

Abstract
Exposure to multiple small doses of hepatitis B virus (HBV) is a frequent occurrence in high-risk groups, including close relatives of infected individuals, primary care givers, and intravenous drug users. It remains uncertain whether such repeated contact may culminate in a symptomatic infection coinciding with hepatitis in individuals not immunoprotected. In this study, we evaluated consequences of multiple exposures to small, liver-nonpathogenic amounts of infectious hepadnavirus in the woodchuck model of hepatitis B. Virus-naïve animals were intravenously injected with 6 weekly doses of 110 DNase digestion-protected virions of woodchuck hepatitis virus (WHV), injected again with 6 weekly 110-virion doses after 7.5 months, and then challenged or not with a liver-pathogenic dose of 1.1 × 106 virions of the same inoculum. The data revealed that two rounds of such repeated exposure did not result in serologically evident infection or hepatitis. However, a low-level WHV DNA-positive infection accompanied by a WHV-specific T cell response in the absence of antiviral antibody reactivity was established. The kinetics of the virus-specific and mitogen-induced (generalized) T cell responses and the inability to induce immunoprotection against challenge with a large, liver-pathogenic virus dose were closely comparable to those previously reported for occult infection initiated by a single liver-nonpathogenic dose of WHV. Thus, repeated exposures to small quantities of hepadnavirus induce molecularly evident but serologically silent infection that does not culminate in hepatitis or generate immune protection. The findings imply that the HBV-specific T cell response encountered in the absence of serological markers of infection likely reflects ongoing occult infection.
INTRODUCTION
Multiple exposures to small amounts of hepatitis B virus (HBV) are of frequent occurrence in both occupational and nonoccupational settings (1–6). Routine vaccination against HBV prevents infection potentially caused by such exposure. However, consequences of repeated contacts with small quantities of HBV of individuals not immunoprotected against the virus are not recognized and it is unknown whether such exposure can culminate in a serologically detectable infection and hepatitis.
The data acquired from the woodchuck model of hepatitis B showed that exposure to a singular low dose (i.e., <1,000 virions) of woodchuck hepatitis virus (WHV), which is a close relative of HBV (7–9), establishes serologically undetectable infection in which the virus genome and its replication are detectable when nucleic acid amplification assays of enhanced sensitivity are applied (10–13). This molecularly evident but immunovirologically silent infection was designated primary occult infection (POI) (12, 14). POI was originally uncovered in offspring born to woodchuck dams convalescent from experimental acute hepatitis (AH) (15). In these animals, WHV DNA was identified in serum and in the immune system but not in the liver and virus replication progressed at a very low level in the absence of detectable serum WHV surface antigen (WHsAg) and antibodies to WHV core antigen (anti-WHc) and in the context of normal liver morphology. This form of silent WHV infection was subsequently reproduced in adult animals by intravenous (i.v.) injection with WHV doses containing less than 1,000 DNase digestion-protected virions (11, 12). WHV replication was again restricted to the lymphatic system and progressed without histologically apparent liver injury. In the most recent study on the lifelong consequences of POI, we uncovered that about 20% of woodchucks injected with a single 100-virion dose of WHV developed hepatocellular carcinoma (HCC) that was preceded by the appearance of the viral genome and its replication markers (i.e., covalently closed circular DNA [cccDNA] and mRNA) in the liver, clearly demonstrating pathogenic relevance of persistent POI (P. M. Mulrooney-Cousins and T. I. Michalak, unpublished data). In general, serum WHV loads during POI do not exceed 100 to 200 virus genome equivalents (vge) or copy numbers/ml, the virus has the wild-type sequence, and it retains liver-pathogenic competence when administered to virus-naïve animals at doses greater than 103 virions (9, 14) (Mulrooney-Cousins and Michalak, unpublished). It was also established that POI induces a WHV-specific T cell response but not an antiviral antibody response and, importantly, that the animals are not protected from reinfection and hepatitis when challenged with liver-pathogenic doses of WHV (i.e., >103 virions) (11, 12, 15).
Although the existence of POI in humans has not yet been thoroughly investigated, the detection of HBV DNA in the blood and/or liver samples from individuals without serological markers of infection, i.e., HBV surface antigen (HBsAg) and/or antibodies to HBV core antigen (anti-HBc), particularly those displaying an HBV-specific T lymphocyte response (16), strongly argues that this type of infection naturally occurs. The prevalence of serum/plasma HBV DNA-reactive and HBsAg- and anti-HBc-negative infection was reported between 0.07% and 7.6% of tested subjects from different areas of endemicity (2, 17–20).
“Classical” occult HBV infection is defined by the presence of HBV DNA in serum, lymphoid cells, and/or liver tissue in the absence of serum HBsAg (14, 21, 22). Resolution of acute hepatitis (AH) is commonly followed by occult HBV infection in which traces of virus persist for decades, if not for life. This essentially silent form of HBV infection is accompanied by the detection of anti-HBc and frequently antibodies to HBsAg (anti-HBs) and by a vigorous HBV-specific T cell response (19, 23–25). Studies of woodchucks followed for life after a self-limited episode of AH delineated virological and immunological characteristics and pathological outcomes of this form of occult infection, subsequently called secondary occult infection (SOI) (9, 13, 14). Among others, it was established that WHV replication in SOI continuing after resolution of symptomatic AH progresses throughout life in both the liver and the immune system, the assembled virions are infectious and liver pathogenic when administered i.v. at doses greater than 1,000 virions, and resolution of hepatitis is rarely complete but normally followed by intermittent minimal to moderate inflammation with periods of nearly normal or normal liver morphology (13). However, HCC develops in approximately 20% of animals with SOI (13, 26). Notably, immunosuppression during SOI can reactivate infection leading to the reappearance of serum WHsAg (27), as is seen in patients with occult HBV infection (28–30). As in POI, the load of circulating WHV is rarely greater than 200 vge/ml, but in contrast to POI, the animals are protected from challenge with liver-pathogenic doses of virus, i.e., >103 virions (10).
Both innate and adaptive immunological responses initiated by hepadnaviral infection are mediators and modulators of associated liver disease. The qualitative attributes of virus-specific T cell responses appear to orchestrate the outcome of hepadnaviral hepatitis. Thus, HBV-infected patients who develop strong and polyclonal virus-specific and T helper type 1 (Th1) responses during AH tend to resolve hepatitis. This coincides with the Th1 cytokine profile, which includes augmented expression of gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and interleukin-2 (IL-2) (31–34). In contrast, patients with a weak and narrowly restricted T cell response tend to develop chronic hepatitis. This is accompanied by expression of type 2 cytokines, such as IL-4 and IL-10 (33, 35, 36). Other studies, including those from this laboratory, delineated the kinetics of antiviral T cell and cytokine responses in POI (11, 16, 37, 38).
The previous studies characterizing molecular and immunological attributes of POI have prompted questions regarding a potential cumulative pathogenic effect of the exposure to multiple small doses of hepadnavirus and the properties of an immune response induced by such exposure. Applying the woodchuck model of HBV infection, we addressed this potentially clinically relevant issue by repeated injections of virus-naïve animals with amounts of WHV known to cause POI.
MATERIALS AND METHODS
Animals.
WHV-naïve, adult woodchucks (Marmota monax) were housed in the Woodchuck Hepatitis Research Facility at Memorial University, St. John's, Newfoundland, Canada. Their WHV-naïve status was verified by the absence of WHV DNA, as analyzed by highly sensitive WHV-specific nested PCR-nucleic acid hybridization (PCR/NAH) assays (sensitivity, <10 vge/ml or <10 vge/μg of total DNA) in randomly selected liver, peripheral blood mononuclear cell (PBMC), and serum samples, and by repeated serum negativity for WHsAg and anti-WHc antibodies as reported previously (10, 13, 15, 39). Experimental protocols were approved by the Institutional President's Committee on Animal Bioethics and Care.
Inoculation, challenge, and rechallenge with WHV.
It was previously established that i.v. injection of a single WHV dose containing less than 1,000 DNase digestion-protected virions invariably induce POI in WHV-naïve, healthy woodchucks (11, 12, 37, 40). This was consistently observed after administration of low doses of different WHV inocula, i.e., WHV/tm3 (GenBank accession number AY334075) (12, 40), WHV/tm4 (GenBank accession number GU734791) (38), and WHV/tm5 (Mulrooney-Cousins and Michalak, unpublished). In the current study, four woodchucks (3 males and 1 female) were i.v. injected with 1.1 × 102 DNase digestion-protected vge (virions) of WHV/tm3 inoculum at weekly intervals for 6 weeks (round 1 of injections) (Fig. 1). After 30 weeks postinjection (w.p.i.) with the first dose of WHV, the same animals were challenged with the same weekly dose of WHV/tm3 for 6 weeks (round 2 of injections) (Fig. 1). This experimental group was designated L6L6 (n = 4). At 49 w.p.i., two of the animals (1/M and 2/F) were i.v. injected with 0.5 ml of sterile phosphate-buffered saline (PBS), pH 7.4 (control subgroup 6L6LN), while the remaining two (3/M and 4/M) were rechallenged with a liver-pathogenic WHV/tm3 dose of 1.1 × 106 virions known to consistently initiate serologically detectable WHV infection and hepatitis (subgroup 6L6LM) (see Fig. 2A). Moreover, two healthy woodchucks (5/F and 6/M) were i.v. infected with 1.1 × 106 WHV/tim3 virions. Subsequently, 5/F was injected with 0.5 ml of PBS (subgroup MN), while 6/M was injected with the same 1.1 × 106 virions of WHV/tm3 (subgroup MM) at 30 w.p.i. (Fig. 2B) In addition, the data available from 8 other woodchucks infected with a single low dose of WHV/tm3 (i.e., 50 virions) and then challenged with a high dose of WHV/tm3 (1.1 × 1010 virions; n = 4) or a low dose of WHV/tm3 (50 virions; n = 3) or injected with PBS (n = 1) served as a comparison.
Fig 1.

Schematic representation of the experimental protocol applied to infect and challenge woodchucks with multiple small, liver-nonpathogenic doses of WHV. Four WHV-naïve animals were i.v. injected at weekly intervals with 6 doses of 1.1 × 102 virions each of WHV/tm3 inoculum (round 1). Thirty weeks after the first virus injection (infection phase), the animals were challenged with another 6 weekly doses of 1.1 × 102 virions each of the same inoculum (round 2). Thirteen weeks following the completion of round 2 (challenge phase), the animals were rechallenged with 1.1 × 106 virions of WHV/tm3 inoculum or injected with 0.5 ml of sterile PBS and followed for up to 75 weeks after the first virus injection (rechallenge phase).
Fig 2.
