

Abstract
This article constructs an argument for using blood chromatin (contained in nucleated blood cells) as a protein biosensor to integrate the ambient epigenetic influences in the internal milieu. An analogy is made to blood glycated hemoglobin (HbA1c) in diabetes as an integrated proxy for glucose levels and body-wide protein glycation. Genome-wide chromatin can serve as an organizing principle that bridges the central and peripheral compartments by entraining commensurable gene networks. Chromatin deposition along these networks will be imposed by the totality of epigenetic influences, which incorporates significant contributions from biochemicals that readily traverse the blood–brain barrier. In a clinical trial, these influences would be dominated by pharmaceuticals designed to override pathophysiological signals. In practice, mRNA readouts would be limited to nonsynaptic gene networks whose critical nodes are occupied by a site-specific chromatin modification. Finally, chromatin measurements in peripheral tissue will retain the influences of a patient’s lifestyle and unique genomic background.
Keywords: blood, brain, chromatin, histone, lymphocyte, neuron, PBMC, remodeling, schizophrenia
Post-mortem brain research has revealed numerous nonsynaptic proteins that distinguish between psychiatric disorders and these offer promising new targets. Many of these targets are involved in gene regulation such as nuclear chromatin, and consequently are not unique to neurons. Furthermore, these proteins are expressed in a broad array of cell types and are functionally regulated by biomolecules that freely traverse the central and peripheral compartments.
This article argues for a clinical (as applied to living human subjects) use of peripheral levels of specifically modified histone proteins, which are structural units of chromatin assemblies (Figure 1). This approach relies on the concept of a ‘blood level’, as opposed to a ‘disease-specific biomarker’. Conceptually this type of blood level would be similar to a physiological or pharmcological blood level. More precisely, blood levels of a modified histone protein are conceptually equivalent to blood levels of another widely used protein biosensor – that is, glycated hemoglobin (HbA1c) blood levels. HbA1c provides an ‘integrated summary’ of blood glucose over a finite time period (Table 1). Extending the analogy, HbA1c is not itself part of the receptor-mediated control of insulin by ambient glucose but is a valuable proxy for protein glycation in many end organs.
Figure 1. Nucleosome structure and histone tail.
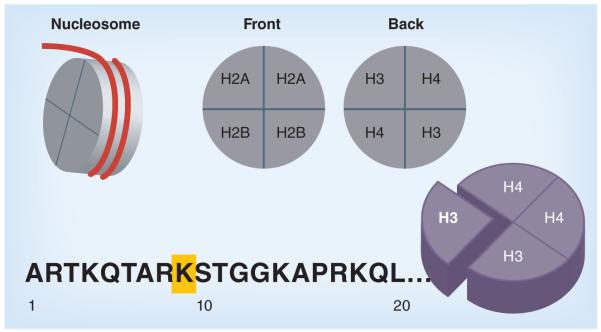
Depiction of DNA strand (red) wrapped around the nucleosome comprised of histone proteins. Amino acid residues along histone ‘tail’ are numbered, and in this representation, the yellow box indicates H3K9, whose acetylation (H3K9ac) or methylation (H3K9me2) comprise a regulatory switch. The H3K9 locus is one of several sites that are known to influence gene transcription.
Table 1.
Comparison of a widely accepted peripheral protein sensor (blood glycated hemoglobin) and a putative peripheral histone sensor for chromatin modifications.
| Characteristics | RBC HbA1c protein | PBMC histone protein |
|---|---|---|
| Chemical bond | Covalent | Covalent |
| Mechanism | Nonenzymatic | Enzymatic |
| Intraindividual variation without perturbation |
<1% [57] | <5% across 12 weeks [49] |
| Measured protein | RBC hemoglobin | PBMC nuclear histones |
| Proxy for | Whole-body glycation of amino acids, AGE and their receptor activity at RAGE |
Whole-body equilibrium between epigenetic environment and the status of histone acetylation or methylation |
| Predictive purpose | Level of nonenzymatic glycation of various proteins and AGE, collagen and the pathological outcomes at RAGE (demyelination, amyloid proteins in the brain, collagen, fibrinogen and acrylamide released) |
Regulation of genome-wide gene expression. The chromatin regulation of nonsynaptic gene networks relating to energy metabolism, inflammation, apoptosis and signaling cascades will be uniformly entrained by the epigenetic milieu and may be comparable |
| Lifespan of host cell; minimum duration of perturbation (days) |
120 | 30 |
| Reversibility of the modification | Advanced glycation products are generally irreversible |
Each modification site may have different temporal dynamics. Acetylation or deacetylation is rapid. Histone methylation is a more stable and durable modification [17] |
| HbA1c and PBMC histones will not predict | Long-term irreversible outcome of AGE in protein crosslinking in numerous protein species |
PBMC chromatin will not proxy for synapse-associated mRNA expression |
AGE: Advanced glycation end products; HbA1c: Glycated hemoglobin; PBMC: Peripheral blood mononuclear cell; RAGE: Receptor for advanced glycation end products; RBC: Red blood cell.
