

Abstract
The involvement of the gut microbiota in metabolic disorders, and the ability of whole grains to affect both host metabolism and gut microbial ecology, suggest that some benefits of whole grains are mediated through their effects on the gut microbiome. Nutritional studies that assess the effect of whole grains on both the gut microbiome and human physiology are needed. We conducted a randomized cross-over trial with four-week treatments in which 28 healthy humans consumed a daily dose of 60 g of whole-grain barley (WGB), brown rice (BR), or an equal mixture of the two (BR+WGB), and characterized their impact on fecal microbial ecology and blood markers of inflammation, glucose and lipid metabolism. All treatments increased microbial diversity, the Firmicutes/Bacteroidetes ratio, and the abundance of the genus Blautia in fecal samples. The inclusion of WGB enriched the genera Roseburia, Bifidobacterium and Dialister, and the species Eubacterium rectale, Roseburia faecis and Roseburia intestinalis. Whole grains, and especially the BR+WGB treatment, reduced plasma interleukin-6 (IL-6) and peak postprandial glucose. Shifts in the abundance of Eubacterium rectale were associated with changes in the glucose and insulin postprandial response. Interestingly, subjects with greater improvements in IL-6 levels harbored significantly higher proportions of Dialister and lower abundance of Coriobacteriaceae. In conclusion, this study revealed that a short-term intake of whole grains induced compositional alterations of the gut microbiota that coincided with improvements in host physiological measures related to metabolic dysfunctions in humans.
Keywords: inflammation, gut microbiota, metabolic disorders, whole grains
Introduction
Obesity is associated with an increased risk in cardiovascular disease, type-2 diabetes, non-alcoholic fatty-liver disease and some cancers, and constitutes a major health concern worldwide (Cornier et al., 2008; Hu, 2011). A diet high in whole grains and dietary fibers has been shown to improve metabolic parameters related to these metabolic disorders (Liu et al., 1999; Fung et al., 2002; Liu et al., 2003; Murtaugh et al., 2003; Jensen et al., 2004; Nettleton et al., 2008). The mechanisms responsible for the benefits of whole grains are not completely understood. It has been proposed that the dietary fiber present in whole grains increases the viscosity of the digesta and binds to bile acids in the small intestine, thus contributing to decreased sugar and lipid (cholesterol) absorption (Behall et al., 2004; Alminger and Eklund-Jonsson, 2008). In addition, phytochemicals and other bioactive compounds in whole grains might provide metabolic benefits (Adom and Liu, 2002; Nilsson et al., 2006; Harris and Kris-Etherton, 2010). Furthermore, the metabolic inflammation associated with obesity and related diseases is now considered to trigger metabolic dysfunctions (Gregor and Hotamisligil, 2011), and the benefits of whole grains might be due to an anti-inflammatory action (Nilsson et al., 2008b; Rosén et al., 2011). In this respect, bacterial fermentation of undigestible constituents of whole grains in the gastrointestinal tract has been suggested to be partly responsible for the benefits of whole grains (Nilsson et al., 2008a; North et al., 2009; Harris and Kris-Etherton, 2010).
A consideration of the gut microbiome in the context of the health effects of whole grains has become especially relevant in light of recent research that indicated an etiological role of gut bacteria in metabolic disorders. Obesity and type 2 diabetes have been linked to alterations in the intestinal microbiota in both the humans and animal models (Ley et al., 2006; Turnbaugh et al., 2006; Cani et al., 2007; Larsen et al., 2010; Vijay-Kumar et al., 2010). If these aberrations contribute to human disease is still unclear, but pathophysiological indicators are reduced in animal models when animals are kept germ-free or when treated with antibiotics, and manifestations of disease can be transmitted through the gut microbiota (Ley et al., 2005; Cani et al., 2008; Vijay-Kumar et al., 2010; Henao-Mejia et al., 2012). Proposed mechanisms by which microbiota contribute to metabolic aberrations are the induction of lipolysis leading to increased fat storage (Bäckhed et al., 2007), hepatic de-novo synthesis of triglycerides (Bäckhed et al., 2004) and the alteration of bile acid metabolites with consequences to lipid metabolism in the host (Claus et al., 2011). Furthermore, the gut microbiome might exacerbate the systemic inflammation associated with obesity and related metabolic disorders (Hotamisligil, 2006; Ding et al., 2010), possibly through the induction of endotoxemia driven by lipopolysaccharide translocation through the intestinal epithelium (Cani et al., 2007; Amar et al., 2008; Cani et al., 2008; Li and Hotamisligil, 2010).
The interplay between the gut microbiota and host metabolism and the ability of whole grains to affect both of these aspects suggest that one mechanism by which whole grains confer their benefits might be through a modulation of the gut microbiome. Recent research has revealed that the composition and metabolism of the gut microbiota can be modulated through prebiotics and fiber (Flint et al., 2007; Louis et al., 2007), and these carbohydrates have been shown to improve metabolic markers in experimental models (Cani et al., 2007; Neyrinck et al., 2011). Despite these encouraging findings, human studies that investigate the effects of whole grains and cereal fibers on host metabolism have neglected, until now, to characterize the gut microbiome and explore its potential contribution to health improvements (Tilg and Kaser, 2011). In addition, although the effect of fiber on the gut microbiota has been recently studied in experimental animals (Neyrinck et al., 2011; Van den Abbeele et al., 2011), information on how whole grains impact human gut microbiome composition is lacking.
The aims of this study were to characterize the impact of the incorporation of whole grains to an otherwise unrestricted diet on gut microbial ecology in healthy human subjects, and to investigate whether a connection with metabolic and immunological improvements exists. For this purpose, we performed a human crossover study with three four-week whole grain treatments, and collected fecal and blood samples at baseline and at the end of each treatment. The effect of whole grains on fecal microbiota composition was characterized by pyrosequencing of 16S rRNA gene tags, and inflammatory and metabolic markers related to metabolic dysfunctions in humans were measured in blood samples. The molecular characterization of fecal microbiota in parallel to host phenotyping allowed an investigation of associations between diet-induced metabolic changes and shifts in the gut microbiome.
Materials and methods
Subjects
The human trial was approved by the Institutional Review Board of the Kansas State University (IRB Approval Number: 5298), and written informed consent was obtained from all subjects. Healthy participants (see Supplementary materials for inclusion/exclusion criteria) were recruited through leaflets distributed on-campus by the College of Human Nutrition at the Kansas State University, Manhattan, KS. Twenty-eight participants, 17 females and 11 males (age 25.9±5.5 years), took part in the study.