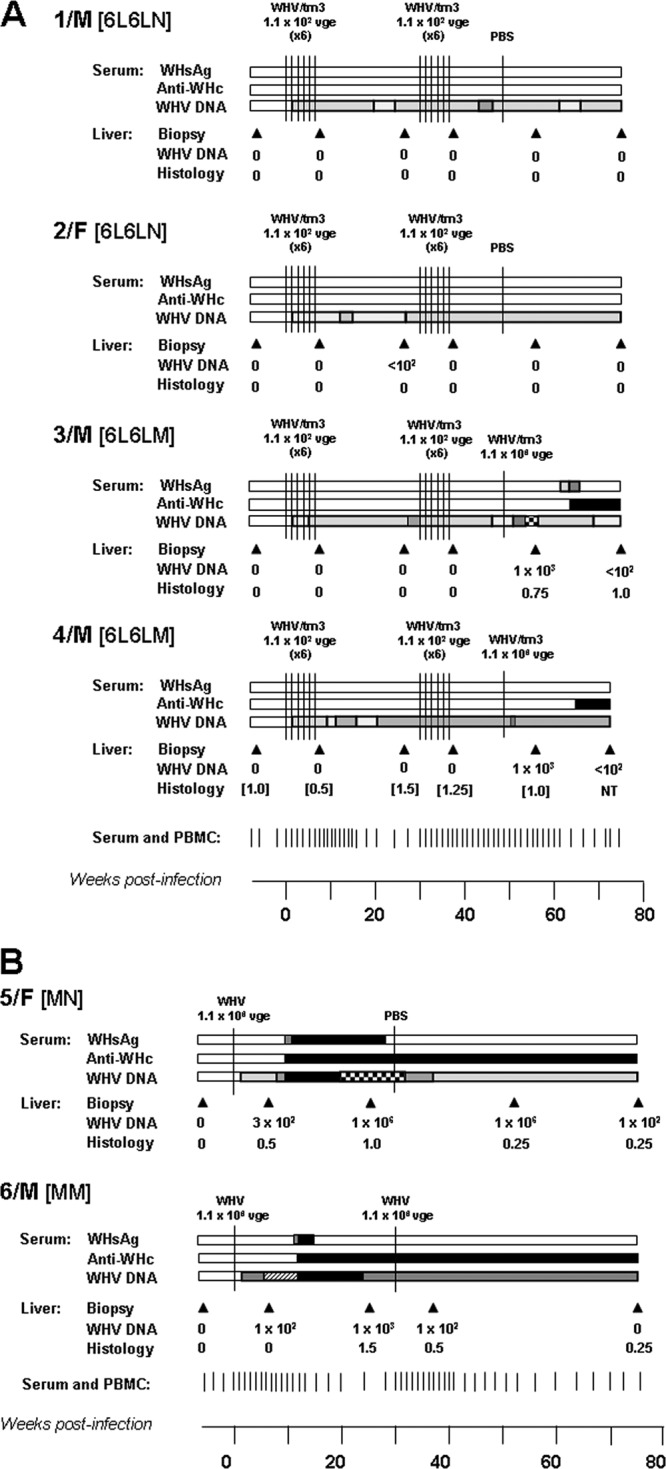
Profiles of serological markers of WHV infection and WHV DNA detection in serum and liver tissue samples and the results on liver histology in woodchucks repeatedly injected and challenged with small, liver-nonpathogenic doses of WHV and in control woodchucks infected with a liver-pathogenic dose of the same virus. (A) Four initially healthy, WHV-naïve animals were i.v. injected with two rounds of 6 weekly doses of 1.1 × 102 virions each and then injected with PBS or challenged with a liver-pathogenic virus dose of 1.1 × 106 virions of WHV/tm3 inoculum. (B) Two initially healthy, control woodchucks were injected with 1.1 × 106 virions of WHV/tm3 inoculum and subsequently injected with PBS or challenged with the same virus dose. For each animal, the time points of WHV or PBS injections are indicated by vertical solid lines. The serum and PBMC samples were collected at the time points marked by the short vertical lines at the bottom of panels A and B. The appearance and duration of the positivity of WHsAg, anti-WHc, and WHV DNA in the serum samples are presented by horizontal bars. The approximate levels of serum WHsAg are depicted as follows: white bars, <3.5 ng/ml; off-white bars, 3.5 to10 ng/ml; light-gray bars, 10 to 200 ng/ml; dark-gray bars, 200 to 500 ng/ml; and black bars, >500 ng/ml. For anti-WHc, the white and black bars reflect the absence and presence of the antibodies, respectively. Anti-WHs were intermittently identified in 3/M, 5/F, and 6/M after the clearance of serum WHsAg (data not shown). The estimated levels of serum WHV DNA are shown as follows: white bars, not detected; off-white bars, <10 vge/ml; light-gray bars, 10 to 102 vge/ml; medium-gray bars, 102 to 2 × 102 vge/ml, dark-gray bars, 2 × 102 to 103 vge/ml; densely hatched bars, 103 to 104 vge/ml; sparsely hatched bars, 104 to 105 vge/ml; checkered bars, 105 to 106 vge/ml; and black bars, >106 vge/ml. The liver biopsy samples were collected at the time points indicated by the solid black arrowheads, and estimated loads of WHV DNA detected are presented in vge/μg of total liver DNA. Liver morphological alterations graded on a scale from 0 to 3 are presented for each liver biopsy sample.
Sample collection and PBMC isolation.
Blood samples were collected on sodium-EDTA or without the addition of anticoagulant prior to infection weekly up to 10 weeks after the last injection with virus, biweekly up to 20 weeks, and then approximately monthly until autopsy. Sera were separated from untreated blood and stored at −20°C. PBMC obtained at the time points indicated in Fig. 2 were isolated from EDTA-treated samples on Ficoll-Paque Plus (Amersham Pharmacia Biotech, AB, Uppsala, Sweden) as reported (41). PBMC viability assessed by the trypan blue dye exclusion method normally exceeded 95%.
Serological and molecular markers of WHV infection.
Serial serum samples were examined for WHsAg, anti-WHc, and antibodies to WHsAg (anti-WHs) using specific enzyme-linked immunosorbent assays reported previously (10, 12, 13). WHV DNA in serum and liver samples was evaluated by nested PCR/NAH assays with WHV core (C), surface (S), and X gene-specific primers (sensitivity, <10 vge/ml or <10 vge/μg of total DNA) (13, 15). Routinely, DNA extracted from 100 μl of serum or one μg of total DNA was used for the first round of PCR amplification. If WHV DNA was not detectable after nested PCR/NAH, DNA isolated from 200 μl of serum or up to 4 μg of total DNA from livers was used for direct PCR. To confirm the status of WHV replication in circulating and organ lymphoid cells and in hepatic tissue, the expression of viral mRNA was evaluated in selected samples by reverse transcription (RT)-PCR/NAH (sensitivity, <10 vge/ml) as described previously (42). Briefly, total RNA was extracted with TRIzol reagent (Invitrogen Life Technologies, Burlington, Ontario, Canada), treated with DNase (an RNase-free DNase digestion kit; Sigma-Aldrich, Oakville, Ontario, Canada), and reversely transcribed to cDNA (10, 42). The resulting cDNA was amplified by PCR with WHV X gene-specific primers. Each test RNA sample treated under the same conditions in the absence of reverse transcriptase served as a DNA contamination control. RNAs extracted from PBMCs and livers of woodchucks prior to infection with WHV and from liver of an animal with serum WHsAg-positive chronic hepatitis were used as negative and positive controls, respectively. For NAH, blots were exposed to radiolabeled, complete recombinant WHV DNA as a probe (13, 15). In addition, the presence of WHV covalently closed circular DNA (cccDNA) was assessed in selected lymphoid cell and liver tissue samples by nested PCR/NAH (sensitivity, ∼100 copies/ml) using conditions and controls as reported in detail previously (10, 12). As a biochemical measure of liver injury, serum levels of sorbitol dehydrogenase (SDH) were determined by an appropriate spectrophotometric assay (11).
Liver tissue samples and histopathology.
Up to six liver biopsy samples were collected from each animal by surgical laparotomy or at autopsy at the time point indicated in Fig. 2. At autopsy, multiple tissue fragments from all lobes of the liver were preserved for analysis. A part of each sample was predestined for nuclei acid extraction and the remaining portion routinely processed to paraffin and stained as described previously (43). Hepatic tissue lesions were graded on the numerical scale of 0 to 3 in 0.25 increments to report the degree of hepatitis (13, 39, 44).
WHV antigens and T cell mitogen.
Recombinant WHV core protein (rWHc), e protein (rWHe), and X protein (rWHx), used to measure WHV-specific T cell proliferative responses, were produced in pET41b(+) Escherichia coli expression system (Novagen, Darmstadt, Germany) and purified using histidine-tagged affinity chromatography (Qiagen, Mississauga, Canada) as described in detail previously (37, 45). These antigens were examined for the presence of potential bacterial contaminants nonspecifically inducing T cell proliferation in vitro and were found free of such impurities as reported (37, 45). In addition, WHsAg was purified from pooled serum of a woodchuck with chronic WHV infection (12, 37), and a synthetic peptide encompassing the WHV T cell immunodominant epitope located between amino acids 97 and 110 of WHV core protein (WHc97-110) (7, 46) was synthesized (Synprep Corporation, Dublin, CA). To assess WHV-specific T cell proliferation, the above-mentioned recombinant WHV antigens and WHsAg were used at concentrations of 1 and 2 μg/ml, while the WHc97-110 peptide was used at concentrations of 5 and 10 μg/ml. Furthermore, the generalized (nonspecific) proliferative capacity of lymphocytes was measured in response to stimulation with concanavalin-A (ConA; Pharmacia Fine Chemicals, Uppsala, Sweden). For this purpose, five serial 2-fold concentrations of ConA ranging from 1.2 to 20 μg/ml were used as reported (11, 37). We have previously established that the carboxyfluorescein diacetate succinimidyl ester (CFSE)-based assay is significantly more sensitive in detecting the WHV-specific immune response due to its ability to visualize even the minute frequency of activated T cells than the previously used adenine incorporation assay (37). On the other hand, the adenine incorporation assay is more convenient than the CFSE-based assay for its analysis of a strong T cell activation usually occurring after stimulation with mitogens, such as ConA. Considering the above, CFSE-based and adenine incorporation assays were used to quantify WHV-specific and mitogen-induced T cell activation, respectively, as in our previous studies (11, 37, 38).
CFSE-based T cell proliferation assay.