Chromatin modifications are multiple & multifunctional
Chromatin is a complex of DNA and protein located in the cell nucleus, and is now known to exert a dominant role in the transcription of genes along the encased DNA strand. The fundamental assembled unit of this complex is the nucleosome depicted in Figure 1. There are numerous sites along the histone amino acid sequence where post-translational modifications are known to impact chromatin configuration and gene expression. Consider the H3 histone protein: acetylation at lysines 9, 14, 18 and 23, and methylation at lysines 4, 36 and 79 results in an open chromatin configuration, while methylation at lysines 9 and 27 results in a closed configuration [1]. The author’s own research has focused on the clinical blood levels of the modified H3K9 residue. [2–5]. The H3K9 residue can either be acetylated or methylated. When acetylated, the local chromatin is in an ‘open’ configuration, and is conducive to gene regulation. When methylated, the local chromatin is in a ‘closed’ configuration and is restrictive to transcription, and is sometimes called ‘heterochromatin’. Thus, heterochromatin is characterized by either the paucity of acetylated H3K9 (H3K9ac) or the presence of a hallmark histone signature, that is, di- or tri-methylation of H3 (H3K9me2/3). H3K9me2 is one example of a theoretically useful modification to identify chromatin repressed gene networks. This derives from the following observations: it is a stable and durable modification; H3K9me2 occupancy is associated predominantly with repression of the gene promoter [6,7]; it is the high-affinity ligand for the attachment of HP1 protein and in this role serves as the gateway to the assembly of an even more restrictive and possibly more resistant type of heterochromatin via the coordination of DNA methylation on site; and it has recently been reported that H3K9me2 is modified by cocaine, a psychostimulant and psychotomimetic [8–10].
Epigenetic modifiers are ambient in both peripheral & central compartments
Chromatin in both peripheral and central compartments is exposed to/regulated by a common array of nonsynaptic biochemical modifiers. Environmental events such as inflammatory responses (cytokines/interleukins and antibodies) [11], viral infections [12], lipids [13] and drugs of abuse (cocaine [14], alcohol [15] and nicotine [16]) have currently known chromatin-remodeling effects. It is clear that biochemicals with intracellular effects such as alcohol or antiepileptics freely traverse both compartments, and in the case of valproic acid, act directly on chromatin levels [2–5]. Nuclear receptor ligands including vitamins, thyroid hormones and steroid hormones (glucocorticoids, estrogens and progesterone) function as ‘chromatin switches’ whose attachment to cognate DNA sequences results in the recruitment of epigenetic modifying enzymes including histone acetyltransferases, histone deactylases (HDACs) and histone methyltransferases [17–20]. This broad array of known epigenetic influences contributes to a ‘total’ epigenetic milieu and is capable of accruing overtime, such as the increasingly chromatin-restricted genome across the developmental trajectory. It should be noted that all epigenetic modifiers may not be present in all cells under all circumstances, but such cell-specific differences are likely to be diminished by the presence of a powerful pharmaceutical agent as in a clinical trial.
Pharmaceuticals such as commonly used psychotropics with receptor-binding properties (all antipsychotics, antidepressants and benzodiazepines) must transit the blood stream before entering the brain. In the blood stream, they must interact with circulating peripheral blood mononuclear cells (PBMCs), which express a wide array of nonsynaptic membrane receptors. These nonsynaptic receptors include those that have demonstrable chromatin-modifying effects, such as the dopamine D2–D5 receptors [21,22], serotonin receptors [23], adrenergic receptors [24], dopamine and serotonin transporters [25], nicotine [26] and opioid receptors [27]. It is almost certain that cellular signaling cascades triggered by these membrane receptors will contribute to the totality of changes in PBMC chromatin [28].