Test meals
Whole grain Prowashonupana Barley (Sustagrain Barley Quick Flakes, ConAgra Mills, Omaha, NE, USA) and whole-grain brown rice (Insta Grains Brown Rice Flakes, Briess, Chilton, WI, USA) flakes were used in this study. Three test meals with different amounts of total dietary fiber were included: a barley treatment (WGB), consisting of 60 g of barley (18.7 g total dietary fiber); a brown rice and barley treatment (BR+WGB), consisting of 30 g each barley and BR (11.5 g total dietary fiber); and a BR treatment, consisting of 60 g of BR (4.4 g total dietary fiber). Subjects were provided with individual bags containing a daily dose of the corresponding treatment (60 g of flakes). Nutritional information of the whole-grain flakes used in the study is available in the Supplementary materials and Supplementary Table S1.
Study design
The study was conducted as a randomized crossover trial over 17 weeks (Figure 1). The first week served as a baseline period, after which each subject underwent three four-week dietary treatments (BR, BR+WGB, WGB) in random order, and interspaced by two-week washout periods. The study was conducted under free-living conditions, and no dietary restrictions were imposed except that subjects were expected to be non-vegetarian. Subjects were instructed to consume the 60 g of flakes daily either plain, with yogurt or with milk, without time restrictions. Weekly symptom diaries were completed by the subjects in which they self-reported bowel movement, discomfort, flatulence, bloating, stool consistency and general well-being on a scale from 1 to 5 (1 being optimal/normal and 5 worst/abnormal).
Figure 1.

Experimental design. Time line of the randomized crossover trial. Three four-week dietary treatments were assessed in succession. The treatments were interspaced by two-week washout (WO) periods. Blood and stool samples (indicated by arrows) were collected during the baseline (BL) and at the end of each treatment period.
Subject parameters and determination of metabolic and immunological markers
Subject parameters were measured at the Human Metabolism Laboratory at Kansas State University. Total body composition was assessed at baseline with dual-energy X-ray absorptiometry (Prodigy GE-Lunar, GE, Waukesha, WI, USA). Blood samples were drawn at baseline and at the end of each dietary treatment after a 12 h overnight fast. An initial blood sample was drawn (time 0). A standard drink containing 75 g of glucose (Fisher Scientific, Pittsburg, PA, USA) was consumed within 10 min, and blood samples were collected at 15, 30, 45, 60, 90 and 120 min for the determination of postprandial glucose and insulin responses. Blood was immediately placed in tubes containing K2-EDTA (Vacutainer, BD, Franklin Lakes, NJ, USA) and centrifuged at 1000–1500x g for 13 min at 5–10 °C. Aliquots of plasma were transferred into tubes for storage at −80 °C until further testing.
Glucose and insulin were measured in plasma samples in duplicate using an automated analyzer (YSI 2300, YSI Life Sciences, Yellow Springs, OH, USA) and the Human Gut Hormone Immunoassay kit (Milliplex, EMD Millipore, Billerica, MA, USA) with a dual laser flow cytometer (Luminex, EMD Millipore, Billerica, MA, USA), respectively. A lipid profile, consisting of total cholesterol, high-density lipoprotein (HDL) and non-HDL cholesterol was performed on the preprandial samples (time 0) using the Cholestech LDX System (Alere, Waltham, MA, USA). Three markers of inflammation were measured in plasma samples by enzyme-linked immunosorbent assays (in duplicate): lipopolysaccharide-binding protein (LBP) (USCN Life Science and Technology, Huston, TX, USA), high-sensitive C-reactive protein (hs-CRP) (Symansis, Timaru, New Zealand), and interleukin 6 (IL-6) (R&D Systems, Minneapolis, MN, USA).
Short-chain fatty acids were quantified in fecal samples by gas chromatography as described in the Supplementary materials.
Compositional analysis of the fecal microbiota by pyrosequencing
Despite the fact that fecal samples represent microbial communities that are shed from the gut and not resident, they provide a good overview over the microbiota present in the distal colon, and are the most practical samples that can be obtained from subjects participating in nutritional trials. Subjects provided fecal samples within 24 h of blood sampling and 2 h of defecation. Fecal material and 1:10 fecal homogenates in phosphate-buffered saline (pH=7) were immediately frozen (−80 °C) and stored until further processing. Bacterial DNA was extracted from fecal homogenates as described by Martínez et al. (2010), using the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) in combination with enzymatic and mechanical cell lysis. Pyrosequencing of amplicons obtained by PCR with universal primers targeting the V1–V3 region of the 16 S rRNA gene was performed as previously described (Martínez et al., 2010), using the 454 Genome Sequencer FLX with GS FLX Titanium series reagents at the Core for Applied Genomics and Ecology (University of Nebraska). Sequences obtained during this study are deposited in the MG-RAST server under the accession numbers 4498555.3, 4498556.3 and 4498557.3.
Sequence processing was performed combining features of QIIME (Caporaso et al., 2010) and the Ribosomal Database Project pipeline (Cole et al., 2009). Three-thousand quality-controlled sequences per sample were randomly selected and used for taxonomic classification. Sequences were assigned to a bacterial phylum, family and genus using the Classifier tool of the Ribosomal Database Project (Wang et al., 2007). In addition, sequences were assigned to operational taxonomic units with 97% sequence homology as described in the Supplementary materials. Chao1 species richness estimator, and Shannon's and Simpson's (defined as 1-Dominance) diversity indices were computed with QIIME.
Query for genes encoding β-glucanases in genomes of human gut microbes
Bacterial genomes available in the Joint Genome Institute database were used to identify large-bowel associated bacteria with β-glucanase encoding activity. The integrated microbial genomes platform of Joint Genome Institute was used to conduct this survey. A list of the strains included in the survey is presented in the Supplementary materials. For the species identified to contain β-glucanase genes, their abundance in the fecal microbiota of our subjects was quantified by BLASTn.
Statistics
Results are presented as means±s.d. Differences in bacterial taxa and host phenotypes among treatments were determined by one-way analysis of variance with repeated measures in combination with Tukey's post-hoc tests, and P<0.05 was considered statistically significant. If the data were not normally distributed, values were subjected to transformations such as square root or logarithm with base 10 to achieve normality. If normality could not be achieved through transformations, the non-parametric Kruskal-Wallis test was performed. When only two groups of data were compared, Student's t-tests were performed. Correlations between host parameters and bacterial populations were assessed by Pearson's correlation tests using GraphPad Prism version 5.00 (GraphPad Software, La Joya, CA, USA). Associations between inflammatory markers and gut microbiome composition were also analyzed through linear models using SAS (SAS Institute Inc., Cary, NC, USA). Additional information on the statistical methods can be found in the Supplementary materials.