The WHV-specific T cell proliferative response after stimulation with WHV antigens or the WHc97–110 peptide was assessed by the CFSE-based flow cytometric assay as described previously (41). Briefly, freshly isolated PBMCs were labeled with 5- and 6-CFSE (Molecular Probes, Eugene, OR) at a final concentration of 1 μM and cultured at a density of 3 × 105 cells/well in 48-well tissue culture plates in triplicate with tested concentrations of WHV antigens or WHc97–110 or with medium alone as a control. The cells were incubated for 5 days at 37°C, harvested, and washed in PBS with 1 mM EDTA (PBS-EDTA). The data were acquired with a FACSCalibur flow cytometer (Becton, Dickinson, Franklin Lakes, NJ). The halving of CFSE fluorescence was deconvoluted using CellQuest Pro software (Becton, Dickinson) (41). The cell division index (CDI) was defined by dividing the percentage of cells with halved CFSE fluorescence after stimulation with a WHV antigen by the percentage of cells with halved CFSE fluorescence cultured in medium only. CDI values of ≥3.1 for rWHc, rWHe, and rWHx and ≥2.1 for WHsAg and the WHc97-110 peptide were considered a measure of the WHV-specific T cell proliferative response (11). However, for clarity of the presentation, only the cutoff value of ≥3.1 was marked in the graphs, showing the WHV-specific T cell response.
Adenine incorporation T lymphocyte proliferation assay.
The generalized proliferative capacity of woodchuck lymphocytes in response to nonspecific (mitogen) stimulation was evaluated using ConA and 3[H]-adenine incorporation assay as described previously (41). Briefly, freshly isolated woodchuck PBMCs were cultured at a density of 1 × 105 cells/well in a 96-well tissue culture plate in the presence of different concentrations of ConA, as indicated above, or medium alone in triplicate. After 5 days in culture, cells were pulsed with 0.1 μCi of [3H]adenine (Amersham Pharmacia Biotech, Uppsala, Sweden) for 12 to 18 h and harvested, and counts per minute (cpm) were measured in each test and in the control wells. The mean cpm values were calculated by averaging the cpm values from the respective triplicate wells. The stimulation index (SI) was calculated by dividing the mean cpm obtained after a mitogenic stimulation by the mean cpm observed without any stimulation (medium only) (37).
Real-time RT-PCR analysis.
Sequential PBMC and liver biopsy samples collected prior to inoculation with WHV and after infection (see Fig. 2) were evaluated for the expression of selected cytokine genes and CD3, as a T cell marker, via real-time reverse transcription (RT)-PCR using a LightCycler (Roche Diagnostics, Mannheim, Germany) (37, 42). In the case of PBMCs, 5 × 105 to 1 × 106 of freshly isolated, unstimulated cells were suspended in TRIzol (Invitrogen) and stored at −20°C. Liver tissue samples were stored at −80°C until RNA isolation using TRIzol reagent. After the collection of all experimental samples, RNA was simultaneously extracted and reversely transcribed to cDNA as reported (37, 42). The expression of woodchuck IFN-α, IFN-γ, TNF-α, IL-2, IL-4, IL-10, and IL-12 was quantified using the equivalent of 50 ng of total RNA and the woodchuck gene-specific primers as reported (37, 42). Transcription of the genes was normalized against expression of woodchuck β-actin. To define the fold increase or decrease, the expression levels detected after inoculation, challenge, or rechallenge with WHV were compared with those found for the same animal prior to infection as reported (37).
Statistical analysis.
Statistical analysis was done using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). The two-tailed, unpaired Student t test, with a 95% confidence interval, was used to compare the means of the groups. P values of ≤0.05 were considered to be statistically significant. The differences marked with one asterisk were significant at a P value of <0.05, with two asterisks were significant at a P value of <0.005, and with three asterisks were significant at a P value of <0.0001.
RESULTS
Repeated exposure to small doses of WHV does not culminate in serologically detectable infection.
As outlined in Fig. 1, woodchucks were first injected weekly with 110 virions of WHV for 6 weeks (round 1), observed for 30 w.p.i. (infection phase), challenged with another six 110-virion doses of the same inoculum (round 2), and followed for several weeks thereafter (challenge phase). Subsequently, the animals were rechallenged with a liver-pathogenic dose of WHV (subgroup 6L6LM) or injected with PBS (subgroup 6L6LN). As shown in Fig. 2A, these repeated exposures to a very low WHV amount established a molecularly evident infection, as was evidenced by the detection of WHV DNA in serum beginning from one w.p.i. The serum WHV DNA load did not increase despite weekly injections and remained at a level not exceeding 2 × 102 vge/ml during follow-up or until challenge with 1.1 × 106 virions (Fig. 2A). In parallel, WHV DNA was detectable in serial PBMC (data not shown) but not in liver biopsy samples (Fig. 2A), while WHV cccDNA and mRNA indicative of virus active replication were detected in PBMCs and lymphatic organs but not, as expected, in liver tissue fragments collected at autopsy, as illustrated in Fig. 3 for 1/M and 2/F, followed for 75 w.p.i. after the first exposure to virus. All animals remained serum WHsAg negative and anti-WHc and anti-WHs nonreactive during the entire observation period or until challenge with a liver-pathogenic virus dose (Fig. 2A). These findings were consistent with those previously reported for POI experimentally induced by a single low dose of WHV (11) and in offspring born to mothers convalescent from WHV hepatitis (15). In contrast, after rechallenge with 1.1 × 106 virions (subgroup 6L6LM), animal 3/M became transiently serum WHsAg positive and permanently anti-WHc reactive from 15 weeks after rechallenge onwards (Fig. 2A). After a decrease of serum WHsAg to an undetectable level, the animal became intermittently reactive for anti-WHs until the end of follow-up (data not shown). Another animal, 4/M, remained WHsAg negative but developed anti-WHc (Fig. 2A). The serum WHV DNA level transiently increased in both woodchucks after rechallenge but decreased to about 1 × 102 vge/ml and persisted at this level to the end of follow-up (Fig. 2A). As anticipated, the injection of 1/M and 2/F with PBS (subgroup 6L6LN) at 50 w.p.i. did not alter the profile of serological and molecular profiles of WHV infection (Fig. 2A).
Fig 3.
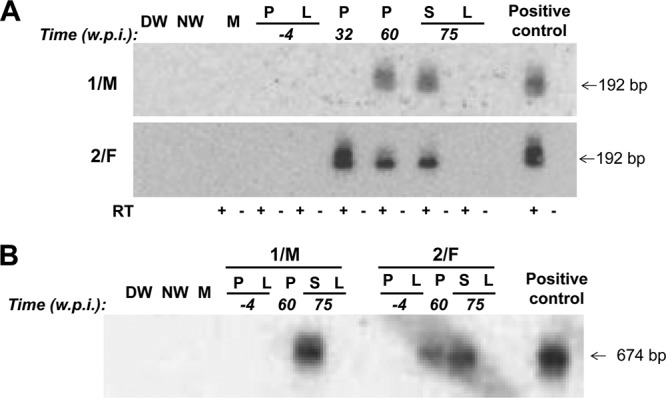
Detection of WHV RNA and cccDNA in lymphoid cells and tissues (P, PBMC; S, spleen) in the absence of liver (L) engagement in the 1/M and 2/F woodchucks injected with two rounds of multiple low doses of WHV and followed for up to 75 weeks after the first virus injection prior to autopsy. (A) Identification of WHV RNA. DNase-treated total RNA was reversely transcribed (RT+) or not reversely transcribed (RT−) prior to PCR amplification with WHV X gene-specific primers. Positive 192-bp amplicons obtained after nested PCR were identified by NAH. (B) Identification of WHV cccDNA. Total DNA was digested with a single-strand-specific nuclease and amplified by nested PCR with primers spanning the nick region of the WHV genome. NAH was used to confirm the specificity of the 674-bp amplicons and validate controls. Contamination controls shown in panels A and B consisted of water added to direct (DW) and nested (NW) amplification reactions instead of test cDNA or DNA, respectively. Mock (M) samples were extracted and amplified in parallel with test samples. RNA and DNA from PBMC and liver samples collected from the woodchucks prior to first exposure to WHV were used as negative controls, while RNA and DNA from the liver of a serum WHsAg-positive animal with chronic infection were included as a positive control.
Animals 5/F and 6/M inoculated with a liver-pathogenic dose of 1.1 × 106 virions developed classical, transiently serum WHsAg-positive infection, coinciding with a transient increase in the serum SDH level, and anti-WHc (Fig. 2B). After the decrease in WHsAg to an undetectable level, anti-WHs were periodically detected in both animals during a 75-w.p.i. observation period (data not shown). Serum WHV DNA was detectable from 1 w.p.i. and persisted to the end of follow-up. Challenge of 5/M with 1.1 × 106 virions of WHV/tm3 (subgroup MM) or injection of 6/F with PBS (subgroup MN) at 30 w.p.i. did not alter serological indicators of WHV infection or the serum WHV DNA load (Fig. 2B).
Repeated small doses of WHV fail to induce hepatitis.
Essentially, all liver biopsy samples collected after the first and second rounds of injections with low WHV doses were WHV DNA negative even when up to 4 μg of total liver DNA was used for a direct round of PCR and despite employment of highly sensitive detection assays (Fig. 2A). A liver tissue sample collected at 27 w.p.i. from 2/F that contained 10 to 1 × 102 vge/μg total DNA was the exception. In contrast, liver biopsy samples from 3/M and 4/M (subgroup 6L6LM) acquired after rechallenge with 1.1 × 106 virions were WHV DNA reactive and contained between 1 × 102 and 1 × 103 vge/μg DNA (Fig. 2A). In 5/M and 6/F controls, the hepatic load of WHV DNA ranged between 1 × 102 and 1 × 106 vge/μg DNA and decreased to an estimated 1 × 102 vge/μg DNA at the end of follow-up despite the fact that 5/M was challenged with another 1.1 × 106-vge dose of WHV/tm3 at 30 w.p.i. (Fig. 2B).
The histological examination of liver biopsy samples from animals injected with weekly low doses of WHV showed an entirely normal morphology (Fig. 2A). This finding was consistent with observations previously reported for either naturally acquired POI (15) or POI experimentally induced with a single small dose of WHV (11, 12). An unexplained moderate to minimal liver inflammation in 4/M that was diagnosed prior to initiation of the infection experiment and protracted throughout the investigation period was the exception (Fig. 2A); however, the serum SDH level in this animal was consistently normal (data not shown). The woodchuck 3/M, which after two rounds of injections with low WHV doses was rechallenged with a liver-pathogenic dose of 1.1 × 106 virions (Fig. 2A), as well as the woodchucks 5/F and 6/M, which were also inoculated with 1.1 × 106 vge of WHV, displayed moderate to minimal liver inflammation (Fig. 2B). Elevated SDH levels were transiently detected at the time of serum WHsAg positivity in 3/M and 5/F (data not shown).
Repeated exposure to small WHV doses induces a virus-specific T cell response but not an antibody response.
