

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) infection is correlated with three human malignancies and can establish lifelong latent infection in multiple cell types within its human host. In order to establish and maintain infection, KSHV utilizes multiple mechanisms to evade the host immune response. One such mechanism is the expression of a family of genes with homology to cellular interferon (IFN) regulatory factors (IRFs), known as viral IRFs (vIRFs). We demonstrate here that KSHV vIRF1, -2, and -3 have a differential ability to block type I interferon signaling mediated by Toll-like receptor 3 (TLR3), a receptor we have previously shown to be activated upon KSHV infection. vIRF1, -2, and -3 inhibited TLR3-driven activation of IFN transcription reporters. However, only vIRF1 and vIRF2 inhibited increases in both IFN-β message and protein levels following TLR3 activation. The expression of vIRF1 and vIRF2 also allowed for increased replication of a virus known to activate TLR3 signaling. Furthermore, vIRF1 and vIRF2 may block TLR3-mediated signaling via different mechanisms. Altogether, this report indicates that vIRFs are able to block IFN mediated by TLRs but that each vIRF has a unique function and mechanism for blocking antiviral IFN responses.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus thought to be the etiological agent of three human malignancies: Kaposi's sarcoma, multicentric Castleman's disease (MCD), and primary effusion lymphoma (PEL) (1–3). Like all herpesviruses, KSHV is capable of establishing lifelong latent infection within the host. In order to establish and maintain infection, KSHV must evade the host immune response. The primary innate antiviral response is mediated by type I interferons (IFN-α and -β) and results in increased antigen presentation, degradation of viral RNA, cessation of protein processing, and apoptosis (4). Type I IFNs are induced following viral detection of pattern-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) (5).
The TLRs most commonly associated with the detection of viral infection include TLR3, -7, -8, and -9 (5). TLR3 resides in cellular endosomes where it recognizes double-stranded RNA (dsRNA) (5). Upon ligand binding, TLR3 signals through the adaptor protein toll/interleukin-1 receptor (TIR)-domain-containing adaptor-inducing beta interferon (TRIF) and ultimately results in the activation of cellular interferon regulatory factors (IRFs). Two major IRFs, IRF3 and IRF7, are responsible for the induction of type I IFN. Upon activation, IRF3 and IRF7 are phosphorylated, can hetero- or homodimerize, and subsequently translocate to the nucleus, where they activate an antiviral transcription program.
We have previously shown that TLR3 is upregulated in response to primary KSHV infection in human monocytes (6). Others have shown that infection of endothelial cells with KSHV suppresses the expression of TLR4, which recognizes lipopolysaccharides (LPSs) (7). Furthermore, our group has demonstrated that the stimulation of TLR7 and TLR8 leads to reactivation and the production of infectious KSHV in latently infected PEL cells (8). Therefore, the interactions between KSHV and TLRs appear to play an important role in KSHV infection and progression.
KSHV encodes multiple homologs of cellular immune proteins, including four homologs of the cellular IRFs, known as viral IRFs (vIRFs), which have been shown to be multifunctional. These four viral proteins are encoded by K9 (vIRF1), K11 and K11.1 (vIRF2), K10.5 and K10.6 (vIRF3), and K10 (vIRF4) (reviewed in reference 9). vIRF1, -2, and -4 are primarily lytic proteins, while vIRF3, also known as latency-associated nuclear antigen 2 (LANA-2), is expressed during latency (9, 10). KSHV vIRFs perform a variety of functions, including inhibition of p53 (10–14), Myc (15, 16), Notch (17), transforming growth factor β (TGF-β) (18), and apoptosis (19–23). Of the four vIRFs, vIRF1, -2, and -3 have been shown to inhibit the IFN response (24–33). In these reports, IFN was activated by the expression of a cellular IRF, treatment with IFN, or viral infection, and blockade of the IFN response was primarily measured by reporter assays. However, it remains unclear whether vIRFs are able to block IFN responses initiated by TLR activation and if the vIRFs are redundant or specific in their abilities to inhibit a particular TLR pathway. Additionally, studies have yet to compare the abilities of vIRF1, -2, and -3 to block IFN responses. Furthermore, while several other groups have examined the ability of KSHV vIRFs to block IFN induction upon viral infection (25, 30, 31, 34, 35), we are the only ones to have described the effect of vIRF expression on levels of viral replication in the context of vIRF2 (36).
Primary KSHV infection of monocytes results in increased TLR3, CXC chemokine ligand 10 (CXCL10), and IFN-β message levels early after infection (6). In this report, we demonstrate that at later time points postinfection, TLR3 and CXCL10 transcripts are downregulated, suggesting that viral genes might contribute to suppression of the innate immune response as the infection process progresses. We demonstrate that KSHV vIRF1, -2, and -3 display differences in their abilities to block IFN signaling mediated by TLR3. TLR3-mediated activation of IFN transcription reporters was inhibited by the expression of vIRF1, -2, and -3. However, only vIRF1 and vIRF2 inhibited IFN-β message and protein levels. Furthermore, vIRF1 and vIRF2 may block TLR3-mediated signaling via different mechanisms. We also compared the ability of each vIRF to inhibit viral replication of encephalomyocarditis virus (EMCV), a virus that is known to be sensitive to IFN. The expression of vIRF1 or vIRF2 rescued EMCV replication. Collectively, our data suggest that the KSHV vIRFs can block TLR3-mediated IFN induction and that each vIRF is unique in its ability to block TLR3-mediated antiviral immune responses.
MATERIALS AND METHODS
Plasmids.
pcDNA3 vIRF1-cMyc and pcDNA3 vIRF3-FLAG were generous gifts of Jae Jung (University of Southern California, Los Angeles, CA), pcDNA4/vIRF2 was as described previously (28), IRF3 and interferon-stimulated regulatory element (ISRE) Luc (37) were generous gifts of Joseph Pagano (University of North Carolina, Chapel Hill), and pIFN-β-luc (38) was a generous gift of Zhijian Chen (University of Texas Southwestern, Dallas TX). The QuikChange XL II site-directed mutagenesis kit (Stratagene) and the pcDNA3.1 directional Topo expression kit (Invitrogen) were utilized to generate the pcDNA3 vIRF1-FLAG and pcDNA3 vIRF2-FLAG constructs.