Results
Physiologic, metabolic and microbiome characteristics of the study population
Twenty-eight volunteers, 11 males and 17 females, participated in the nutritional trial, and subjects' parameters are presented in Table 1. Based on percent body fat, 13 subjects were classified as overweight, using as cutoff values >31% body fat for women and >25% for men. This Metabolic and immunological markers included in the study were plasma fasting glucose and insulin levels, glycemic and insulin postprandial response, a lipid panel (total cholesterol, HDL, non-HDL), and inflammatory markers (hs-CRP, IL-6 and LBP). The rationale for the inclusion of these markers is their suitability in determining the progression of metabolic aberrancies and the risk of cardiovascular disease (Schumann et al., 1990; Spranger et al., 2003; Cardellini et al., 2005; Ridker, 2009), and their association with obesity (Sun et al., 2010). Accordingly, positive correlations between body fat and all three inflammatory markers were observed (Figures 2a–c). LBP and hs-CRP were highly correlated (r=0.90, P<0.0001) (Figure 2d). The linear model identified body fat as a significant factor affecting IL-6 (P<0.01), hs-CRP (P<0.0001) and LBP (P<0.0001). Furthermore, significant positive correlations existed between IL-6 and postprandial glucose response (Figure 2a). Together, these associations substantiate the link between adiposity, a low-grade systemic inflammation, and glucose metabolism (Hotamisligil, 2006).
Table 1. Baseline characteristics of the 28 subjects, and differentiated by gender and percent body fat (values are presented as mean±s.d.).
| Overall |
Gender |
Body fata |
|||||
|---|---|---|---|---|---|---|---|
| All subjects (n=28) | Male (n=11) | Female (n=17) | P-value | Overweight (n=13) | Normoweight (n=15) | P-value | |
| Age | 25.9±5.4 | 26.7±5.4 | 25.4±5.8 | NS | 28.6±6.6 | 23.6±3.0 | < 0.05 |
| Weight (kg) | 72.3±18.3 | 87.7±17.1 | 62.3±10.5 | < 0.001 | 79.7±19.5 | 65.9±14.9 | < 0.05 |
| BMI (kg m−2) | 25.1±4.5 | 27.4±4.8 | 23.6±3.7 | < 0.05 | 27.9±4.4 | 22.7±3.0 | < 0.001 |
| Body fat mass (kg) | 20.8±10.3 | 20.2±11.6 | 21.2±9.8 | NS | 29.7±8.1 | 13.1±3.6 | < 0.001 |
| Body fat (%) | 29.6±11.0 | 22.8±8.5 | 34.0±10.8 | < 0.01 | 39.2±7.4 | 21.3±6.3 | < 0.001 |
| Cholesterol (mmol l−1) | |||||||
| Total cholesterol | 4.86±1.12 | 4.07±0.69 | 5.22±1.67 | < 0.01 | 4.76±1.26 | 4.75±1.07 | NS |
| Non-high-density lipoprotein | 3.13±1.03 | 2.78±0.74 | 3.29±1.38 | NS | 3.23±1.21 | 2.94±0.85 | NS |
| High-density lipoprotein | 1.65±0.42 | 1.30±0.28 | 1.84±0.57 | < 0.001 | 1.53±0.45 | 1.65±0.42 | NS |
| Fasting plasma glucose (mmol l−1) | 5.17±0.74 | 5.14±0.72 | 5.15±1.42 | NS | 5.44±0.93 | 4.94±0.40 | NS |
| Fasting plasma insulin (μlU ml−1) | 43.44±18.86 | 42.76±20.55 | 44.93±21.14 | NS | 49.77±19.92 | 40.34±18.38 | NS |
| Inflammatory markers | |||||||
| IL-6 (pg ml−1) (min–max) | 1.75±1.43 (0.06–5.17) | 1.18±0.81 (0.33–2.59) | 2.01±1.60 (0.06–5.17) | NS | 2.29±1.34 (0.65–4.89) | 1.28±1.33 (0.06–5.17) | NS |
| Hs-CRP (mg l−1) (min–max) | 1.69±2.24 (0.002–7.039) | 0.32±0.22 (0.052–0.805) | 2.38±2.46 (0.002–7.039) | < 0.01 | 2.47±2.36 (0.115–6.696) | 0.97±1.87 (0.002–7.039) | NS |
| LBP (μg ml−1) (min–max) | 15.09±19.85 (0.12–88.05) | 4.42±3.03 (1.00–9.61) | 20.91±22.41 (0.12–88.05) | < 0.01 | 23.98±24.90 (2.12–88.05) | 7.48±8.60 (0.12–28.51) | NS |
Abbreviations: hs-CRP, high-sensitive C-reactive protein; IL, interleukin; LBP, lipopolysaccharide-binding protein; NS, not significant.
Women with over 31% body fat, and men with over 25% body fat were considered as overweight individuals. All others were considered lean.
Figure 2.

Associations among host physiological characteristics and their correlation with bacterial populations in fecal samples at baseline. Heatmap displaying correlation coefficients between metabolic and physiological parameters of the study population at baseline (a). Correlations between hs-CRP with body fat (b), LBP with body fat (c), hs-CRP and LBP (d), hs-CRP and Ruminococcaceae (e), LBP with Ruminococcaceae (f) and Oscillibacter with postprandial AUC glucose (g). Pearson's correlation (r) and the corresponding P-values are presented.
Pyrosequencing revealed that the baseline fecal microbiota of the participants was dominated by the phyla Firmicutes and Bacteroidetes, with lower proportions of Verrucomicrobia and Actinobacteria, in agreement with previous molecular characterizations of the human fecal microbiota (Ley et al., 2006; Martínez et al., 2010). We investigated whether associations between host phenotypes and microbial populations existed (Supplementary Figure S1). No significant correlation was observed between any bacterial group and body fat or BMI, although overweight subjects harbored significantly lower abundances of Ruminococcaceae (10.8±5.4% versus 17.9±9.9%, P<0.05) and Faecalibacterium (1.8±1.8% versus 3.7±2.5%, P<0.05). The analysis revealed negative correlations between the family Ruminococcaceae and all the three inflammatory markers at baseline (Figures 2e and f, and Supplementary Figure S1). Within this family, the genera Faecalibacterium and Ruminococcus displayed negative correlations with hs-CRP (r=−0.48, P<0.05, and r=−0.60, P<0.01, respectively). The analysis also revealed a negative association between Oscillibacter and postprandial glucose area under the curve (Figure 2f). Regarding the markers of lipid metabolism, proportions of Bacteroidetes, Bacteroidaceae and Bacteroides were positively correlated to plasma HDL values (r=0.54, P<0.05; r=0.56, P<0.05; r=0.56, P<0.05; respectively) (Supplementary Figure S2).