We have previously reported that the injection of virus-naïve woodchucks to a single low (50 vge), medium (∼1 × 106 vge), or high (>1 × 1010 vge) dose of WHV induces a WHV-specific T cell response of essentially comparable kinetics and magnitude (11, 37); however, only liver-pathogenic doses of WHV equal to or greater than 1 × 103 virions caused infection accompanied by hepatitis and the appearance of virus-specific antibodies (11, 37). Hence, it was of interest to determine whether multiple exposures to small amounts of WHV induce a T cell immune response with the kinetics comparable to that induced by injection with a single dose of virus and alter T cell reactivity following challenge with WHV. For this purpose, serial PBMC samples collected throughout the experiment were stimulated with different WHV antigens and analyzed in the CFSE-based T cell proliferative assay. As shown in Fig. 4A and also in Fig. S1A in the supplemental material, all woodchucks in the 6L6L group mounted a WHV-specific T cell response, which was clearly apparent from 7 to 8 w.p.i. This WHV-specific T cell reactivity fluctuated during the 30-w.p.i. follow-up, occasionally falling to undetectable levels. Similar to the first round, a WHV-specific T cell response reappeared in 6 to 7 weeks after administering the first dose of the second round of injections and intermittently persisted during the whole observation period up to 75 w.p.i. (subgroup 6L6LN; 1/M and 2/F) or until rechallenge with a liver-pathogenic dose of WHV (subgroup 6L6LM; 3/M and 4/M) (Fig. 4A; see also Fig. S1A). As already indicated, the animals did not develop an anti-WHc or anti-WHs antibody response despite the presence of evident virus-specific T cell reactivity.
Fig 4.

Kinetics of WHV-specific T cell proliferative response in woodchucks exposed to two rounds of multiple low doses of WHV and in control animals infected with a liver-pathogenic dose of the same virus. Serial PBMC samples were stimulated in vitro with four different WHV antigens or the WHc97–110 peptide and analyzed for resultant proliferation using a CFSE-based proliferation assay to define the cell division index (CDI). The dashed horizontal line represents the cutoff value for the positive WHV-specific T response, the solid gray shaded area and the groups of 6 arrows under each panel show the time points at which animals were repeatedly injected with 1.1 × 102 virions, and a singular arrow shows the time point at which 1.1 × 106 virions or PBS was administered. (A) Profiles of WHV-specific T cell responses in individual animals injected with two rounds of 6 weekly 1.1 × 102-virion doses of WHV and then challenged or not challenged with a liver-pathogenic WHV dose of 1.1 × 106 virions. (B) Profiles of WHV-specific T cell proliferative responses in two woodchucks injected with a single liver-pathogenic dose of 1.1 × 106 virions and challenged with the same virus dose or injected with PBS. The results are presented as CDIs where the highest CDI value given by stimulation with any of the WHV antigens tested or the WHc97–110 peptide at a given time point was considered a measure of the WHV-specific T cell response. The subgroup designations are explained in Materials and Methods.
Woodchucks 3/M and 4/M, which were rechallenged with 1.1 × 106 vge, showed a strong and multispecific WHV-specific T cell response from 6 to 8 weeks after rechallenge. The response was delayed, as observed after round 1 and round 2 of WHV injections and reported previously (11). Of note is that the profile of the WHV-specific T cell response in 4/M, which demonstrated histological evidence of preexisting, WHV-unrelated liver inflammation, was similar to that in 3/M.
Control animals 5/F and 6/M, inoculated with a single dose of 1 × 106 virions, showed a virus-specific T cell response from 6 to 8 w.p.i., which was protracted until 18 w.p.i. and subsequently decreased, but remained intermittently detectable until the end of follow-up. 6/M, challenged with the same dose of WHV/tm3 given at 30 w.p.i., displayed, as expected, an increase in a WHV-specific T cell response, which peaked between 4 and 6 weeks postchallenge (Fig. 4B; see also Fig. S1B in the supplemental material). Both animals developed anti-WHc and remained antibody reactive and, as indicated previously, periodically displayed detectable anti-WHs until the end of follow-up. Taken together, the data showed that the kinetics and magnitude of a virus-specific T cell proliferative response following repeated exposure to small amounts of WHV was in fact closely comparable to that previously seen in the animals infected with a single low, medium, or high dose of WHV (11, 37). Therefore, the results implied that, irrespective of the quantity and number of exposures to infectious virus, a stereotypic specific T cell response develops that appears late, between 6 to 8 weeks postexposure, and persists intermittently for a long time after exposure.
The first virus dose in a series of multiple small-dose injections of WHV augments a nonspecific lymphocyte proliferative response.
In previous studies, the generalized (mitogen-induced) lymphocyte proliferative response was found to be significantly augmented immediately following primary exposure to WHV independently of virus dose, and this event is associated with the activation-induced death in lymphocytes (37, 38). In the current study, freshly isolated serial PBMCs collected during the infection, challenge, and rechallenge phases (Fig. 1) were mitogen stimulated and their proliferation was measured using an adenine incorporation assay. As shown in Fig. 5A, immediately after the first of six injections with WHV in round 1, lymphocytes from all four woodchucks mounted a significantly greater (P > 0.001) response to ConA than during the preinfection period. This heightened reactivity was not enhanced further by administration of subsequent 1.1 × 102-virus doses (Fig. 5A; see also Fig. S2A in the supplemental material). This generalized T cell response was evident in all 4 animals between 1 and 3 w.p.i. and then subsided, although not uniformly, between 4 and 6 w.p.i. Similarly, as it was previously found (11, 37), a decrease in the nonspecific T cell response preceded the appearance of WHV-specific T cell reactivity (Fig. 5A; see also Fig. S2A). A similar profile of a mitogen-induced T cell response was observed after the first of six injections given in round 2 (Fig. 5A; see also Fig. S2A).
Fig 5.

Comparative kinetics of mitogen-induced (generalized) and WHV-specific T cell responses in woodchucks injected with multiple liver-nonpathogenic doses of WHV and in control animals infected with a liver-pathogenic dose of WHV. PBMCs were stimulated with five serial concentrations of ConA ranging from 1.2 to 20 μg/ml, and the resultant proliferation was measured by an adenine incorporation proliferation assay to determine the stimulation index (SI). The SI values obtained for all five concentrations of ConA were averaged to define the mean mitogenic stimulation index (MMSI), which are presented by the solid black lines. The dashed horizontal line represents the cutoff value for the positive WHV-specific T response, and a shaded area under a solid gray line outlines the profile of this response in each animal (the data imported from Fig. 4). The CDI values for WHV-specific T cell response and MMSI values after mitogenic stimulation are shown on the left and right y axes, respectively. (A) Comparative profiles of a lymphocyte proliferative response induced by ConA and WHV antigens in individual animals injected with two rounds of 6 weekly doses of 1.1 × 102 virions and then challenged or not challenged with a liver-pathogenic dose of 1.1 × 106 virions. (B) Profiles of ConA-induced and WHV-specific T cell proliferative responses in two woodchucks injected with a single liver-pathogenic dose of 1.1 × 106 virions and challenged with the same virus dose or injected with PBS. Other markings and the subgroup designations are explained in the legend to Fig. 1.
When 3/M and 4/M were rechallenged with 1.1 × 106 virions of WHV, they displayed comparable, although not identical, profiles of generalized T cell reactivity. Thus, 3/M showed a heightened proliferative capacity between 2 and 4 weeks after rechallenge, which subsided prior to the increase in a WHV-specific T cell response. 4/M displayed augmented ConA-induced T cell proliferation at 1 week after rechallenge. This reactivity subsequently decreased and then again temporarily increased, and each time the fall was followed by an increase in the WHV-specific T cell response. This profile of augmented nonspecific T cell proliferation was not seen after injection with PBS (subgroup 6L6LN) (Fig. 5A; see also Fig. S2A in the supplemental material), as observed previously (11). Overall, an elevated generalized lymphocyte proliferative response coincided with decreased or absent WHV-specific T cell reactivity. Collectively, the data showed that the first of multiple exposures to small quantities of hepadnavirus significantly increased the virus-nonspecific T cell response and that a decrease preceded the appearance or reappearance of virus-specific T lymphocyte reactivity.
Peripheral lymphoid cells display aberrant cytokine gene expression following multiple exposure to WHV.
Considering our findings that both high and low doses of hepadnavirus modify the expression of cytokines in circulating immune cells (11, 37), we evaluated whether multiple exposure to small amounts of WHV can similarly affect expression of the previously examined cytokine genes in peripheral lymphoid cells. To assess that possibility, freshly isolated PBMCs not subjected to any ex vivo stimulation with either WHV antigens or ConA were analyzed by real-time RT-PCR. Taking into consideration the opposing profiles of mitogen-induced and WHV-specific T cell responses, different phases of the infection experiment (Fig. 1), and our previous observations (11, 37), the investigation period was divided into the following four phases: (i) virus naïve (before the first WHV injection), (ii) heightened mitogen-induced T cell reactivity (between 1 and 3 weeks after injection with the first dose in round 1 or round 2 or after rechallenge with 1.1 × 106 virions), (iii) lowered mitogen-induced and absent WHV-specific T cell reactivity (4 to 6 weeks, as defined above), and (iv) the appearance and heightened levels of the WHV-specific T cell response (7 to 15 weeks, as defined above). As shown in Fig. 6 (the first from the left vertical panel), phase B, after the first injection in round 1, coincided with elevated expression of IFN-α and IFN-γ, along with IL-2, IL-4, and IL-12, and unaltered transcription of TNF-α and IL-10 compared to the expression levels found for these genes in phase A. These elevated levels decreased during phase C for IFN-α, IFN-γ, IL-2, and IL-12 to levels not significantly different from those prior to infection. Finally, phase D was accompanied by significantly greater expression of IFN-α, IFN-γ, TNF-α, IL-2, and IL-12, but not IL-4 and IL-10, than the levels observed during phases A and C. The synchronous elevated expression of the cytokines during phase D coincided with the appearance and augmented levels of the WHV-specific T cell response.
Fig 6.