What to measure in living patients: global modifications, gene networks or single promoters?
Global changes in histone modifications, which can be measured in total nuclear extracts, are induced by most physiological/pharmaceutical influences [2–5,11,29], as well as by knockouts of key enzymes [8,30]. Global histone measures are correlated with well-defined cellular or neurobiological functioning [31]. Therefore, global levels of a particular histone modification(s) would represent a raw summary of the epigenetic influences operative during a period and could be used to monitor experimental pharmacology in a clinical treatment trial.
The open or closed nature of chromatin assemblies can also be assessed by measuring gene expression levels in peripheral tissue. This is because mRNA transcription is the ‘readout’ of chromatin assemblies. Network or single-gene promoter studies usually conducted with chromatin immunoprecipitation (ChIP) have already demonstrated involvement of chromatin remodeling in single-gene expression relevant to CNS disease [32]. We have shown that GAD67, as an example of an epigenetically regulated prototypic schizophrenia gene, can be transcribed in a dose-dependent manner by an HDAC inhibitor in PBMCs from living patients [4]. Progressing from single-gene readouts to nodal patterns of selected gene networks is intuitive because any given chromatin modification has a genome-wide distribution and is likely to occupy promoter networks common to all cells across the central–peripheral divide (see next section). Consequently, gene-expression comparisons must be limited to chromatin-entrained network(s) that are commensurably regulated in both central and peripheral compartments (Figure 2). Importantly, the poised states of chromatin not being reflected by expression must be considered when measuring the ‘epigenetic temperature’ at a particular promoter or genomic sequence (see next section).
Figure 2. H3K9 dimethylation-entrained gene networks revealed by ChIP-Seq.
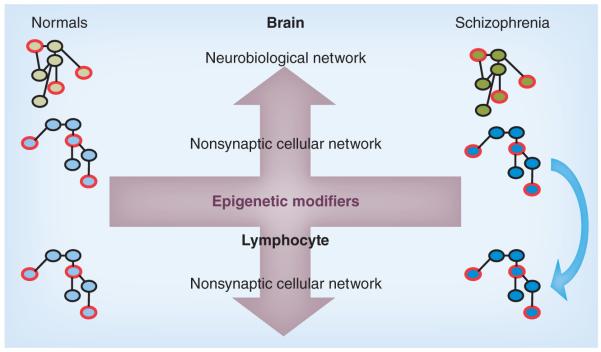
Promoter networks that are entrained by the restrictive H3K9 dimethylation (H3K9me2) mark are hypothesized to be enriched in schizophrenia brain (represented as the darker colors) [10]. Two types of networks can be excavated based on promoter occupancy and ontologies/networks. A neurobiological network with the densest occupancy by H3K9me2 can be utilized for neurobiological comparison in brain samples (green network). A parallel network based on common cell physiology (blue network) that shows enrichment in both brain and lymphocyte (peripheral blood mononuclear cell; arrow) from schizophrenia patients can be utilized to monitor H3K9-based network regulation in living patients and with pharmacological treatment. A finite selection of hubs/nodes (red borders) can be utilized for comparison of mRNA expression.
Genome-wide transcription & chromatin-entrained networks
Chromatin-containing PBMCs are not neurons and clearly cannot be used to monitor synaptic proteins. Consequently, the use of peripheral tissue is best supported if mRNA measurements are limited to nonsynaptic proteins whose both expression and function are common to PBMCs and neurons. In this common domain, there are three categories of chromatin occupancy that could theoretically be similar between PBMCs and neurons: open chromatin with active expression; open chromatin with poised but inactive expression; and repressed chromatin with silent expression. There is strong evidence of similar chromatin-expression patterns across tissue types as noted below. As a backdrop, we note that patient-specific PBMCs carry the full complement of genetic variation (e.g., promoter SNPs) and chromatin enzymes also present in the neuron.