Cell cultures.
KSHV-THP1 cell lines were generated and maintained as described previously (39). Briefly, THP1 monocytes were infected with recombinant KSHV.219 (rKSHV.219) and maintained in 0.1 μg/ml puromycin. 293-TLR3 cells and control 293 cells (InvivoGen) were cultured at 37°C with 5% CO2 in Dulbecco's modified Eagle medium (DMEM) (Cellgro) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Cellgro), 100 U/ml each of penicillin and streptomycin, 2 mM l-glutamine, and 10 μg/ml blasticidin S (InvivoGen). To activate TLR3 signaling, cells were treated with high-molecular-weight poly(I·C) (InvivoGen) at 20 μg/ml for the indicated times. Cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Immunoblotting.
Upon harvesting, cells were washed once with phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay (RIPA) buffer with the addition of protease inhibitor complete tabs (Roche) and phosphatase inhibitor cocktail I and II (Sigma). Protein concentrations were determined by Bradford assays (Bio-Rad), and equal amounts of proteins were separated using SDS-PAGE and transferred onto Hybond-ECL nitrocellulose membranes (GE Healthcare). The antibodies we used included anti-Myc-horseradish peroxidase (HRP) (Invitrogen), anti-Xpress (Invitrogen), anti-ECS-HRP (Bethyl), anti-phospho-IRF3 (Ser396) (Cell Signaling Technology), IRF3 (Cell Signaling Technology), and actin (Santa Cruz Biotechnology). Blots were exposed to the appropriate secondary antibodies conjugated to horseradish peroxidase when necessary and visualized using chemiluminescence (GE Corporation).
Quantitative real-time PCR.
RNA was isolated with the RNeasy Plus minikit or the RNeasy microkit (Qiagen) according to the manufacturer's instructions and as described previously (6, 40). Reverse transcription was performed using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) and random hexamer primers (Invitrogen). Real-time PCR (RT-PCR) was performed on an ABI 7300 using SYBR green PCR master mix (Applied Biosystems). PCR was carried out with 1 cycle of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All fold activations were normalized to that of β-actin. The primer sequences used were 5′-GATCTGTCTCATAATGGCTTG (forward) and 5′-GACAGATTCCGAATGCTTGTG (reverse) for TLR3, 5′-GGAACCTCCAGTCTCAGCACC (forward) and 5′-CAGCCTCTGTGTGGTCCAATCC (reverse) for CXCL10, 5′-CAGCAATTTTCAGTGTCAGAAGC (forward) and 5′-TCATCCTGTCCTTGAGGCAGT (reverse) for IFN-β, 5′-GTCGTGAAGGAAGCAGT (forward) and 5′-CACGTGGCTTTTGGCCGCAGAGGC (reverse) for EMCV, and 5′-TCATGAAGTGTGACGTGGACATC (forward) and 5′-CAGGAGGAGCAATGATCTTGATCT (reverse) for β-actin (41).
Luciferase assays.
Cells were lysed with reporter lysis buffer (Promega) for 1 h on ice, and luciferase assays were performed with a luciferase assay system (Promega) according to the manufacturer's instructions. The results were read using a FLUOstar Optima (BMG Labtech) microplate reader and normalized to total protein as determined by the Bradford assay (Bio-Rad).
IFN-β ELISA.
Supernatants were exposed to an enzyme-linked immunosorbent assay (ELISA) specific for IFN-β (human origin; PBL Interferon Source) per the manufacturer's instructions. Absorbance at 450 nm was read on a VersaMax tunable microplate reader (Molecular Devices). The standard provided by the manufacturer was plotted with a 4-parameter fit, and the concentration for each sample was determined based on the standard curve.
Immunofluorescence assay.
293-TLR3 cells were plated directly onto coverslips placed in 6-well dishes. After the indicated treatment, the coverslips were washed with PBS and fixed for 15 min with 3% paraformaldehyde. The cells were then washed 3 times in PBS for 5 min each and placed in 10% goat serum (Vector Laboratories) and 0.2% Triton X-100 in PBS for 1 h at room temperature. Then, cells were washed once in 2% bovine serum albumin (BSA) (Sigma) in PBS before the addition of a 1:100 dilution of anti-IRF3 (Cell Signaling Technology) in 10% goat serum in PBS. Primary antibody incubation was performed at 4°C overnight. The coverslips were then washed 3 times for 5 min each in 2% BSA in PBS, and anti-rabbit Alexa Fluor 488 (Invitrogen) was added at a dilution of 1:500. Following secondary antibody incubation for 1 h at room temperature, cells were washed 3 times in 2% BSA in PBS for 5 min each, and the coverslips were allowed to air dry before being mounted onto slides with Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen).
Slides were examined on a Nikon Eclipse Ti microscope using Nikon Elements software. Images were taken using the same settings, and contrast adjustments were applied equally to all images.
Viral infections and plaque assays.
KSHV was produced as described previously using Vero cells containing latent rKSHV.219 (KSHV-Vero) and a recombinant baculovirus KSHV Orf50 (Bac50), kindly provided by Jeffrey Vieira (42). KSHV infection was performed as described previously (39, 40).
To produce EMCV, a baby hamster kidney (BHK)-21 cell monolayer was infected at a multiplicity of infection (MOI) of 10 with EMCV (ATCC VR-1762) for 24 h. Supernatants were harvested, subjected to centrifugation for 30 min at 10,000 rpm, aliquoted, and stored at −80°C. Viral titers were quantified on L929 cells by standard plaque assays as described previously (43). Infection of 293-TLR3 cells was performed in 12-well CellBind dishes (Corning) by applying 200 μl virus at the indicated MOI in complete medium for 1 h, after which an additional 800 μl complete medium was added to the cells. Twenty-four hours postinfection, cells and supernatants were harvested. EMCV plaque assays were performed on L929 cells by the addition of 200 μl serially diluted viral supernatant and incubation for 1 h prior to the addition of 2× MEMα medium mixed 1:1 with 2.5% carboxymethylcellulose (Sigma). Cells were incubated for 24 h, and then plaques were washed 1 time with water before being stained with 0.25% crystal violet and counted.