Effects of whole grains on fecal microbial communities
Sequence data obtained by pyrosequencing were used to establish the effects of whole grains on the gut microbiota composition. This analysis revealed that whole grains had a measurable effect on gut microbiota composition. All three treatments significantly increased the bacterial diversity measured by Shannon's and Simpson's indices but not by Chao1 (Supplementary Figure S3). These results indicated an increase in community evenness (Shannon's and Simpson's), but not in total species richness (Chao1).
In accordance to previous studies that assessed the effect of diet on the gut microbiome (Martínez et al., 2010; Davis et al., 2011), substantial inter-individual variation was observed in response to whole grains (Supplementary Table 2). Despite this variability, several diet-induced shifts reached statistical significance in the entire study population. The proportion of the phylum Firmicutes increased, while Bacteroidetes were reduced (Table 2). The decrease in Bacteroidetes was largely caused by a reduction of the genus Bacteroides (Table 2).
Table 2. Abundance of dominant bacterial taxa (% of total microbiota) in fecal samples as determined by 454 pyrosequencing (values are presented as mean±s.d.).
| Baseline | BR | BR+WGB | WGB | P-value | Confirmation by linear model | |
|---|---|---|---|---|---|---|
| Phylum | ||||||
| Firmicutes | 57.30±14.13 | 65.06±11.40a | 65.53±10.64a | 65.42±12.05a | 0.003 | Yes |
| Bacteroidetes | 37.99±14.35 | 30.74±11.62a | 29.85±11.93a | 30.32±12.22a | 0.01 | Yes |
| Verrucomicrobia | 1.82±1.98 | 1.34±1.53 | 0.68±0.80 | 0.59±0.80 | NS | Yes |
| Actinobacteria | 1.24±0.97 | 1.42±1.78 | 2.23±3.32 | 2.05±2.73 | NS | Yes |
| Family | ||||||
| Bacteroidaceae | 28.55±15.73 | 22.89±10.37 | 21.19±11.87a | 23.48±12.62 | 0.013 | Yes |
| Lachnospiraceae | 22.21±7.90 | 22.62±7.91 | 23.11±6.56 | 22.65±7.63 | NS | Yes |
| Ruminococcaceae | 14.64±8.76 | 17.32±8.90 | 16.53±8.06 | 15.82±8.32 | NS | Yes |
| Incertae Sedis XIV | 5.79±3.15 | 7.63±4.47 | 8.16±3.97b | 8.62±4.32b | 0.001 | Yes |
| Porphyromonadaceae | 3.40±3.07 | 2.69±3.42 | 2.76±3.10 | 1.95±1.55a | 0.022 | No |
| Prevotellaceae | 2.97±9.24 | 2.34±6.56 | 3.59±10.10 | 2.39±6.50 | NS | Yes |
| Verrucromicrobiaceae | 1.85±4.58 | 0.77±1.53 | 0.68±1.28 | 0.59±0.80 | NS | Yes |
| Rikenellaceae | 1.77±2.09 | 1.68±1.85 | 1.12±1.06 | 1.35±1.68 | NS | Yes |
| Veillonellaceae | 1.59±1.13 | 1.52±1.19 | 1.86±1.19 | 1.97±1.60 | NS | Yes |
| Genus | ||||||
| Bacteroides | 28.55±15.73 | 22.89±10.37 | 21.19±11.87a | 23.48±12.62 | 0.022 | Yes |
| Blautia | 5.68±3.15 | 7.61±4.47 | 8.14±3.97b | 8.61±4.32b | 0.001 | Yes |
| Ruminococcus | 4.20±4.91 | 5.35±5.05 | 4.171±5.75 | 3.46±4.32 | NS | Yes |
| Faecealibacterium | 2.82±2.38 | 3.06±2.29 | 3.86±3.22 | 3.86±3.19 | NS | Yes |
| Prevotella | 2.79±8.89 | 1.99±6.24 | 3.34±9.84 | 2.02±6.30 | NS | Yes |
| Dorea | 2.59±2.01 | 3.12±2.22 | 3.08±1.80 | 2.75±1.86 | NS | Yes |
| Parabacteroides | 2.58±3.05 | 2.06±3.23 | 2.10±3.14 | 1.59±1.44 | NS | Yes |
| Roseburia | 1.98±1.35 | 1.70±1.25 | 2.42±1.58 | 3.06±2.91e | 0.01 | Yes |
| Akkermansia | 1.85±4.58 | 0.77±1.53 | 0.68±1.28 | 0.59±0.80 | NS | Yes |
| Coprococcus | 1.82±2.09 | 1.91±2.08 | 1.47±2.22 | 1.35±1.78 | NS | Yes |
| Alistipes | 1.76±2.08 | 1.67±1.85 | 1.11±1.05 | 1.34±1.67 | NS | Yes |
| Oscillibacter | 1.27±1.04 | 1.24±1.00 | 1.08±0.83 | 0.96±0.61 | NS | Yes |
| Bifidobacterium | 0.99±1.88 | 1.02±1.64 | 1.95±3.16 | 1.84±2.54d | 0.011 | No |
| Subdoligranulum | 0.94±1.03 | 1.17±1.43 | 1.42±1.73 | 1.09±1.02 | NS | Yes |
| Dialister | 0.75±1.17 | 0.60±0.89 | 0.94±1.21 | 1.14±1.69d | 0.027 | No |
| Odoribacter | 0.26±0.24 | 0.28±0.35 | 0.28±0.41 | 0.15±0.18b | 0.002 | No |
| Operational Taxonomic Units (OTU number, closest hit in database, % identity with 16S rRNA gene) | ||||||
| 1737, Odoribacter splanchnicus, 99% | 0.15±0.14 | 0.13±0.18 | 0.15±0.24 | 0.07±0.10b | 0.001 | No |
| 679, Eubacterium rectale, 94% | 0.25±0.32 | 0.31±0.42 | 0.43±0.57 | 0.57±0.63b,e | < 0.0001 | Yes |
| 956, Roseburia faecis, 99% | 0.12±0.17 | 0.06±0.07 | 0.26±0.31 | 0.53±0.92b,f | < 0.0001 | Yes |
| 770, Roseburia intestinalis, 100% | 0.09±0.12 | 0.04±0.05 | 0.17±0.18d | 0.30±0.42a,f | < 0.0001 | Yes |
| 3, Blautia wexlerae, 100% | 1.07±0.78 | 1.58±1.11 | 1.49±0.98 | 1.82±1.14c,g | < 0.0001 | Yes |
| 179-188, Blautia spp. | 1.81±1.13 | 2.38±1.69 | 2.75±1.75a | 2.80±2.04b | 0.006 | Yes |
| 44-19-1999-93, Eubacterium rectale, 98% | 2.48±2.67 | 2.75±3.27 | 3.65±3.45 | 4.83±3.98a,e,g | 0.001 | Yes |
Abbreviations: BR, brown rice; NS, not significant; OUT, operational taxonomic unit; WGB, whole grain barley.