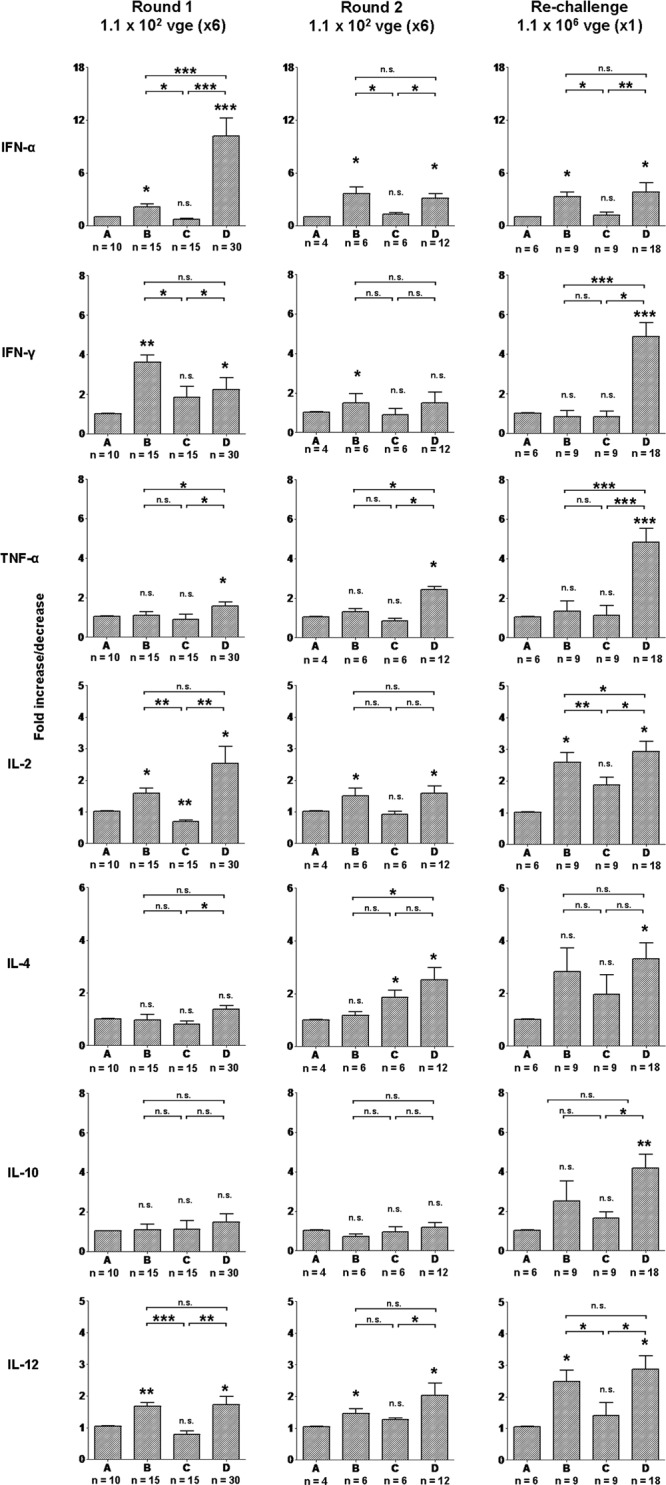
Quantitative analysis of cytokine gene expression in circulating lymphoid cells in woodchucks injected with two rounds of multiple liver-nonpathogenic doses of WHV and then with a single liver-pathogenic dose of the same virus. The expression of IFN-α, IFN-γ, TNF-α, IL-2, IL-4, IL-10, and IL-12 was analyzed in freshly isolated, nonstimulated PBMCs collected from woodchucks injected with 6 doses of 1.1 × 102 virions each (n = 4), challenged with another six 1.1 × 102-virion doses (n = 4), and rechallenged with 1.1 × 106 virions (n = 2) of the same WHV inoculum. After each primary or subsequent exposure to WHV, the timeline of infection was divided into four phases (phases A, B, C, and D) according to the distinct profiles of WHV-specific and mitogen-induced T cell responses as described in Results and reported previously (11, 37). The number (n) under each bar shows the number of PBMC samples examined. The level of expression of each cytokine was normalized against the level of β-actin and then compared with the level found prior to primary infection (phase A) to calculate the increase or decrease in expression. The P values above each bar represented by asterisks were obtained by comparing the mean expression level of a given gene during phase B, C, or D, with the mean level of expression determined for the gene in phase A, while the P values shown at the top of each panel were obtained by comparing each mean expression level of a given gene determined for phases B, C, and D. Values marked with one asterisk were significantly different at a P value of ≤0.05, values with two asterisks were significantly different at a P value of ≤0.005, and values with three asterisks were significantly different at a P value of ≤0.0001. n.s., not significantly different.
Similar evaluations were performed after challenge with round 2 of low-dose virus injections. As presented in Fig. 6 (the middle vertical panel), phase B again was characterized by high expression of IFN-α, IFN-γ, IL-2, and IL-12, but not TNF-α, all of which subsequently decreased during phase C to preinfection levels. Similar to the first round of multiple injections, in the second round, phase D was accompanied by elevated levels of IFN-α, IFN-γ, and TNF-α, as well as IL-2 and IL-12, compared to the expression levels detected in phase C.
Finally, when the animals, after two rounds of multiple low-dose injections, were rechallenged with a liver-pathogenic dose of 1 × 106 virions (Fig. 6), they displayed the pattern of cytokine expression that was essentially identical to that initiated following the primary exposure to a WHV dose capable of producing serologically evident infection and hepatitis as reported (11, 37) and also observed in woodchucks 5/F and 6/M in the current study (data not shown). Thus, the significantly increased levels of IFN-α, IL-2, and IL-12, but not other cytokines tested, were evident during phase B compared to the levels found before rechallenge. The elevated levels of IFN-α, IL-2, and IL-12 decreased during phase C compared to phase B and then rose along with the expression of IFN-γ, TNF-α, IL-4, and IL-10 during phase D compared to the levels detected in phase A. Collectively, the data showed that the POI established by repeated exposure to small amounts of WHV or exposure of such POI bearing animals to a medium, liver-pathogenic dose of WHV was accompanied by an aberrant cytokine expression in PBMCs which has already been recognized as a characteristic of WHV infection (11, 37).
Hepatic expression of IFN-α is augmented alone after multiple exposures to low doses of WHV.
The liver samples collected during infection, challenge, and rechallenge phases were analyzed for the expression of IFN-α, IFN-γ, TNF-α, IL-4, and CD3 and compared to the levels found in liver biopsy samples collected prior to round 1 of WHV injections. As shown in Fig. 7, the hepatic expression of IFN-α was significantly greater in liver biopsy samples acquired at 6 w.p.i. and subsided in those obtained at 27 w.p.i. compared to the levels identified in virus-naïve animals. However, the same liver samples did not display any meaningful changes in transcription of IFN-γ, TNF-α, IL-4, or CD3. Interestingly, liver samples collected at 6 weeks after the first of 6 injections in round 2 (or 36 w.p.i.) also demonstrated a significantly higher expression of IFN-α in the context of normal liver histology but unchanged levels of IFN-γ, TNF-α, IL-4, and CD3 (Fig. 2 and 7). As anticipated, hepatic expression of the cytokines tested and CD3 were the same in liver biopsy samples obtained at 6 and 28 weeks after injection with PBS (Fig. 7). However, the expression of TNF-α, IFN-γ, and CD3 was significantly enhanced in samples collected at 6 and 28 weeks after rechallenge with the liver-pathogenic 1.1 × 106-virion dose (Fig. 7). In summary, the results suggest that POI established due to multiple injections with low doses of hepadnavirus triggered the hepatic expression of IFN-α, but not that of IFN-γ or TNF-α, in the absence of detectable WHV DNA in the liver and histological and molecular (i.e., CD3 expression) evidence of inflammation.
Fig 7.
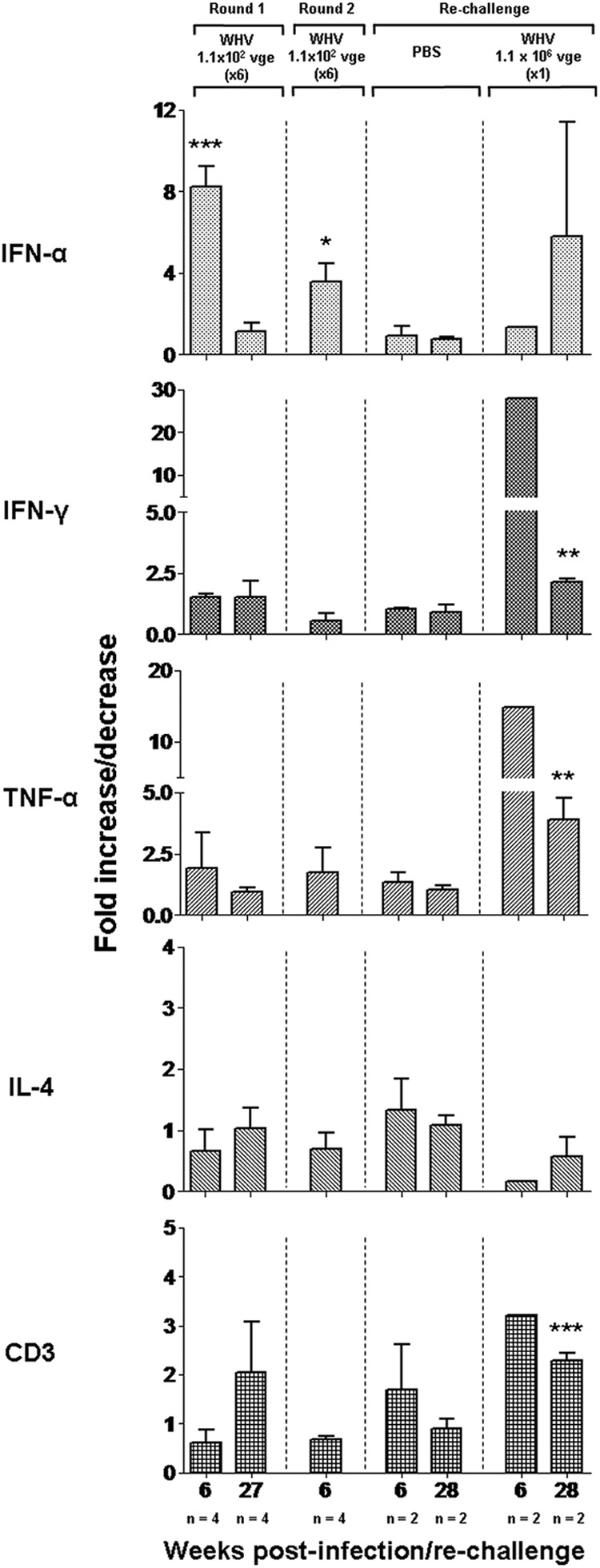
Hepatic expression of selected cytokines and CD3 in woodchucks after two rounds of repeated injections with liver-nonpathogenic doses of WHV and subsequent challenge with a high, liver-pathogenic dose of WHV or injection with PBS. The levels of IFN-α, IFN-γ, TNF-α, IL-4, and CD3 mRNA were measured by real-time RT-PCR in liver tissue samples collected after the first round (n = 4) and second round (n = 4) of injections with 1.1 × 102 virions and then injected with PBS (n = 2) or rechallenged with 1.1 × 106 virions (n = 2). The numbers (n) under each bar at the bottom of the figure indicate, respectively, the number of liver biopsy samples analyzed and the time of their collection, presented in weeks after the first injection of WHV in round 1 or round 2 or in weeks after rechallenge. The mean expression level of particular genes was normalized against β-actin expression and then compared with the mean determined in the liver samples collected prior to infection. Values marked with one asterisk were significant at a P value of ≤0.05, values with two asterisks were significant at a P value of ≤0.005, and values with three asterisks were significant at a P value of ≤0.0001.