RESULTS
TLR3 levels are modulated by KSHV infection in monocytic cells.
Our previous work indicated that upon primary KSHV infection of the THP1 monocytic cell line, expression of TLR3 mRNA is upregulated approximately 8- to 15-fold compared to that for mock-infected cells (6). This increase in TLR3 expression was concomitant with increases in downstream TLR3 effectors, CXCL10, IFN-β, and CC chemokine ligand 2 (CCL2), at 16 h postinfection (6). In order to determine whether TLR3 upregulation was maintained postinfection, THP1 monocytic cells were infected with KSHV, and the relative levels of TLR3 message were determined by quantitative RT-PCR at multiple time points. TLR3 message levels initially increased, as seen at 16 h postinfection (6); however, at 48 h postinfection, TLR3 message levels declined (Fig. 1A). By 120 h postinfection, TLR3 message levels had decreased to a level similar to that seen in latently infected KSHV-THP1 cells (39). The same pattern was seen with CXCL10, a chemokine downstream of TLR3 (Fig. 1B). These data suggest that KSHV infection induces a host response, as reflected by increases in TLR3 and CXCL10 transcripts early after infection, but that the levels of these transcripts are downregulated over time.
Fig 1.
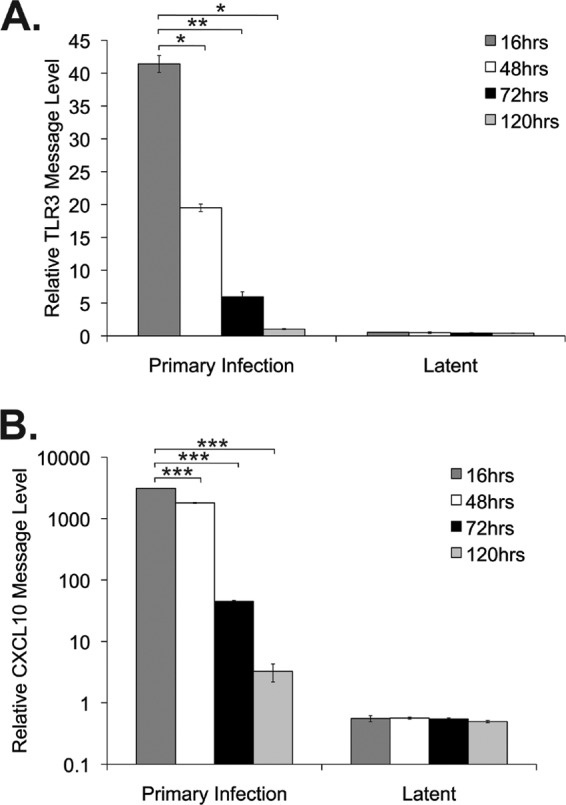
KSHV infection leads to decreased TLR3 and CXCL10 message levels. THP1 monocytic cells were infected with KSHV and harvested at the indicated time points. Latently infected KSHV-THP1 cells maintained under selection were harvested concurrently as controls. RNA was isolated from cells, and relative message levels were determined by quantitative RT-PCR. TLR3 (A) and CXCL10 (B) message levels were normalized to that of β-actin and are represented as fold increases over those of mock-infected monocytes after normalization. Values represent the means plus or minus the standard deviations of the means from duplicate biological replicates. *, P < 0.02; **, P < 0.005; and ***, P < 0.002 (by Student's t test).
vIRF1, -2, and -3 inhibit TLR3-activated interferon promoter elements.
KSHV downregulation of TLR3 and its downstream responses suggests that TLR3 may be a target of KSHV immune inhibition. As both vIRF1 and vIRF3 transcripts can be detected in latently infected monocytes (39), we hypothesized that KSHV vIRFs may antagonize TLR3 signaling to reduce immune detection. Furthermore, it remains unclear if the vIRFs are redundant or have unique functions in blocking IFN signaling. To address and compare the ability of vIRFs to inhibit TLR3 signaling, 293 cells stably expressing TLR3 and control 293 cells were cotransfected with the control vector or vectors expressing vIRF1, -2, or -3 together with an ISRE luciferase reporter. Twenty-four hours posttransfection, cells were stimulated with poly(I·C), a double-stranded RNA analogue and ligand for TLR3. All three vIRFs blocked the ISRE reporter activity induced by TLR3 (Fig. 2A). To confirm this result, a different reporter construct containing the entire human IFN-β promoter was cotransfected with the vIRFs (38). Expression of all three vIRFs decreased transcription of this reporter compared to transcription in vector control-expressing cells (Fig. 2B). All three vIRF proteins were adequately expressed prior to assessment of promoter activity (Fig. 2C). These data indicate that vIRF1, -2, and -3 inhibit TLR3-driven IFN promoter activation.
Fig 2.