Significantly different to baseline: aP<0.05, bP<0.01, cP<0.001.
Significantly different to BR: dP<0.05, eP<0.01, fP<0.001.
Significantly different to BR+WGB: gP<0.05.
The increase in Firmicutes was more comprehensive and shifts in the abundance of several taxa were detected. All three dietary treatments increased the abundance of the genus Blautia and two operational taxonomic units within this genus (Table 2), although significance was only achieved when WGB was included in the treatment. Several compositional shifts were strictly associated with the consumption of WGB, namely the genera Roseburia, Bifidobacterium and Dialister and the species E. rectale, R. faecis and R. intestinalis (Table 2), and many of these taxa increased gradually with WGB intake. The linear regression model confirmed all of these significant changes except for the species Bifidobacterium, and Dialister. Other taxa clearly responded to WGB, but because of inter-individual variation, these shifts did not reach statistical significance. For example, Bacteroides coprocola, which was only detected in three subjects, showed a 10-fold increase with WGB consumption in only two of the subjects (Supplementary Table S2). Although both whole grains led to an equivalent increase in the Firmicutes/Bacteroides ratio, no family or genus showed a significant increase for BR, suggesting that this test meal induced diverse alterations in the gut microbiome that are not consistent among subjects.
No significant differences were detected in the amounts of short-chain fatty acids for any of the treatments. It is possible that an increase in short-chain fatty acids could not be detected in fecal samples they are for the most part absorbed in the gastrointestinal tract (Millet et al., 2010).
Distribution of β-glucanase genes in human gut microbes
WGB contains a high amount of β-glucans (14.1%), while none were detected in BR (Supplementary Table S1). In order to test if the ability to hydrolyze β-glucans could explain the specific shifts in the fecal microbiota induced through WGB, we investigated distribution of β-glucanase genes in 112 strains originating from the human gut. This analysis revealed that β-glucanase genes are present in a variety of gut bacterial species from a broad taxonomic range, including ten Bacteroides, four Bifidobacterium, three Collinsella, two Clostridium, two Coproccus, two Eubacterium, one Ruminococcus, two Roseburia, and one Akkermansia species (Supplementary Table S2). Of these species, only E. rectale, R. faecis and R. intestinalis were significantly increased through WGB, indicating that the mere presence of β-glucanase encoding genes does not predict the changes in community composition in response to the diet.
Whole grain-induced metabolic and immunological changes
The daily consumption of 60 g of whole grains for 4 weeks improved immunological and metabolic markers in the human subjects. The findings for the entire study population are shown in Supplementary Table S3, and differentiated by gender and body fat in Supplementary Tables S4 and S5. A significant decrease in plasma IL-6 levels for the BR+WGB treatment versus baseline values was detected (Figure 3a). Quantitatively, this reduction was highest in overweight subjects (Figure 3b). In women, all three treatments significantly reduced IL-6 (Figure 3c). The linear model analysis confirmed the anti-inflammatory effect of whole grains and revealed a significant reduction of IL-6 for BR+WGB and WGB treatments (P<0.01, P<0.05). Despite not achieving statistical significance due to high inter-individual variation, hs-CRP plasma levels were halved during the BR+WGB period compared with the baseline (Supplementary Tables S3-S5).
Figure 3.

Immunological and metabolic improvements induced through whole-grain consumption. Plasma IL-6 levels in the entire subject population (a), in overweight participants (b), and in females (c). Maximum postprandial glucose levels in the entire subject population (d) and overweight subjects (e) during the three treatments (BR, BR+WGB, WGB) and at baseline. *P< 0.05, **P< 0.01, §P< 0.1.
Whole-grain consumption significantly improved glucose and lipid metabolism. Postprandial peak glucose levels were significantly lowered in overweight subjects during the BR+WGB period (P<0.05), and the reduction approached significance in the entire study population (P<0.1) (Figures 3d and e). Fasting glucose levels were significantly decreased in women and overweight subjects, and in females, total cholesterol was significantly reduced (Supplementary Tables S4 and S5).
Links between whole grain-induced metabolic improvements and fecal microbial community structure
To determine whether effects of whole grains were related to the gut microbiome, a correlation analysis was performed between bacterial shifts and changes in the metabolic markers that occurred during the BR+WGB period. We focused the analysis on the BR+WGB treatment as it induced the most significant metabolic improvements (Figure 3). This analysis revealed that increases in the abundance of E. rectale were associated with improvements in the postprandial glucose and insulin response (Supplementary Figures S4A and SB). The association between E. rectale and maximum postprandial glucose levels approached significance (Supplementary Figure S4C).
In addition, we categorized subjects into the three groups (terciles) according to the magnitude of the improvements in IL-6, hs-CRP, fasting glucose and glucose peak through BR+WGB. The baseline proportions of the bacterial groups between the three groups were compared. This analysis revealed that the gut microbiota of subjects with the highest improvement in IL-6 (3rd tercile) contained significantly higher percentages of Veillonellaceae (Figure 4a), and within this family, the genus Dialister (Figure 4b). Conversely, Coriobacteriaceae were significantly decreased in subjects with the highest improvement in IL-6 (Figure 4c). No significant differences in microbiome composition were detected between the terciles generated for hs-CRP, fasting glucose and postprandial glucose peak.
Figure 4.
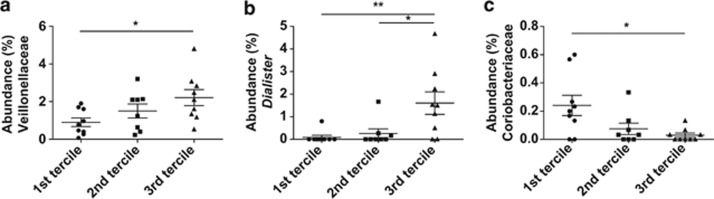
Abundance of specific taxa in subjects that showed differences in their IL-6 response to whole grains. Subjects were classified into terciles according to the magnitude of the change in plasma IL-6 levels induced by whole-grain consumption (BR+B treatment versus baseline). The proportions of bacterial taxa in fecal samples during the baseline were compared in the three terciles and significant differences existed in the proportions of Veillonellaceae (a), Dialister (b) and Coriobacteriaceae (c) in fecal samples during baseline. *P< 0.05, **P< 0.01.