DISCUSSION
The pathogenic consequences of repeated exposure to small amounts of infectious hepadnavirus were poorly recognized. The overall objective of this study was to determine virological and immunological characteristics, as well as potential pathological outcomes of multiple i.v. injections with liver-nonpathogenic doses of WHV, in the woodchuck model of HBV infection. Overall, the results revealed that such repeated exposure does not culminate in serologically identifiable infection or cause histologically evident hepatitis; however, a molecularly evident infection detectable by nucleic acid amplification assays of enhanced sensitivity was established. Furthermore, the profiles of the induced immune responses were essentially identical to those initiated by a singular low dose of WHV causing POI (11, 37). These responses were characterized by (i) a delayed appearance of virus-specific T cell reactivity against multiple epitopes of structural and nonstructural proteins of WHV, (ii) a heightened capacity of T cells to proliferate in response to mitogenic stimuli appearing immediately after exposure to WHV, (iii) an impaired early innate immune response, as revealed by analysis of the cytokine expression in sequential PBMC samples, (iv) the lack of induction of virus-specific antibodies, and (v) the inability to generate protection against infection caused by liver-pathogenic doses of WHV.
It was previously shown that a very low (50 virions) (11), medium (∼106 virions) (11), or massive (>1010 virions) (11, 37, 46, 47) dose of WHV induces a virus-specific T cell response that appears late (i.e., 6 to 8 w.p.i.), as in humans and chimpanzees infected with HBV (31, 48, 49). This is in contrast to infections with almost all other viruses in which this type of immune response became detectable in about 2 w.p.i. (50–52), e.g., infections with lymphocytic choriomeningitis virus (LCMV) (53, 54), influenza virus (55), or cytomegalovirus (56, 57). Independently of the dose of WHV, the virus-specific T cell response peaked between 6 and 15 weeks after primary exposure and was intermittently detectable at low levels to the end of the observation period or until challenge with WHV (11, 37). A similar delayed appearance of WHV-specific T cell reactivity was detected in the current study (i.e., 7 to 8 w.p.i.), and the period of the augmented T cell proliferative response remained transient despite repeated injections with virus. Challenge of the woodchucks with the second round of small doses of WHV (see Fig. 1) resulted again in the increase of a delayed virus-specific T cell response (i.e., 6 to 7 weeks after injecting the first dose in round 2), which was intermittently detectable during the 75-w.p.i. follow-up or until challenge with a liver-pathogenic dose of virus. These results were closely comparable to those reported for animals infected with a single small (50 virions), medium (∼106 virions), or large (>1010 virions) dose of WHV and rechallenged with a single small, medium, or large quantity of the same inoculum (11). Following rechallenge with 1.1 × 106 virions, a serologically evident infection and hepatitis coinciding with the detection of WHV in liver tissue was established. This demonstrated that the animals, after two rounds of multiple injections with small amounts of WHV, were not protected against a pathogenic effect caused by a larger quantity of WHV. These findings were identical to those previously reported for animals exposed to three 50-virion doses of WHV given at approximately 6-month intervals and subsequently challenged with 1.1 × 106 virions of the same inoculum (11).
In regard to the above, for the successful activation of T cell response, CD4+ T cells require two signals: antigen and costimulation. Not surprisingly, the strength and duration of both signals during initial stimulation affect the quantity and quality of the resultant T cell response. Evidence obtained thus far suggests that the dose of antigen determines the strength (magnitude) and breadth (clonality) of the antigen-specific response (58, 59). T cell responses induced following exposure to low doses of antigen are impaired, i.e., they are rather oligoclonal and fail to provide protection against subsequent challenge (60, 61). Similarly, low or suboptimal costimulation of antigen-presenting cells (APCs) induces a tolerant phenotype in interacting T cells (62, 63). Thus, it can be surmised that low doses of antigen can induce a T cell response with impaired functional capacities. Our data concur with that and indicate that T cell immune responses generated following the exposure to small amounts of hepadnavirus have functionally defective attributes.
In concordance with previous findings (11, 37), the data from the present study showed that the postponement of the WHV-specific T cell response was preceded by the heightened virus-nonspecific proliferation of T cells in response to mitogenic stimulus. In this regard, we have previously demonstrated that this type of elevated proliferative capability was equally evident after stimulation of lymphocytes with ConA, phytohemagglutinin (PHA), or pokeweed mitogen (PWM) (11, 37), indicating that the response was rather pan-T cell wide but not limited to the activation of a particular subtype of lymphocytes. Taken together, the results imply that the exposure to hepadnavirus either directly bestows upon the lymphocytes an ability to undergo increased cell division or indirectly orchestrates the environment that sustains the higher rates of lymphocyte proliferation in response to stimulation. The implications of this heightened capability of lymphocytes to undergo a rapid expansion shortly after exposure to hepadnavirus are not yet fully understood. However, it was found that this event preceded the augmented apoptotic death of the activated cells (37, 38). We postulated that such an aberrant activation accompanied by activation-induced apoptotic death immediately following infection with hepadnavirus may render lymphocytes temporarily insensitive to virus-specific activation. In consequence, this early inertness may contribute to the failure of the prompt development of a virus-specific T cell response. It is of note that this immediate generalized T cell activation is independent of the status of the virus-specific memory immune response, since this event was equally evident after primary infection caused by different doses of WHV, as well as following challenge of woodchucks convalescent from serologically evident self-limiting AH and those with established POI.
As in POI induced by a single low dose of WHV (11), the repeated injections with similar small amounts of virus in the current study did not initiate any detectable virus-specific antibody response. Thus, even though the magnitude of the WHV-specific T cell responses induced by the multiple injections was comparable to that seen in serum WHsAg-positive infection coinciding with hepatitis and the detection of anti-WHc, the traits of the B cell response appeared to be different. As indicated, this discordance between anti-WHV T cell and antibody responses was associated with the lack of protection against infection with a liver-pathogenic dose of the same virus, suggesting that the antibodies played an essential role in preventing symptomatic infection. This concurs with the observations made in asymptomatic infections caused by other viruses, such as hepatitis C virus (HCV) (64–66), human immunodeficiency virus (HIV) (67), or simian immunodeficiency virus (SIV) (68), wherein virus-specific T cell proliferative and cytotoxic T lymphocyte (CTL) reactivity occur in the absence of serological or pathological evidence of infection with the respective virus.
Our previous studies demonstrated that the delayed anti-WHV T cell response in either symptomatic infection or POI is associated with the preceding aberrant expression of cytokines priming this response (11, 37). In the present study, the cytokine expression profiles in intact (i.e., mitogen or WHV antigen nonstimulated) PBMCs collected after multiple small dose injections showed that, after exposure to virus, the cells displayed transiently elevated IFN-α and IFN-γ expression in the absence of TNF-α upregulation for up to 6 w.p.i. (Fig. 6). Thus, similar to the kinetics of the WHV-specific T cell response, the expression pattern of the above-mentioned cytokines was similar to that previously identified after exposure to a single low, medium, or high dose of WHV (11, 37). This profile of cytokine transcription was contrastingly different from that seen in infections with other viral pathogens where virus invasion promptly induces expression of both type I interferons and TNF-α (69–71). From the functional point of view, the expression of TNF-α triggered by virus drives the differentiation of immature dendritic cells (DCs) from their precursors (72, 73), while type I IFNs and TNF-α together activate natural killer (NK) cells and APCs that subsequently aid in the initiation of the effective virus-specific T cell response. On the contrary, the absence of TNF-α expression induces improper maturation and functional impairment of APCs. DCs display impaired antigen-specific and allostimulatory capacities in the absence of TNF-α (74, 75), while APCs cultured in the presence of IFN-α and TNF-α show a strong expression of CD80, CD83, and CD86 (76, 77), similar to those generated after in vitro infection with herpes simplex virus (HSV), suggesting the synergistic potential of these two cytokines in activating APCs. Similar maturation of DCs after LCMV infection can be abolished by the administration of anti-TNF-α antibodies in infected mice, suggesting the vital role of TNF-α during the differentiation and maturation of DCs (77). Overall, the prompt expression of TNF-α after infection is accompanied by proper activation of APCs and is subsequently followed by an effective antigen-specific immune response. However, along with IFN-α and TNF-α, the initiation of effective antiviral immunity also requires synchronous expression of other cytokines, such as IL-12, IL-2, IL-4, and IL-10 (69). Taken together, our data, in conjunction with those reported by others, suggest that the absence of TNF-α, coupled with nonsynchronous expression of other cytokines early in hepadnaviral infection, including infection established after multiple exposures to low doses to WHV, could translate into inappropriate differentiation and activation of DCs, which then fails to timely prime antiviral adaptive immunity as evidenced by the delayed appearance of the WHV-specific T cell response. The postponed appearance of a virus-specific response is also a distinctive characteristic of HBV infection, which may suggest a similar underlying mechanism.
The proliferative capacity of the lymphocytes in response to either antigenic or mitogenic stimulus is profoundly influenced by the cytokine microenvironment. To better understand the events underlying the discordant profiles of proliferation following multiple exposures to hepadnavirus, we further analyzed the expression of selected cytokines in PBMCs collected during different phases of infection, which were defined by the prevailing response to either mitogenic or viral stimulus following the first and the second rounds of injections with small amounts of WHV (see Fig. 6). The quantitative analysis showed that this discordant proliferative response to WHV or mitogen coexisted with distinct profiles of cytokine expression, as we have previously uncovered for WHV infection with a single liver-pathogenic or -nonpathogenic virus dose (11, 37). In brief, the results showed that the higher proliferative capacity of lymphocytes in response to mitogen stimulation immediately after infection (i.e., 1 to 3 w.p.i.; phase B) could be a consequence of increased expression of progrowth cytokines, such as IFN-α, IL-2, and IL-12. The subsequent reduced proliferative responsiveness to mitogen coincided with the reduced expression of these cytokines (i.e., 4 to 6 w.p.i.; phase C). Thus, the absence of IL-2 and IFN-α/IL-12, which are essential for sustaining lymphocyte growth, could halt the enhanced proliferative response of the cells, as was evidenced by lower rates of adenine incorporation. Subsequently, the appearance of a WHV-specific T cell response was associated with a rebound in the expression of IFN-α, IL-2, and IL-12 and significantly heightened the transcription of IFN-γ, as well as TNF-α. Again, the expression profiles of these cytokines closely resembled those described for the equivalent phase of POI induced with a single liver-nonpathogenic dose of WHV (11). In regard to the cytokine response, the results obtained in the current work showed for the first time that (i) the primary exposure to a very small and otherwise liver-nonpathogenic dose of hepadnavirus promptly induces the cytokine response and the subsequent exposure to similar amounts of infectious virus is without apparent effect on the profile of cytokine expression (as well as the patterns of T and B cell responses), (ii) there was no noticeable difference between the cytokine expression profile induced by the first and the second rounds of injections with small, liver-nonpathogenic doses of WHV, and (iii) the cytokine expression profile induced by a large, liver-pathogenic amount of virus given after repeated exposure to liver-nonpathogenic doses is essentially identical to that initiated by administration of a single liver-pathogenic quantity of virus.