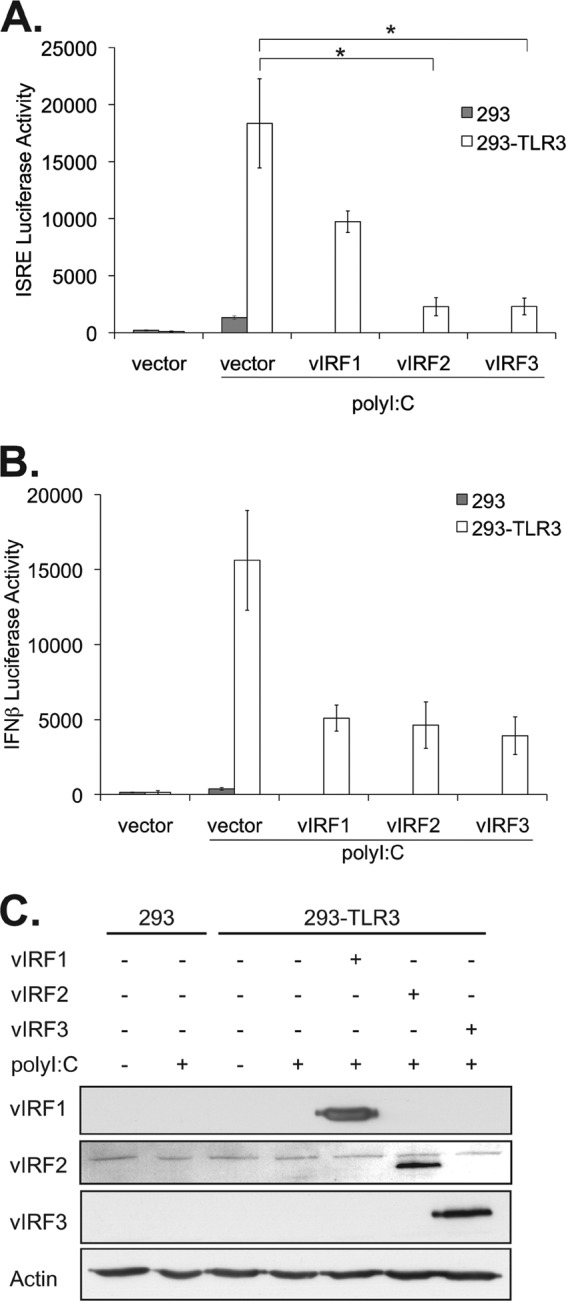
vIRF1, -2, and -3 block activation of IFN-responsive promoters. 293 and 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3 and cotransfected with ISRE (A) or pIFN-β (B) luciferase reporters. Twenty-four hours posttransfection, cells were treated with poly(I·C) for 24 h and harvested. Luciferase activity was measured and normalized to total protein content. (C) Lysates harvested 24 h after poly(I·C) treatment were subjected to SDS-PAGE and immunoblotted with the epitope tags Myc (vIRF1), Xpress (vIRF2), and FLAG (vIRF3) to determine the expression levels of vIRFs posttransfection. Values represent the means plus or minus the standard deviations of the means from triplicate samples. *, P < 0.02 (by Student's t test).
vIRF1 and vIRF2 block production of IFN-β message and protein.
To further assess vIRF1, -2, and -3 blockade of TLR3-mediated interferon responses, we examined the transcript levels of CXCL10, IFN-α, and IFN-β activated by TLR3 ligation. 293-TLR3 cells were transfected with an empty vector control or with vectors expressing vIRF1, -2, or -3 for 24 h followed by stimulation with poly(I·C). Response to the ligand was assessed by quantitative RT-PCR measurement of CXCL10, IFN-α, and IFN-β message levels normalized to that of the β-actin control and compared to vector-untreated cells. Expression of vIRF1 and vIRF2 resulted in decreased levels of CXCL10 message compared to those of the vector control and vIRF3 poly(I·C)-stimulated cells (Fig. 3A). Similarly, while IFN-α was only marginally induced by TLR3 ligation, vIRF1 was able to suppress this increase (Fig. 3B). Furthermore, vIRF1 and vIRF2 inhibited the increase in IFN-β message levels mediated by poly(I·C) treatment, while vIRF3 expression did not reduce IFN-β message levels (Fig. 3C). Next, we examined production and secretion of IFN-β by ELISA. To eliminate potential vector effects, all vIRFs were expressed from the same pcDNA3 vector and tagged with the FLAG epitope. Expression of either vIRF1 or vIRF2 significantly decreased TLR3-mediated IFN-β production, while vIRF3 expression had little effect (Fig. 3D). The inability of vIRF3 to block poly(I·C)-mediated IFN-β production in this cell type was not due to low vIRF3 expression levels, as vIRF2 expression appeared to be lower than that of vIRF3 (Fig. 3E). These data demonstrate that the vIRFs have a differential ability to block TLR3-induced IFN responses.
Fig 3.

vIRF1 and vIRF2 block TLR3-mediated transcription and production of IFN-β. 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3. Twenty-four hours posttransfection, cells were treated with poly(I·C) for 14 h, and cells were harvested for analysis by quantitative RT-PCR. Relative CXCL10 (A), IFN-α (B), and IFN-β (C) message levels were normalized to that of β-actin and represented as fold increases over those of untreated cells. (D and E) Cells were transfected with pcDNA3 vIRF-FLAG constructs or empty pcDNA3 vector as the control. Twenty-four hours posttransfection, cells were treated with poly(I·C), and cells and supernatants were harvested 24 h later. (D) Supernatants were quantitated for secreted IFN-β levels by ELISA. (E) Lysed cells were subjected to SDS-PAGE and immunoblotted with anti-FLAG to determine expression levels of vIRFs posttransfection. The asterisks denote nonspecific bands. (F) 293-TLR3 cells were transfected with control vector or vIRF1, -2, or -3. One day posttransfection, cells were infected with EMCV at an MOI of 0.1 for 24 h before harvesting. Quantitative RT-PCR was performed on RNA isolated from EMCV-infected cells to assess IFN-β message levels normalized to that of β-actin, represented as fold increase over control vector mock-infected cells. Values represent the means plus or minus the standard deviations of the means from triplicate samples. *, P < 0.03, and **, P < 0.02 (by Student's t test).
Next, we assessed the ability of vIRFs to block induction of TLR3 from a biological ligand. For these studies, we utilized the encephalomyocarditis virus, a single-stranded RNA virus within the Picornaviridae family. During EMCV replication, dsRNA is produced (44), which can activate TLR3 (45). 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3. Twenty-four hours after transfection, cells were infected with EMCV; 24 h postinfection, cells were harvested, and the production of IFN-β message was determined by quantitative RT-PCR. EMCV infection resulted in a >100-fold increase in the IFN-β message level compared to that of the mock-infected cells; expression of vIRF1 and vIRF2 inhibited this increase in IFN-β message levels, while vIRF3 did not have a significant effect on IFN mRNA induction (Fig. 3F).
vIRF1 inhibits IFN response by inhibiting IRF3.