Gastrointestinal symptoms
Self-reported symptoms diaries revealed that 60 g of WGB significantly increased all the gastrointestinal symptoms surveyed, especially flatulence, while 30 g caused only a slight increase in flatulence (Supplementary Table S6). The addition of BR to the diet did not result in any reported changes in symptoms.
Discussion
The metabolic and immunological benefits of whole grains have been shown in various studies (Fung et al., 2002; Behall et al., 2004; Jensen et al., 2004; Nilsson et al., 2006, 2008b), and a contribution of the gut microbiome to these effects has been suggested (North et al., 2009). However, the assessment of bacterial participation in these processes has been limited to hydrogen breath measurements, and the effects of whole grains on the gut microbiome structure have not been investigated. In this study, we showed that whole grains have a significant effect on the composition of the fecal microbiota that coincided with metabolic and immunological improvements in healthy human individuals.
All whole-grain test meals caused an increase in community diversity within the subjects, driven by an increase in evenness of bacterial species. Therefore, WGB and BR seem to differ in their effects on the gut microbiota when compared with prebiotics and dietary fibers, which have not been shown to increase community diversity (Martínez et al., 2010; Davis et al., 2011; Van den Abbeele et al., 2011). These differences might be due to compositional complexity of whole grains, which contain a variety of carbohydrates, potentially affecting a wider scope of bacterial taxa. Interestingly, a higher microbial diversity in fecal samples was also observed in children from Burkina Faso, who consumed a diet high in whole grains, legumes and vegetables, when compared with Europeans (De Filippo et al., 2010). In addition, weaning in human infants leads to a drastic increase in diversity likely caused by the incorporation of more diverse arrays of dietary carbohydrates (reviewed in Koropatkin et al., 2012). Therefore, it appears that bacterial diversity in the gut can be increased by providing a broader range of undigestible substrates, and our findings showed that this can be achieved by intake of whole grains.
This study revealed shifts in the fecal microbiota that were induced by both BR and WGB, while others were specific to WGB intake. Both whole grains increased the Firmicutes/Bacteroidetes ratio and the abundance of the genus Blautia. The overall shift in microbiota structure in favor of an expansion of Firmicutes could be the result of an increased carbohydrate intake (Duncan et al., 2008). However, in a previous study, we did not observe an increase in the Firmicutes/Bacteroidetes ratio with the consumption of crackers containing resistant starches (Martínez et al., 2010), although the dose of carbohydrates and fiber in these crackers exceeded that of the whole-grain test meals. Interestingly, a decrease of Bacteroides was also shown to be associated with a long-term consumption of diets rich in whole grains, dietary fibers and vegetables in African children and US individuals (De Filippo et al., 2010; Wu et al., 2011). These and our findings suggest that other components included in whole grains and other plant-derived food products might influence community structure at the phylum level, specifically decreasing Bacteroidetes. The reason for the increase in the genus Blautia through whole grains might be due to a syntrophic effect. Blautia species are acetogenic and might benefit from the production of hydrogen, which is a product of glycan fermentation, and, therefore, likely induced by whole grain consumption (Nakamura et al., 2010; Koropatkin et al., 2012).
We detected several bacterial taxa that displayed a specific increase with WGB, several with a clear dose response. This is likely due to its high content of β-glucans. Accordingly, the bacteria that specifically responded to WGB harbor genes encoding for β-glucanases and utilize the substrate in vitro (Hughes et al., 2008; Tasse et al., 2010). However, the in vivo findings cannot solely be explained based on functional and genomic attributes of community members, as Bacteroides species decreased during WGB consumption, but possess β-glucanase genes and can utilize β-glucans in vitro (Crittenden et al., 2002; Tasse et al., 2010; Zhao and Cheung, 2011). A possible explanation for the in vivo findings could entail preferences towards distinct β-glucan structures and molecular weights. The β(1–4) to β(1–3) linkage ratio in barley is 2.3-3, while Bacteroides species have been shown to especially possess β(1–3)-glucanase activity (Salyers et al., 1977). Moreover, barley-derived β-glucan fractions of high molecular weight have also been shown to be poorly fermented by Bacteroides (Hughes et al., 2008). However, previous human trials with prebiotics and resistant starches have also revealed that the ability of a species to utilize substrates in vitro does not predict population shifts in vivo (Martínez et al., 2010; Davis et al., 2011; Koropatkin et al., 2012). Therefore, although the findings obtained suggest that β-glucans are the main cause for the shifts in composition induced by WGB, the exact mechanisms by which these changes are restricted to only a small number of taxa are likely to be due to competitive interactions.
A main objective of this study was to determine whether the effects of whole grains on the gut microbiome are associated with physiological benefits. The whole grains used in our study led to immunological and metabolic improvements, especially when BR+WGB was consumed. Plasma IL-6 was reduced, and a tendency for a decrease in plasma hs-CRP was detected. In addition to this anti-inflammatory effect, an improvement in the glycemic response during BR+WGB treatment was detected. Our findings are in agreement with previous research that established the immunological and metabolic benefits of whole grains (Casiraghi et al., 2006; Kallio et al., 2008; Nilsson et al., 2008b; Rosén et al., 2011). Most importantly, inflammation has been identified as a main cause of metabolic disorders (Hotamisligil, 2006), and the anti-inflammatory effect could provide a mechanism by which whole grains improve glucose metabolism.
The anti-inflammatory effect of whole grains might be mediated through its effect on the gut microbiota. A remarkable positive correlation between LBP and hs-CRP was identified in our study population, supporting a link between bacterial lipopolysaccharide and systemic inflammation. The associations of these markers with body-fat support the hypothesis that endotoxemia could contribute to obesity (Cani et al., 2007; Delzenne and Cani, 2011). WGB led to an increase of bacterial taxa such as bifidobacteria and Roseburia, which have been suggested to affect immune/inflammatory and metabolic functions in animal models (Cani et al., 2008; Neyrinck et al., 2011). Although one could envision that these shifts might underlie the anti-inflammatory effect of whole grains, no significant correlations between these taxa and inflammatory markers were observed. However, shifts in the abundance of E. rectale induced through the BR+WGB diet correlated with decreased postprandial glucose and insulin responses. This organism produces butyrate, which might contribute to the immunological benefits of whole grain consumption through its anti-inflammatory effects.