In regard to the cytokine expression in the liver, a strongly augmented expression of IFN-α, in the absence of increase in expression of IFN-γ, TNF-α, IL-4, or CD3, was detected after each round of multiple injections with low doses of WHV. This finding was surprising, considering the fact that, even after multiple injections of small amounts of virus, the initiated infection was restricted to the lymphatic system and did not involve hepatic tissue, as observed in our previous studies (11, 12, 37, 40). However, it is of note that the significantly heightened expression of IFN-α alone was also found during POI in woodchucks injected with a single low dose of WHV (11). We believe that this strong intrahepatic IFN-α response initiated following either singular or multiple exposures to very small amounts of WHV could be sufficient in protecting hepatocytes against the trace quantities of invading virus. In contrast, such augmented expression of IFN-α was not detectable in the livers of initially healthy, WHV-naïve animals or woodchucks with POI challenged with a large, liver-pathogenic amount of WHV (11), supporting the notion that infection with a liver-pathogenic dose of hepadnavirus initiates an impaired innate response in the liver (42).
In conclusion, our study has shown that repeated exposure to small amounts of otherwise pathogenic hepadnavirus establishes molecularly evident but serologically undetectable infection which initiation coincides with the appearance of a virus-specific T cell response. The current findings imply that multiple contacts with very small quantities of infectious HBV unlikely can culminate in serum HBsAg and anti-HBc-positive infection and hepatitis; however, the detection of virus-specific T cell reactivity should be expected. This corresponds well with the situation encountered in patients with occult HBV infection progressing in the absence of virus-specific antibodies and carrying virus-specific CD8+ T cells (22–25). A recent discovery that HCC can be a long-term pathological consequence of POI in the woodchuck model of HBV infection (Mulrooney-Cousins and Michalak, unpublished) raises the possibility that this form of infection initiated by either single or multiple exposures to trace amounts of HBV may lead to a similar outcome in humans.
Supplementary Material
ACKNOWLEDGMENTS
We thank Norma D. Churchill and Colleen L. Trelegan for their expert technical assistance.
This study was supported by operating grant MOP-14818 from the Canadian Institutes of Health Research. T.I.M. is the Canada Research Chair (Tier 1) in Viral Hepatitis/Immunology sponsored by the Canada Research Chair Program and funded by the Canadian Institutes of Health Research and by the Canada Foundation for Innovation.
We declare that no competing interests exist.
Footnotes
Published ahead of print 7 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01363-12.
REFERENCES
- 1. Bes M, Vargas V, Piron M, Casamitjana N, Esteban JI, Vilanova N, Pinacho A, Quer J, Puig L, Guardia J, Sauleda S. 2012. T cell responses and viral variability in blood donation candidates with occult hepatitis B infection. J. Hepatol. 56:765–774 [DOI] [PubMed] [Google Scholar]
- 2. Fang Y, Shang QL, Liu JY, Li D, Xu WZ, Teng X, Zhao HW, Fu LJ, Zhang FM, Gu HX. 2009. Prevalence of occult hepatitis B virus infection among hepatopathy patients and healthy people in China. J. Infect. 58:383–388 [DOI] [PubMed] [Google Scholar]
- 3. FitzSimons D, Francois G, De Carli G, Shouval D, Pruss-Ustun A, Puro V, Williams I, Lavanchy D, De Schryver A, Kopka A, Ncube F, Ippolito G, Van Damme P. 2008. Hepatitis B virus, hepatitis C virus and other blood-borne infections in healthcare workers: guidelines for prevention and management in industrialised countries. Occup. Environ. Med. 65:446–451 [DOI] [PubMed] [Google Scholar]
- 4. MacCannell T, Laramie AK, Gomaa A, Perz JF. 2010. Occupational exposure of health care personnel to hepatitis B and hepatitis C: prevention and surveillance strategies. Clin. Liver Dis. 14:23–36 [DOI] [PubMed] [Google Scholar]
- 5. Michelin A, Henderson DK. 2010. Infection control guidelines for prevention of health care-associated transmission of hepatitis B and C viruses. Clin. Liver Dis. 14:119–136 [DOI] [PubMed] [Google Scholar]
- 6. Torbenson M, Kannangai R, Astemborski J, Strathdee SA, Vlahov D, Thomas DL. 2004. High prevalence of occult hepatitis B in Baltimore injection drug users. Hepatology 39:51–57 [DOI] [PubMed] [Google Scholar]
- 7. Menne S, Cote PJ. 2007. The woodchuck as an animal model for pathogenesis and therapy of chronic hepatitis B virus infection. World J. Gastroenterol. 13:104–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michalak TI. 2000. Occult persistence and lymphotropism of hepadnaviral infection: insights from the woodchuck viral hepatitis model. Immunol. Rev. 174:98–111 [DOI] [PubMed] [Google Scholar]
- 9. Mulrooney-Cousins PM, Michalak TI. 2011. New aspects of natural history and pathogenicity of hepadnaviral infection and hepatocyte function revealed by the woodchuck model of hepatitis B, p 355–378 In Mizuguchi Y. (ed), Liver biopsy in medicine. Intech, Rijeka, Croatia [Google Scholar]
- 10. Coffin CS, Pham TN, Mulrooney PM, Churchill ND, Michalak TI. 2004. Persistence of isolated antibodies to woodchuck hepatitis virus core antigen is indicative of occult infection. Hepatology 40:1053–1061 [DOI] [PubMed] [Google Scholar]
- 11. Gujar SA, Michalak TI. 2009. Primary occult hepadnavirus infection induces virus-specific T-cell and aberrant cytokine responses in the absence of antiviral antibody reactivity in the woodchuck model of hepatitis B virus infection. J. Virol. 83:3861–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Michalak TI, Mulrooney PM, Coffin CS. 2004. Low doses of hepadnavirus induce infection of the lymphatic system that does not engage the liver. J. Virol. 78:1730–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michalak TI, Pardoe IU, Coffin CS, Churchill ND, Freake DS, Smith P, Trelegan CL. 1999. Occult lifelong persistence of infectious hepadnavirus and residual liver inflammation in woodchucks convalescent from acute viral hepatitis. Hepatology 29:928–938 [DOI] [PubMed] [Google Scholar]
- 14. Mulrooney-Cousins PM, Michalak TI. 2007. Persistent occult hepatitis B virus infection: experimental findings and clinical implications. World J. Gastroenterol. 13:5682–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coffin CS, Michalak TI. 1999. Persistence of infectious hepadnavirus in the offspring of woodchuck mothers recovered from viral hepatitis. J. Clin. Invest. 104:203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zerbini A, Pilli M, Boni C, Fisicaro P, Penna A, Di Vincenzo P, Giuberti T, Orlandini A, Raffa G, Pollicino T, Raimondo G, Ferrari C, Missale G. 2008. The characteristics of the cell-mediated immune response identify different profiles of occult hepatitis B virus infection. Gastroenterology 134:1470–1481 [DOI] [PubMed] [Google Scholar]
- 17. Challine D, Chevaliez S, Pawlotsky JM. 2008. Efficacy of serologic marker screening in identifying hepatitis B virus infection in organ, tissue, and cell donors. Gastroenterology 135:1185–1191 [DOI] [PubMed] [Google Scholar]
- 18. Firnhaber C, Chen CY, Evans D, Maskew M, Schulz D, Reyneke A, Kramvis A. 2012. Prevalence of hepatitis B virus (HBV) co-infection in HBV serologically-negative South African HIV patients and retrospective evaluation of the clinical course of mono- and co-infection. Int. J. Infect. Dis. 16:e268–e272 [DOI] [PubMed] [Google Scholar]
- 19. Raimondo G, Pollicino T, Cacciola I, Squadrito G. 2007. Occult hepatitis B virus infection. J. Hepatol. 46:160–170 [DOI] [PubMed] [Google Scholar]
- 20. Song EY, Yun YM, Park MH, Seo DH. 2009. Prevalence of occult hepatitis B virus infection in a general adult population in Korea. Intervirology 52:57–62 [DOI] [PubMed] [Google Scholar]
- 21. de la Fuente RA, Gutierrez ML, Garcia-Samaniego J, Fernandez-Rodriguez C, Lledo JL, Castellano G. 2011. Pathogenesis of occult chronic hepatitis B virus infection. World J. Gastroenterol. 17:1543–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raimondo G, Allain JP, Brunetto MR, Buendia MA, Chen DS, Colombo M, Craxi A, Donato F, Ferrari C, Gaeta GB, Gerlich WH, Levrero M, Locarnini S, Michalak T, Mondelli MU, Pawlotsky JM, Pollicino T, Prati D, Puoti M, Samuel D, Shouval D, Smedile A, Squadrito G, Trepo C, Villa E, Will H, Zanetti AR, Zoulim F. 2008. Statements from the Taormina expert meeting on occult hepatitis B virus infection. J. Hepatol. 49:652–657 [DOI] [PubMed] [Google Scholar]
- 23. Michalak TI, Pasquinelli C, Guilhot S, Chisari FV. 1994. Hepatitis B virus persistence after recovery from acute viral hepatitis. J. Clin. Invest. 93:230–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. 1996. The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 2:1104–1108 [DOI] [PubMed] [Google Scholar]
- 25. Yuki N, Nagaoka T, Yamashiro M, Mochizuki K, Kaneko A, Yamamoto K, Omura M, Hikiji K, Kato M. 2003. Long-term histologic and virologic outcomes of acute self-limited hepatitis B. Hepatology 37:1172–1179 [DOI] [PubMed] [Google Scholar]
- 26. Korba BE, Wells FV, Baldwin B, Cote PJ, Tennant BC, Popper H, Gerin JL. 1989. Hepatocellular carcinoma in woodchuck hepatitis virus-infected woodchucks: presence of viral DNA in tumor tissue from chronic carriers and animals serologically recovered from acute infections. Hepatology 9:461–470 [DOI] [PubMed] [Google Scholar]
- 27. Menne S, Cotte PI, Butler SD, Toshkov IA, Gerin JL, Tennant BC. 2007. Immunosuppression reactivates viral replication long after resolution of woodchuck hepatitis virus infection. Hepatology 54:614–622 [DOI] [PubMed] [Google Scholar]
- 28. Hoofnagle JH. 2009. Reactivation of hepatitis B. Hepatology 49:S156–S165 [DOI] [PubMed] [Google Scholar]
- 29. Mastroianni CM, Lichtner M, Citton R, Del Borgo C, Rago A, Martini H, Cimino G, Vullo V. 2011. Current trends in management of hepatitis B virus reactivation in the biologic therapy era. World J. Gastroenterol. 17:3881–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Papamichalis P, Alexiou A, Boulbou M, Dalekos GN, Rigopoulou EI. 2012. Reactivation of resolved hepatitis B virus infection after immunosuppression: is it time to adopt pre-emptive therapy? Clin. Res. Hepatol. Gastroenterol. 36:84–93 [DOI] [PubMed] [Google Scholar]