The data presented above indicate that not all vIRFs block TLR3-mediated IFN responses equally, suggesting that the mechanisms by which the vIRFs inhibit IFN may be different. Currently, the mechanism of vIRF1 inhibition of IFN is best understood. vIRF1 is known to bind to cellular IRF1 (25, 33), IRF3, and IRF7 (34). Most notably, vIRF1 interaction with IRF3 inhibited IRF3-mediated transcription (34). This seems to occur through vIRF1 blockade of IRF3 cotranscription factors p300 and p300/CBP-associated factor (PCAF) (25, 34, 46, 47). To elucidate how vIRFs block TLR3-mediated IFN expression, we examined phosphorylation of IRF3 in 293-TLR3 cells expressing vIRFs or the control vector. Following poly(I·C) treatment, phosphorylation of IRF3 increased dramatically, and this increase was reduced to unstimulated levels by expression of vIRF1 but not vIRF2 or vIRF3 (Fig. 4A). Changes in phosphorylation were not due to changes in total IRF3 protein, as IRF3 levels remained unchanged regardless of poly(I·C) treatment. Furthermore, the expression of vIRF1 seemed to reduce IRF3 nuclear localization upon poly(I·C) treatment compared to the vector, vIRF2, and vIRF3 (Fig. 4B). These data suggest that while vIRF1 and vIRF2 both inhibit TLR3-mediated increases in IFN-β, they may accomplish this via different mechanisms.
Fig 4.

vIRF1 expression inhibits IRF3 downstream of TLR3. (A) 293 and 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3. Twenty-four hours posttransfection, cells were treated with poly(I·C) for 16 h before cells were harvested, lysed, and subjected to immunoblotting. (B) 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3, and 4 h posttransfection, cells were trypsinized and moved to 6-well dishes containing coverslips. Twenty hours after replating, cells were treated with poly(I·C) for 1 h before coverslips were harvested and stained for IRF3 (shown in green). DAPI staining is shown in blue. Arrows point to vIRF-expressing cells. (C) 293-TLR3 cells were transfected with a control vector or vIRF1, -2, or -3 and cotransfected with control vector or cellular IRF3. One day posttransfection, cells were treated with poly(I·C) for 24 h before supernatants were harvested and subjected to IFN-β ELISA. Values represent the means plus or minus the standard deviations of the means of results from triplicate samples. *, P < 0.05; **, P < 0.04; and ***, P < 0.01 (by Student's t test).
To address the ability of vIRF1 to block activation of cellular IRF3, cells were cotransfected with the control vector or vectors expressing vIRF1, -2, or -3 and an IRF3 expression plasmid or an empty control. Upon poly(I·C) treatment, all three vIRFs were able to significantly reduce IFN-β production when IRF3 was expressed (Fig. 4C). vIRF1 was able to decrease IFN-β expression to the highest degree. These data suggest that vIRF1 inhibits TLR3-mediated interferon responses by preventing IRF3 phosphorylation and nuclear translocation.
Expression of vIRF1 and vIRF2 results in increased EMCV replication.
To address the significance of vIRF blockade of IFN signaling and viral replication, we utilized EMCV-mediated activation of TLR3. 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3. One day after transfection, cells were infected with EMCV, and cells and supernatants were harvested 24 h postinfection to assess EMCV viral genomic load by RT-PCR and infectious titers by plaque assay. Expression of the vIRFs alone did not affect EMCV replication (Fig. 5A and B), and their expression was confirmed by immunoblotting (Fig. 5C). This result is in concordance with that in our previous report, demonstrating that vIRF2 had no effect on EMCV replication compared to that of the control (36).
Fig 5.
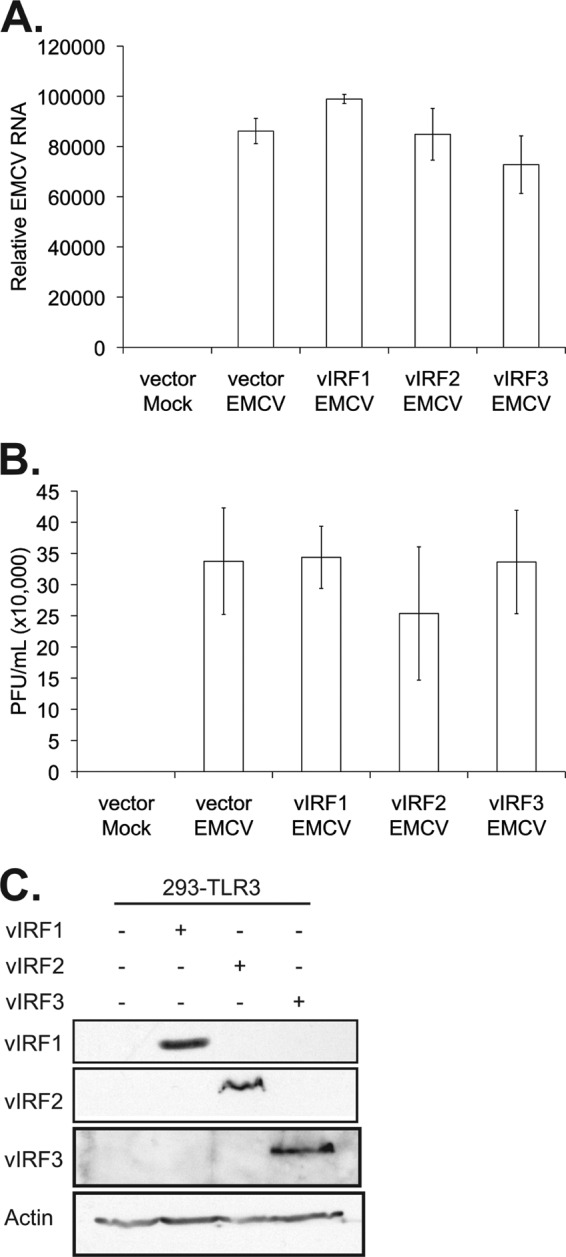
vIRF expression does not alter EMCV infectivity. 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3. Twenty-four hours posttransfection, cells were infected with EMCV at an MOI of 0.001 for 24 h before harvesting. (A) Quantitative RT-PCR was performed on RNA to measure EMCV genetic material, normalized to that of β-actin, represented as the fold increase over vector mock-infected cells. (B) Plaque assays were performed on L929 cells with six different 1:10 serial dilutions of viral supernatant. (C) Lysates harvested at the time of EMCV infection were subjected to SDS-PAGE and immunoblotted with the epitope tags Myc (vIRF1), Xpress (vIRF2), and FLAG (vIRF3) to determine the expression levels of vIRFs posttransfection. Values represent the means plus or minus the standard deviations of the means from triplicate biological replicates.