Interestingly, compositional differences at baseline were detected in the gut microbiome of subjects that differed in the magnitude of their anti-inflammatory response to whole grains. Subjects with the greatest reduction in plasma IL-6 concentration had significantly higher proportions of Dialister and a lower abundance of Coriobacteriaceae. These bacterial groups have been linked to chronic inflammation in previous studies. D. invisus and Coriobacteriaceae have been shown to be reduced and increased in patients with Crohn's disease and colitic mice, respectively (Clavel et al., 2009; Würdemann et al., 2009; Willing et al., 2010; Joossens et al., 2011). The association of Dialister and Coriobacteriaceae with IL-6 response suggests that these taxa may condition the capability of an individual to be immunologically responsive to whole grains.
Before the start of the treatments, associations between bacterial groups, inflammatory state and host metabolism were observed (Figure 3). Ruminococcaceae negatively correlated with markers of inflammation and were more dominant in normoweight individuals. In addition, Bacteroidetes positively correlated with HDL cholesterol. These observations could result from an impact of these taxa on host physiology, and these associations provide a rationale to develop dietary strategies that target Ruminococcaceae and Bacteroidetes to improve human metabolic and immunological functions. However, host physiology (inflammatory state, cholesterol/bile acid metabolism) might also shape the microbiome composition. If systemic inflammation and cholesterol metabolism impact levels of Ruminococcaceae and Bacteroidetes, respectively, then these interactions could explain the discrepancies related to an altered microbiome in obese versus normoweight individuals (Ley et al., 2006; Duncan et al., 2008; Schwiertz et al., 2010). Not obesity per se, but the associated inflammatory and metabolic aberrations could shape microbiome composition and might cause variable and more complex patterns of dysbiosis.
This study has provided novel information about the relationship between whole grains, the gut microbiota and host metabolism. Whole grain-induced alterations in the characteristics and composition of the fecal microbiota coincided with immunological and metabolic benefits, and the clear associations between the reduction of IL-6 and the presence of certain bacterial taxa (Dialister, Coriobacteriaceae) indicate an important functional role of gut bacteria in the physiologic effects of whole grains.
Acknowledgments
The dedication of the subjects is greatly appreciated. The project was supported by ConAgra Foods (Omaha, Nebraska) and matching funds through the United States Department of Agriculture, Midwest Advanced Food Manufacturing Alliance program.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Adom KK, Liu RH. Antioxidant activity of grains. J Agric Food Chem. 2002;50:6182–6187. doi: 10.1021/jf0205099. [DOI] [PubMed] [Google Scholar]
- Alminger M, Eklund-Jonsson C. Whole-grain cereal products based on a high-fibre barley or oat genotype lower post-prandial glucose and insulin responses in healthy humans. Eur J Nutr. 2008;47:294–300. doi: 10.1007/s00394-008-0724-9. [DOI] [PubMed] [Google Scholar]
- Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behall KM, Scholfield DJ, Hallfrisch J. Lipids significantly reduced by diets containing barley in moderately hypercholesterolemic men. J Am Coll Nutr. 2004;23:55–62. doi: 10.1080/07315724.2004.10719343. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardellini M, Perego L, D'Adamo M, Marini MA, Procopio C, Hribal ML, et al. C-174G polymorphism in the promoter of the interleukin-6 gene is associated with insulin resistance. Diabetes Care. 2005;28:2007–2012. doi: 10.2337/diacare.28.8.2007. [DOI] [PubMed] [Google Scholar]
- Casiraghi MC, Garsetti M, Testolin G, Brighenti F. Post-prandial responses to cereal products enriched with barley beta-glucan. J Am Coll Nutr. 2006;25:313–320. doi: 10.1080/07315724.2006.10719541. [DOI] [PubMed] [Google Scholar]
- Claus SP, Ellero SL, Berger B, Krause L, Bruttin A, Molina J, et al. Colonization-induced host-gut microbial metabolic interaction. MBio. 2011;2:e00271–10. doi: 10.1128/mBio.00271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel T, Charrier C, Braune A, Wenning M, Blaut M, Haller D. Isolation of bacteria from the ileal mucosa of TNFdeltaARE mice and description of Enterorhabdus mucosicola gen. nov., sp. nov. Int J Syst Evol Microbiol. 2009;59:1805–1812. doi: 10.1099/ijs.0.003087-0. [DOI] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, et al. The metabolic syndrome. Endocr Rev. 2008;29:777–822. doi: 10.1210/er.2008-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden R, Karppinen S, Ojanen S, Tenkanen M, Faerstrom R, Matto J, et al. In vitro fermentation of cereal dietary fibre carbohydrates by probiotics and intestinal bacteria. J Sci Food and Agric. 2002;8:781–789. [Google Scholar]
- Delzenne NM, Cani PD. Interaction between obesity and the gut microbiota: relevance in nutrition. Annu Rev Nutr. 2011;31:15–31. doi: 10.1146/annurev-nutr-072610-145146. [DOI] [PubMed] [Google Scholar]
- Davis LMG, Martínez I, Walter J, Goin C, Hutkins RW. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One. 2011;6:e25200. doi: 10.1371/journal.pone.0025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, et al. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32:1720–1724. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- Flint HJ, Duncan SH, Scott KP, Louis P. Interactions and competition within the microbial community of the human colon: links between diet and health. Environ Microbiol. 2007;9:1101–1111. doi: 10.1111/j.1462-2920.2007.01281.x. [DOI] [PubMed] [Google Scholar]
- Fung TT, Hu FB, Pereira MA, Liu S, Stampfer MJ, Colditz GA, et al. Whole-grain intake and the risk of type 2 diabetes: a prospective study in men. Am J Clin Nutr. 2002;76:535–540. doi: 10.1093/ajcn/76.3.535. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Harris KA, Kris-Etherton PM. Effects of whole grains on coronary heart disease risk. Curr Atheroscler Rep. 2010;12:368–376. doi: 10.1007/s11883-010-0136-1. [DOI] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diab Care. 2011;34:1249–1257. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SA, Shewry PR, Gibson GR, McCleary BV, Rastall RA. In vitro fermentation of oat and barley derived beta-glucans by human faecal microbiota. FEMS Microbiol Ecol. 2008;64:482–493. doi: 10.1111/j.1574-6941.2008.00478.x. [DOI] [PubMed] [Google Scholar]
