus gattii meningoencephalitis. J. Antimicrob. Chemother. 67:1432–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. NAS 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 25. Kim JH, Jung SY, Lee YJ, Song KJ, Kwon D, Kim K, Park S, Im KI, Shin HJ. 2008. Effect of therapeutic chemical agents in vitro and on experimental meningoencephalitis due to Naegleria fowleri. Antimicrob. Agents Chemother. 52:4010–4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuo X, Djordjevic JT, Bijosono Oei J, Desmarini D, Schibeci SD, Jolliffe KA, Sorrell TC. 2011. Miltefosine induces apoptosis-like cell death in yeast via Cox9p in cytochrome c oxidase. Mol. Pharmacol. 80:476–485 [DOI] [PubMed] [Google Scholar]
- 27. Berman J, Bryceson AD, Croft S, Engel J, Gutteridge W, Karbwang J, Sindermann H, Soto J, Sundar S, Urbina JA. 2006. Miltefosine: issues to be addressed in the future. Trans. R. Soc. Trop. Med. Hyg. 100(Suppl 1):S41–S44 [DOI] [PubMed] [Google Scholar]
- 28. Dorlo TP, van Thiel PP, Huitema AD, Keizer RJ, de Vries HJ, Beijnen JH, de Vries PJ. 2008. Pharmacokinetics of miltefosine in Old World cutaneous leishmaniasis patients. Antimicrob. Agents Chemother. 52:2855–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arndt D, Zeisig R, Fichtner I, Teppke AD, Fahr A. 1999. Pharmacokinetics of sterically stabilized hexadecylphosphocholine liposomes versus conventional liposomes and free hexadecylphosphocholine in tumor-free and human breast carcinoma bearing mice. Breast Cancer Res. Treat 58:71–80 [DOI] [PubMed] [Google Scholar]
- 30. Breiser A, Kim DJ, Fleer EA, Damenz W, Drube A, Berger M, Nagel GA, Eibl H, Unger C. 1987. Distribution and metabolism of hexadecylphosphocholine in mice. Lipids 22:925–926 [DOI] [PubMed] [Google Scholar]