While the expression of the individual KSHV vIRFs had no effect on EMCV replication, it remained a possibility that the infection overwhelmed vIRF inhibition of immune responses. Therefore, cells were treated with poly(I·C) for 6 h prior to infection with EMCV to activate TLR3-mediated immune signaling. EMCV replication in cells pretreated with poly(I·C) and expressing either vIRF1 or vIRF2 was rescued as determined by increased viral RNA and production of infectious particles compared to those of the vector control (Fig. 6A and B). In contrast, cells expressing vIRF3 exhibited a statistically significant decrease in viral RNA and infectious particle production (Fig. 6A and B). This alteration in EMCV replication was not due to altered expression of the vIRFs (Fig. 6C). Together, these data suggest that vIRF1- and vIRF2-mediated blockade of the immune response may allow for more efficient infection by pathogens that activate TLR3. In contrast, vIRF3 expression did not affect EMCV viral replication.
Fig 6.
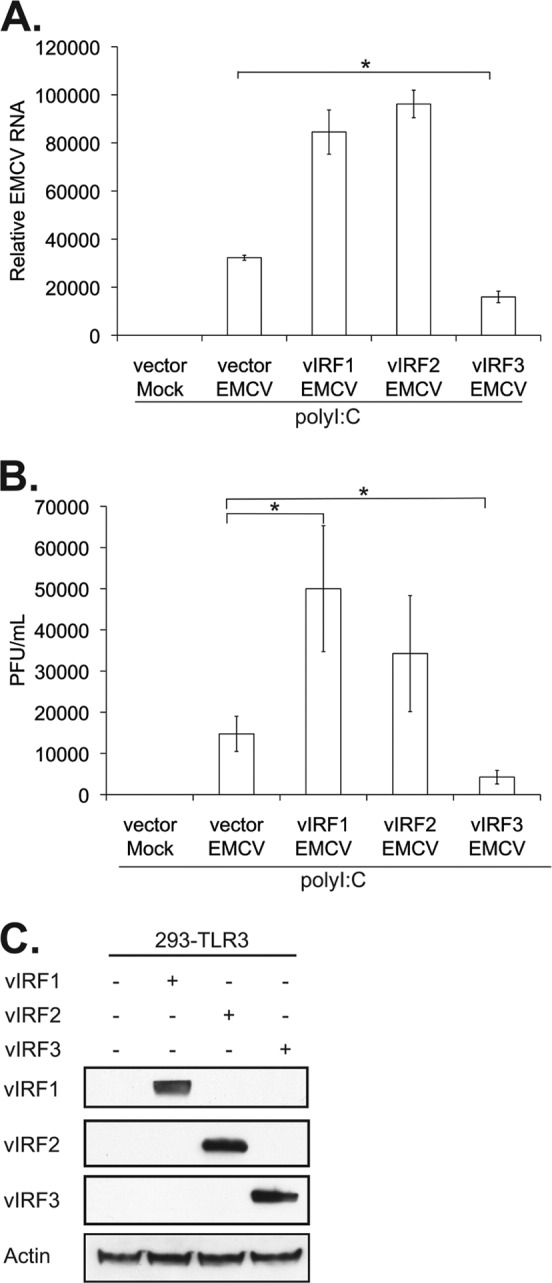
vIRF1 or vIRF2 expression leads to increased EMCV viral production following poly(I·C) pretreatment. 293-TLR3 cells were transfected with the control vector or vectors expressing vIRF1, -2, or -3. Twenty-four hours posttransfection, cells were treated with poly(I·C) for 6 h prior to infection with EMCV at an MOI of 0.001 for 24 h before harvesting. (A) Quantitative RT-PCR was performed on RNA to investigate EMCV genetic material, normalized to that of β-actin, represented as the fold increase over vector mock-infected cells. (B) Plaque assays were performed on L929 cells with six different 1:10 serial dilutions of viral supernatant. (C) Lysates harvested at the time of EMCV infection were subjected to SDS-PAGE and immunoblotted with the epitope tags Myc (vIRF1), Xpress (vIRF2), and FLAG (vIRF3) to determine the expression levels of vIRFs posttransfection. Values represent the means plus or minus the standard deviations of the means from triplicate biological replicates. *, P < 0.02 (Student's t test).
DISCUSSION
In summary, KSHV vIRF1, -2, and -3 displayed differences in their abilities to block interferon signaling induced by TLR3. At early time points following primary KSHV infection, TLR3 and CXCL10 message levels were increased in monocytes (6), but at later times these message levels decreased, suggesting that KSHV is capable of counteracting the host innate immune response to viral infection. Expression of KSHV vIRF1, -2, or -3 resulted in a blockade of TLR3-mediated activation of IFN-responsive promoter reporters. However, only vIRF1 and vIRF2 inhibited increases in both IFN-β message and protein levels in response to TLR3 activation. We found that vIRF1 but not vIRF2 or vIRF3 led to decreased phosphorylation and nuclear translocation of IRF3 in response to TLR3 activation. This suggests that the mechanisms by which the vIRFs reduce IFN activation may be different. Furthermore, vIRF1 and vIRF2 may block TLR3-mediating signaling via different mechanisms. Following infection with EMCV, the expression of vIRF1 and vIRF2 but not vIRF3 resulted in decreased production of IFN-β and corresponded with increased viral production when cells were pretreated with poly(I·C). While others have previously determined the ability of vIRFs to inhibit the induction of IFN upon viral infection (25, 30, 31, 34, 35), this study is the first to compare the impact of expression of different vIRFs on viral replication.