- 31. Bertoletti A, Gehring AJ. 2006. The immune response during hepatitis B virus infection. J. Gen. Virol. 87:1439–1449 [DOI] [PubMed] [Google Scholar]
- 32. Chisari FV. 1997. Cytotoxic T cells and viral hepatitis. J. Clin. Invest. 99:1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dandri M, Locarnini S. 2012. New insight in the pathobiology of hepatitis B virus infection. Gut 61(Suppl 1):i6–i17 [DOI] [PubMed] [Google Scholar]
- 34. Tan AT, Koh S, Goh V, Bertoletti A. 2008. Understanding the immunopathogenesis of chronic hepatitis B virus: an Asian prospective. J. Gastroenterol. Hepatol. 23:833–843 [DOI] [PubMed] [Google Scholar]
- 35. Bertoletti A, Ferrari C. 2003. Kinetics of the immune responses during HBV and HCV infection. Hepatology 38:4–13 [DOI] [PubMed] [Google Scholar]
- 36. Guidotti LG, Chisari FV. 2006. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 1:23–61 [DOI] [PubMed] [Google Scholar]
- 37. Gujar SA, Jenkins AK, Guy CS, Wang J, Michalak TI. 2008. Aberrant lymphocyte activation precedes delayed virus-specific T-cell response after both primary infection and secondary exposure to hepadnavirus in the woodchuck model of hepatitis B virus infection. J. Virol. 82:6992–7008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gujar SA, Jenkins AK, MacParland SA, Michalak TI. 2010. Pre-acute hepadnaviral infection is associated with activation-induced apoptotic death of lymphocytes in the woodchuck (Marmota monax) model of hepatitis B. Dev. Comp. Immunol. 34:999–1008 [DOI] [PubMed] [Google Scholar]
- 39. Hodgson PD, Michalak TI. 2001. Augmented hepatic interferon gamma expression and T-cell influx characterize acute hepatitis progressing to recovery and residual lifelong virus persistence in experimental adult woodchuck hepatitis virus infection. Hepatology 34:1049–1059 [DOI] [PubMed] [Google Scholar]
- 40. Mulrooney-Cousins PM, Michalak TI. 2008. Repeated passage of wild-type woodchuck hepatitis virus in lymphoid cells does not generate cell type-specific variants or alter virus infectivity. J. Virol. 82:7540–7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gujar SA, Michalak TI. 2005. Flow cytometric quantification of T cell proliferation and division kinetics in woodchuck model of hepatitis B. Immunol. Invest. 34:215–236 [PubMed] [Google Scholar]
- 42. Guy CS, Mulrooney-Cousins PM, Churchill ND, Michalak TI. 2008. Intrahepatic expression of genes affiliated with innate and adaptive immune responses immediately after invasion and during acute infection with woodchuck hepadnavirus. J. Virol. 82:8579–8591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Michalak TI, Hodgson PD, Churchill ND. 2000. Posttranscriptional inhibition of class I major histocompatibility complex presentation on hepatocytes and lymphoid cells in chronic woodchuck hepatitis virus infection. J. Virol. 74:4483–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Michalak TI, Lin B, Churchill ND, Dzwonkowski P, Desousa JR. 1990. Hepadna virus nucleocapsid and surface antigens and the antigen-specific antibodies associated with hepatocyte plasma membranes in experimental woodchuck acute hepatitis. Lab. Invest. 62:680–689 [PubMed] [Google Scholar]
- 45. Wang J, Gujar SA, Cova L, Michalak TI. 2007. Bicistronic woodchuck hepatitis virus core and gamma interferon DNA vaccine can protect from hepatitis but does not elicit sterilizing antiviral immunity. J. Virol. 81:903–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Menne S, Maschke J, Lu M, Grosse-Wilde H, Roggendorf M. 1998. T-cell response to woodchuck hepatitis virus (WHV) antigens during acute self-limited WHV infection and convalescence and after viral challenge. J. Virol. 72:6083–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Menne S, Maschke J, Tolle T, Kreuzfelder E, Grosse-Wilde H, Roggendorf M. 1997. Determination of peripheral blood mononuclear cell responses to mitogens and woodchuck hepatitis virus core antigen in woodchucks by 5-bromo-2′-deoxyuridine or 2[3H]adenine incorporation. Arch. Virol. 142:511–521 [DOI] [PubMed] [Google Scholar]
- 48. Webster GJ, Reignat S, Brown D, Ogg GS, Jones L, Seneviratne SL, Williams R, Dusheiko G, Bertoletti A. 2004. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J. Virol. 78:5707–5719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Webster GJ, Reignat S, Maini MK, Whalley SA, Ogg GS, King A, Brown D, Amlot PL, Williams R, Vergani D, Dusheiko GM, Bertoletti A. 2000. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology 32:1117–1124 [DOI] [PubMed] [Google Scholar]
- 50. Castellino F, Germain RN. 2006. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu. Rev. Immunol. 24:519–540 [DOI] [PubMed] [Google Scholar]
- 51. Gourley TS, Wherry EJ, Masopust D, Ahmed R. 2004. Generation and maintenance of immunological memory. Semin. Immunol. 16:323–333 [DOI] [PubMed] [Google Scholar]
- 52. Kaech SM, Wherry EJ, Ahmed R. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2:251–262 [DOI] [PubMed] [Google Scholar]
- 53. De Boer RJ, Homann D, Perelson AS. 2003. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 171:3928–3935 [DOI] [PubMed] [Google Scholar]
- 54. Khanolkar A, Fuller MJ, Zajac AJ. 2002. T cell responses to viral infections: lessons from lymphocytic choriomeningitis virus. Immunol. Res. 26:309–321 [DOI] [PubMed] [Google Scholar]
- 55. Brown DM, Roman E, Swain SL. 2004. CD4 T cell responses to influenza infection. Semin. Immunol. 16:171–177 [DOI] [PubMed] [Google Scholar]
- 56. Gamadia LE, Remmerswaal EB, Weel JF, Bemelman F, van Lier RA, Ten Berge IJ. 2003. Primary immune responses to human CMV: a critical role for IFN-gamma-producing CD4+ T cells in protection against CMV disease. Blood 101:2686–2692 [DOI] [PubMed] [Google Scholar]
- 57. Schlub TE, Sun JC, Walton SM, Robbins SH, Pinto AK, Munks MW, Hill AB, Brossay L, Oxenius A, Davenport MP. 2011. Comparing the kinetics of NK cells, CD4, and CD8 T cells in murine cytomegalovirus infection. J. Immunol. 187:1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J. 2011. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc. Natl. Acad. Sci. U. S. A. 108:9089–9094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zehn D, Lee SY, Bevan MJ. 2009. Complete but curtailed T-cell response to very low-affinity antigen. Nature 458:211–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tseng KE, Chung CY, H'ng WS, Wang SL. 2009. Early infection termination affects number of CD8+ memory T cells and protective capacities in listeria monocytogenes-infected mice upon rechallenge. J. Immunol. 182:4590–4600 [DOI] [PubMed] [Google Scholar]
- 61. Williams MA, Bevan MJ. 2004. Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J. Immunol. 173:6694–6702 [DOI] [PubMed] [Google Scholar]
- 62. Manicassamy S, Pulendran B. 2011. Dendritic cell control of tolerogenic responses. Immunol. Rev. 241:206–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pulendran B, Tang H, Manicassamy S. 2010. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat. Immunol. 11:647–655 [DOI] [PubMed] [Google Scholar]
- 64. Kamal SM, Amin A, Madwar M, Graham CS, He Q, Al Tawil A, Rasenack J, Nakano T, Robertson B, Ismail A, Koziel MJ. 2004. Cellular immune responses in seronegative sexual contacts of acute hepatitis C patients. J. Virol. 78:12252–12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Koziel MJ, Wong DK, Dudley D, Houghton M, Walker BD. 1997. Hepatitis C virus-specific cytolytic T lymphocyte and T helper cell responses in seronegative persons. J. Infect. Dis. 176:859–866 [DOI] [PubMed] [Google Scholar]
- 66. Quiroga JA, Llorente S, Castillo I, Rodriguez-Inigo E, Pardo M, Carreno V. 2006. Cellular immune responses associated with occult hepatitis C virus infection of the liver. J. Virol. 80:10972–10979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Promadej N, Costello C, Wernett MM, Kulkarni PS, Robison VA, Nelson KE, Hodge TW, Suriyanon V, Duerr A, McNicholl JM. 2003. Broad human immunodeficiency virus (HIV)-specific T cell responses to conserved HIV proteins in HIV-seronegative women highly exposed to a single HIV-infected partner. J. Infect. Dis. 187:1053–1063 [DOI] [PubMed] [Google Scholar]
- 68. Tasca S, Tsai L, Trunova N, Gettie A, Saifuddin M, Bohm R, Chakrabarti L, Cheng-Mayer C. 2007. Induction of potent local cellular immunity with low dose X4 SHIV(SF33A) vaginal exposure. Virology 367:196–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposti MA. 2005. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 6:1011–1019 [DOI] [PubMed] [Google Scholar]
- 70. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 71. Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Iwamoto S, Iwai S, Tsujiyama K, Kurahashi C, Takeshita K, Naoe M, Masunaga A, Ogawa Y, Oguchi K, Miyazaki A. 2007. TNF-alpha drives human CD14+ monocytes to differentiate into CD70+ dendritic cells evoking Th1 and Th17 responses. J. Immunol. 179:1449–1457 [DOI] [PubMed] [Google Scholar]
- 73. Reis e Sousa C, Sher A, Kaye P. 1999. The role of dendritic cells in the induction and regulation of immunity to microbial infection. Curr. Opin. Immunol. 11:392–399 [DOI] [PubMed] [Google Scholar]
- 74. Dauer M, Pohl K, Obermaier B, Meskendahl T, Robe J, Schnurr M, Endres S, Eigler A. 2003. Interferon-alpha disables dendritic cell precursors: dendritic cells derived from interferon-alpha-treated monocytes are defective in maturation and T-cell stimulation. Immunology 110:38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McRae BL, Nagal T, Semnani RT, van Seventer JM, van Seventer GA. 2000. Interferon-alpha and -beta inhibit the in vitro differentiation of immunocompetent human dendritic cells from CD14(+) precursors. Blood 96:210–217 [PubMed] [Google Scholar]
- 76. Chen B, Shi Y, Smith JD, Choi D, Geiger JD, Mule JJ. 1998. The role of tumor necrosis factor alpha in modulating the quantity of peripheral blood-derived, cytokine-driven human dendritic cells and its role in enhancing the quality of dendritic cell function in presenting soluble antigens to CD4+ T cells in vitro. Blood 91:4652–4661 [PubMed] [Google Scholar]
- 77. Kadowaki N, Antonenko S, Lau JY, Liu YJ. 2000. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 192:219–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.