- Jensen MK, Koh-Banerjee P, Hu FB, Franz M, Sampson L, Grønbaek M, et al. Intakes of whole grains, bran, and germ and the risk of coronary heart disease in men. Am J Clin Nutr. 2004;80:1492–1499. doi: 10.1093/ajcn/80.6.1492. [DOI] [PubMed] [Google Scholar]
- Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut. 2011;60:631–6377. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- Kallio P, Kolehmainen M, Laaksonen DE, Pulkkinen L, Atalay M, Mykkänen H, et al. Inflammation markers are modulated by responses to diets differing in postprandial insulin responses in individuals with the metabolic syndrome. Am J Clin Nutr. 2008;87:1497–1503. doi: 10.1093/ajcn/87.5.1497. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Li P, Hotamisligil GS. Metabolism: host and microbes in a pickle. Nature. 2010;464:1287–1288. doi: 10.1038/4641287a. [DOI] [PubMed] [Google Scholar]
- Liu S, Stampfer MJ, Hu FB, Giovannucci E, Rimm E, Manson JE, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses' Health Study. Am J Clin Nutr. 1999;70:412–419. doi: 10.1093/ajcn/70.3.412. [DOI] [PubMed] [Google Scholar]
- Liu S, Willett WC, Manson JE, Hu FB, Rosner B, Colditz G. Relation between changes in intakes of dietary fiber and grain products and changes in weight and development of obesity among middle-aged women. Am J Clin Nutr. 2003;78:920–927. doi: 10.1093/ajcn/78.5.920. [DOI] [PubMed] [Google Scholar]
- Louis P, Scott KP, Duncan SH, Flint HJ. Understanding the effects of diet on bacterial metabolism in the large intestine. J Appl Microbiol. 2007;102:1197–1208. doi: 10.1111/j.1365-2672.2007.03322.x. [DOI] [PubMed] [Google Scholar]
- Martínez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One. 2010;5:e15046. doi: 10.1371/journal.pone.0015046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet S, Van Oeckel MJ, Aluwé M, Delezie E, De Brabander DL. Prediction of in vivo short-chain fatty acid production in hindgut fermenting mammals: problems and pitfalls. Crit Rev Food Sci Nutr. 2010;50:605–619. doi: 10.1080/10408390802565939. [DOI] [PubMed] [Google Scholar]
- Murtaugh MA, Jacobs DR, Jacob B, Steffen LM, Marquart L. Epidemiological support for the protection of whole grains against diabetes. Proc Nutr Soc. 2003;62:143–149. doi: 10.1079/pns2002223. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Lin HC, McSeeney S, Mackie RI, Gaskins HR. Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Annu Rev Food Sci Technol. 2010;1:363–395. doi: 10.1146/annurev.food.102308.124101. [DOI] [PubMed] [Google Scholar]
- Nettleton JA, Steffen LM, Loehr LR, Rosamond WD, Folsom AR. Incident heart failure is associated with lower whole-grain intake and greater high-fat dairy and egg intake in the Atherosclerosis Risk in Communities (ARIC) study. J Am Diet Assoc. 2008;108:1881–1887. doi: 10.1016/j.jada.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyrinck AM, Possemiers S, Verstraete W, De Backer F, Cani PD, Delzenne NM. Dietary modulation of clostridial cluster XIVa gut bacteria (Roseburia spp.) by chitin-glucan fiber improves host metabolic alterations induced by high-fat diet in mice. J Nutr Biochem. 2011;23:51–59. doi: 10.1016/j.jnutbio.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Nilsson A, Granfeldt Y, Ostman E, Preston T, Björck I. Effects of GI and content of indigestible carbohydrates of cereal-based evening meals on glucose tolerance at a subsequent standardised breakfast. Eur J Clin Nutr. 2006;60:1092–1099. doi: 10.1038/sj.ejcn.1602423. [DOI] [PubMed] [Google Scholar]
- Nilsson AC, Ostman EM, Granfeldt Y, Björck IM. Effect of cereal test breakfasts differing in glycemic index and content of indigestible carbohydrates on daylong glucose tolerance in healthy subjects. Am J Clin Nutr. 2008a;87:645–654. doi: 10.1093/ajcn/87.3.645. [DOI] [PubMed] [Google Scholar]
- Nilsson AC, Ostman EM, Holst JJ, Björck IM. Including indigestible carbohydrates in the evening meal of healthy subjects improves glucose tolerance, lowers inflammatory markers, and increases satiety after a subsequent standardized breakfast. J Nutr. 2008b;138:732–739. doi: 10.1093/jn/138.4.732. [DOI] [PubMed] [Google Scholar]
- North CJ, Venter CS, Jerling JC. The effects of dietary fibre on C-reactive protein, an inflammation marker predicting cardiovascular disease. Eur J Clin Nutr. 2009;63:921–933. doi: 10.1038/ejcn.2009.8. [DOI] [PubMed] [Google Scholar]
- Ridker PM. C-reactive protein: eighty years from discovery to emergence as a major risk marker for cardiovascular disease. Clin Chem. 2009;55:209–215. doi: 10.1373/clinchem.2008.119214. [DOI] [PubMed] [Google Scholar]
- Rosén LA, Ostman EM, Björck IM. Effects of cereal breakfasts on postprandial glucose, appetite regulation and voluntary energy intake at a subsequent standardized lunch; focusing on rye products. Nutr J. 2011;19:10–17. doi: 10.1186/1475-2891-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salyers AA, Palmer JK, Wilkins TD. Laminarinase (beta-glucanase) activity in Bacteroides from the human colon. Appl Environ Microbiol. 1977;33:1118–1124. doi: 10.1128/aem.33.5.1118-1124.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, et al. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- Schwiertz A, Tars D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18:190–195. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- Sun L, Yu Z, Ye X, Zou S, Li H, Yu D, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diab Care. 2010;33:1925–1932. doi: 10.2337/dc10-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasse L, Bercovici J, Pizzut-Serin S, Robe P, Tap J, Klopp C, et al. Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 2010;20:1605–1612. doi: 10.1101/gr.108332.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121:2126–2132. doi: 10.1172/JCI58109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Van den Abbeele P, Gérard P, Rabot S, Bruneau A, El Aidy S, Derrien M, et al. Arabinoxylans and inulin differentially modulate the mucosal and luminal gut microbiota and mucin-degradation in humanized rats. Environ Microbiol. 2011;13:2667–2680. doi: 10.1111/j.1462-2920.2011.02533.x. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würdemann D, Tindall BJ, Pukall R, Lünsdorf H, Strömpl C, Namuth T, et al. Gordonibacter pamelaeae gen. nov., sp. nov., a new member of the Coriobacteriaceae isolated from a patient with Crohn's disease, and reclassification of Eggerthella hongkongensis Lau et al. 2006 as Paraeggerthella hongkongensis gen. nov., comb. nov. Int J Syst Evol Microbiol. 2009;59:1405–1415. doi: 10.1099/ijs.0.005900-0. [DOI] [PubMed] [Google Scholar]
- Zhao J, Cheung PC. Fermentation of β-Glucans derived from different sources by bifidobacteria: evaluation of their bifidogenic effect. J Agric Food Chem. 2011;59:5986–5992. doi: 10.1021/jf200621y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.