KSHV and its macaque homolog, rhesus macaque rhadinovirus (RRV), are the only viruses known to encode proteins with homology to cellular IRFs (48–50). Recently, an RRV knockout virus was generated in which all copies of vIRFs were removed (51). Infection of rhesus macaque peripheral blood mononuclear cells or fibroblasts with vIRF knockout RRV resulted in the increased induction of type I and II IFN compared to infection with wild-type RRV (51). Furthermore, IRF3 phosphorylation and nuclear accumulation were increased in knockout virus infection, suggesting that at least one RRV vIRF targets IRF3 to modulate type I IFN responses (51). Rhesus macaques infected with an RRV deleted for the vIRFs exhibited sustained production of proinflammatory cytokines, decreased viral load, and diminished B cell hyperplasia compared to those infected with the wild-type RRV (52). A similar result was observed when PEL cells with silenced KSHV vIRF1 expression were transferred into immunocompromised mice. The mice that received cells with decreased vIRF1 expression exhibited higher levels of IRF3 and signal-transducing activators of transcription 1 (STAT1) than mice given control cells (53). These data indicate that vIRFs may have a profound impact on viral infection and virus-related malignancies. Collectively, our data suggest that KSHV vIRF1, -2, and -3 are able to differentially inhibit IFN induction in a context-dependent TLR-specific manner. We found that KSHV vIRF1 and vIRF2 but not vIRF3 could inhibit TLR3-mediated interferon induction. These data raise the possibility that each vIRF may show specificity in inhibiting interferon signaling activated by a specific TLR.
ACKNOWLEDGMENTS
We thank members of the Damania and Dittmer labs for their helpful discussions.
This work was supported by NIH grant DE018281 (to B.D.). S.R.J. was supported by 5T32-CA09156 and 5T32-AI007151. B.D. is a Leukemia & Lymphoma Society Scholar and Burroughs Wellcome Fund Investigator in Infectious Disease. M.T.H. was supported by grant number R01 AI 067641. D.J.B. was supported by grants from the Medical Research Council (MRC) (G0400408 and G0800154), Cancer Research United Kingdom (C7934), and the University Hospital Birmingham Charities (17-3-694).
Footnotes
Published ahead of print 31 October 2012
REFERENCES
- 1. Ablashi DV, Chatlynne LG, Whitman JE, Jr, Cesarman E. 2002. Spectrum of Kaposi's sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin. Microbiol. Rev. 15:439–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 3. Schulz TF. 1998. Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8). J. Gen. Virol. 79:1573–1591 [DOI] [PubMed] [Google Scholar]
- 4. Alsharifi M, Mullbacher A, Regner M. 2008. Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 86:239–245 [DOI] [PubMed] [Google Scholar]
- 5. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 6. West J, Damania B. 2008. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J. Virol. 82:5440–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lagos D, Vart RJ, Gratrix F, Westrop SJ, Emuss V, Wong PP, Robey R, Imami N, Bower M, Gotch F, Boshoff C. 2008. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 4:470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. 2009. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U. S. A. 106:11725–11730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jacobs SR, Damania B. 2011. The viral interferon regulatory factors of KSHV: immunosuppressors or oncogenes? Front. Immun. 2:19 doi:10.3389/fimmu.2011.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. 2001. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75:429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee HR, Toth Z, Shin YC, Lee JS, Chang H, Gu W, Oh TK, Kim MH, Jung JU. 2009. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 83:6739–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakamura H, Li M, Zarycki J, Jung JU. 2001. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 75:7572–7582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seo T, Park J, Lee D, Hwang SG, Choe J. 2001. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 75:6193–6198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shin YC, Nakamura H, Liang X, Feng P, Chang H, Kowalik TF, Jung JU. 2006. Inhibition of the ATM/p53 signal transduction pathway by Kaposi's sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 80:2257–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baresova P, Pitha PM, Lubyova B. 2012. The Kaposi's sarcoma-associated herpesvirus vIRF-3 protein binds to F-box of Skp2 and acts as a regulator of c-Myc function and stability. J. Biol. Chem. 287:16199–16208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lubyova B, Kellum MJ, Frisancho JA, Pitha PM. 2007. Stimulation of c-Myc transcriptional activity by vIRF-3 of Kaposi sarcoma-associated herpesvirus. J. Biol. Chem. 282:31944–31953 [DOI] [PubMed] [Google Scholar]
- 17. Heinzelmann K, Scholz BA, Nowak A, Fossum E, Kremmer E, Haas J, Frank R, Kempkes B. 2010. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4/K10) is a novel interaction partner of CSL/CBF1, the major downstream effector of Notch signaling. J. Virol. 84:12255–12264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seo T, Park J, Choe J. 2005. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 1 inhibits transforming growth factor-beta signaling. Cancer Res. 65:1738–1747 [DOI] [PubMed] [Google Scholar]
- 19. Choi YB, Nicholas J. 2010. Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 6:e1001031 doi:10.1371/journal.ppat.1001031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Esteban M, Garcia MA, Domingo-Gil E, Arroyo J, Nombela C, Rivas C. 2003. The latency protein LANA2 from Kaposi's sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 84:1463–1470 [DOI] [PubMed] [Google Scholar]
- 21. Kirchhoff S, Sebens T, Baumann S, Krueger A, Zawatzky R, Li-Weber M, Meinl E, Neipel F, Fleckenstein B, Krammer PH. 2002. Viral IFN-regulatory factors inhibit activation-induced cell death via two positive regulatory IFN-regulatory factor 1-dependent domains in the CD95 ligand promoter. J. Immunol. 168:1226–1234 [DOI] [PubMed] [Google Scholar]
- 22. Seo T, Lee D, Shim YS, Angell JE, Chidambaram NV, Kalvakolanu DV, Choe J. 2002. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus interacts with a cell death regulator, GRIM19, and inhibits interferon/retinoic acid-induced cell death. J. Virol. 76:8797–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wies E, Mori Y, Hahn A, Kremmer E, Sturzl M, Fleckenstein B, Neipel F. 2008. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 111:320–327 [DOI] [PubMed] [Google Scholar]
- 24. Aresté C, Mutocheluh M, Blackbourn DJ. 2009. Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J. Biol. Chem. 284:23272–23285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burýsek L, Yeow WS, Lubyova B, Kellum M, Schafer SL, Huang YQ, Pitha PM. 1999. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 73:7334–7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burysek L, Yeow WS, Pitha PM. 1999. Unique properties of a second human herpesvirus 8-encoded interferon regulatory factor (vIRF-2). J. Hum. Virol. 2:19–32 [PubMed] [Google Scholar]
- 27. Flowers CC, Flowers SP, Nabel GJ. 1998. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor confers resistance to the antiproliferative effect of interferon-alpha. Mol. Med. 4:402–412 [PMC free article] [PubMed] [Google Scholar]
- 28. Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ. 2006. Inhibition of interferon signaling by the Kaposi's sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 80:3092–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao SJ, Boshoff C, Jayachandra S, Weiss RA, Chang Y, Moore PS. 1997. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 15:1979–1985 [DOI] [PubMed] [Google Scholar]
- 30. Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. 2007. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 81:8282–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lubyova B, Pitha PM. 2000. Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors. J. Virol. 74:8194–8201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wies E, Hahn AS, Schmidt K, Viebahn C, Rohland N, Lux A, Schellhorn T, Holzer A, Jung JU, Neipel F. 2009. The Kaposi's sarcoma-associated herpesvirus-encoded vIRF-3 inhibits cellular IRF-5. J. Biol. Chem. 284:8525–8538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zimring JC, Goodbourn S, Offermann MK. 1998. Human herpesvirus 8 encodes an interferon regulatory factor (IRF) homolog that represses IRF-1-mediated transcription. J. Virol. 72:701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ, Jr., Barber GN, Hiscott J. 2001. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 20:800–811 [DOI] [PubMed] [Google Scholar]
- 35. Lubyova B, Kellum MJ, Frisancho AJ, Pitha PM. 2004. Kaposi's sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J. Biol. Chem. 279:7643–7654 [DOI] [PubMed] [Google Scholar]
- 36. Mutocheluh M, Hindle L, Areste C, Chanas SA, Butler LM, Lowry K, Shah K, Evans DJ, Blackbourn DJ. 2011. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J. Gen. Virol. 92:2394–2398 [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Pagano JS. 1997. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 17:5748–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fredericksen B, Akkaraju GR, Foy E, Wang C, Pflugheber J, Chen ZJ, Gale M., Jr 2002. Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol. 15:29–40 [DOI] [PubMed] [Google Scholar]
- 39. Gregory SM, Wang L, West JA, Dittmer DP, Damania B. 2012. Latent Kaposi's sarcoma-associated herpesvirus infection of monocytes downregulates expression of adaptive immune response costimulatory receptors and proinflammatory cytokines.J. Virol. 86:3916–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. West JA, Gregory SM, Sivaramanan V, Su L, Damania B. 2011. Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin P, Hu SW, Chang TH. 2003. Correlation between gene expression of aryl hydrocarbon receptor (AhR), hydrocarbon receptor nuclear translocator (Arnt), cytochromes P4501A1 (CYP1A1) and 1B1 (CYP1B1), and inducibility of CYP1A1 and CYP1B1 in human lymphocytes. Toxicol. Sci. 71:20–26 [DOI] [PubMed] [Google Scholar]
- 42. Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240 [DOI] [PubMed] [Google Scholar]
- 43. Simpson DA, Davis NL, Lin SC, Russell D, Johnston RE. 1996. Complete nucleotide sequence and full-length cDNA clone of S.A.AR86 a South African alphavirus related to Sindbis. Virology 222:464–469 [DOI] [PubMed] [Google Scholar]
- 44. Aminev AG, Amineva SP, Palmenberg AC. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 95:59–73 [DOI] [PubMed] [Google Scholar]
- 45. Hardarson HS, Baker JS, Yang Z, Purevjav E, Huang CH, Alexopoulou L, Li N, Flavell RA, Bowles NE, Vallejo JG. 2007. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 292:H251–H258 [DOI] [PubMed] [Google Scholar]
- 46. Li M, Damania B, Alvarez X, Ogryzko V, Ozato K, Jung JU. 2000. Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell. Biol. 20:8254–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seo T, Lee D, Lee B, Chung JH, Choe J. 2000. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) binds to, and inhibits transactivation of, CREB-binding protein. Biochem. Biophys. Res. Commun. 270:23–27 [DOI] [PubMed] [Google Scholar]
- 48. Alexander L, Denekamp L, Knapp A, Auerbach MR, Damania B, Desrosiers RC. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26–95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Searles RP, Bergquam EP, Axthelm MK, Wong SW. 1999. Sequence and genomic analysis of a rhesus macaque rhadinovirus with similarity to Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 73:3040–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robinson BA, Estep RD, Messaoudi I, Rogers KS, Wong SW. 2012. Viral interferon regulatory factors decrease the induction of type I and type II interferon during rhesus macaque rhadinovirus infection. J. Virol. 86:2197–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robinson BA, O'Connor MA, Li H, Engelmann F, Poland B, Grant R, DeFilippis V, Estep RD, Axthelm MK, Messaoudi I, Wong SW. 2012. Viral interferon regulatory factors are critical for delay of the host immune response against rhesus macaque rhadinovirus infection. J. Virol. 86:2769–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang YJ, Patel D, Nan Y, Fan S. 2011. Inhibition of primary effusion lymphoma engraftment in SCID mice by morpholino oligomers against early lytic genes of Kaposi's sarcoma-associated herpesvirus. Antivir. Ther. 16:657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]