■ Overlapping patterns Genome-wide mRNA expression across tissues
Genome-wide mRNA expression data, now known as the transcriptome map, have striking similarities in gene expression across tissues such as brain and mesodermal tissue [33,34]. Furthermore, Middleton et al. have now demonstrated that mRNA-expression patterns in PBMCs can distinguish between psychiatric diagnostic groups, which by definition are diseases of brain tissue [35]. In this study, using mRNA-expression differences from PBMCs for predictive classification, greater than 85% diagnostic accuracy was achieved. Among the identified signals were proteins linked to chromatin remodeling (e.g., Sin3 and the SWI/SNF complex) [35]. In addition, the cytogenetic loci of differentially expressed genes mapped remarkably well to the genetic-linkage map locations identified by population genetic studies from both schizophrenia and bipolar sibling-pair analyses. Sullivan et al. report that 50% of a set of schizophrenia candidate genes (including GAD67) were comparably expressed in PBMCs and the brain [2,36,37].
Genome-wide chromatin maps across tissues
How similar is the genome-wide chromatin map between neuronal and non-neuronal tissue? This question is informed by two studies charting the occupancy of two separate histone modifications, namely H3k4me3 (a mark of active promoters [38]) and H3K9me2 (a mark of restrictive chromatin [39]). Findings applicable to our proposal are that only 6213 out of 15,204 annotated promoters (40.8%) were neuron-specific compared with PBMCs or conversely almost 60% of active genes marked by H3K4me3 are common to both neurons and PBMCs. Likewise, Wen et al. have compared adult mouse brain versus liver using H3K9me2 ChIP and DNA sequencing, and demonstrate that identical gene promoters occupied by H3K9me2 are silenced in both tissues, suggesting an equivalent chromatin–mRNA relationship for many genes in either neuronal versus non-neuronal tissue [39]. We have demonstrated this equivalence at the single candidate-gene level with the GAD67 promoter showing epigenetic regulation in both brain and PBMCs [2,37].
Repressed promoters are insulated from tissue-specific transcription factors, and have significantly less variability in expression across tissues [39]. Consequently, chromatin deposited on repressed cellular networks maybe a better proxy for the effects of systemic biomolecules as noted above, and particularly the response to a pharmaceutical modifier such as an HDAC inhibitor(Figure 2). The ‘opening’ of a previously repressed promoter should be a clearly interpretable readout and, hypothetically, the clinical response could be linked to the ‘lighting up’ of commensurate nonsynaptic networks. Genes involved in energy metabolism are an example of a nonsynaptic network that could be monitored [40]. The measurement of this readout in circulating PBMCs would approximate changes in CNS chromatin exposed to a common pharmaceutical modifier (Figure 2).
■ Nonsynaptic gene networks in post-mortem brain of psychiatric disorders
If chromatin levels in the PBMC are to be a useful proxy in clinical psychiatric studies, two requirements must be satisfied. The first requirement would be to identify abnormally regulated gene networks in the diseased brain. The second requirement would be to select, from among these ‘diseased’ networks, one that is chromatin entrained and commensurately regulated in both neuron and PBMC. By necessity, these would need to be nonsynaptic networks. Towards the first requirement, several gene networks not primarily linked to synaptic neurotransmission are now identified to be differentially regulated in the brains of psychiatric subjects. These include genes connected to inflammation and apoptosis [41], immune function [42,43], energy metabolism [40], arachidonic acid signaling and prostaglandins [44], housekeeping functions (translation, transcription, energy conversion and metabolism) and stress response (heat shock and biotic stress) [40].
What these studies clearly establish are differences in expression between both the brain and PBMCs of psychiatric patients. What is now required is a mapping function between these two tissues.
■ Chromatin: a mapping function between central & peripheral compartments
Transcriptional irregularities in schizophrenia are documented in both the brain and the periphery; however, no mapping transform of these compartments has been possible, primarily due to the lack of a mapping function. Gene expression is regulated by transcription factors via a variety of DNA–protein interactions, including modifications to the chromatin complex. Specific chromatin modifications and the enzymes that catalyze their formation are a common biological platform in all cells. Superimposed on this transcription factor-based regulation, histone-modifying enzymes will respond to influences from the common epigenetic milieu, especially if pharmaceuticals are introduced. The peripheral and central compartments become especially comparable with the introduction of powerful pharmaceutical enzyme inhibitors that are designed to override aberrant transcriptional signals. Under these conditions, the intensity of a specific modification, such as the repressive H3K9me2/3 modification along the nodes of a network, will be the result of the effects of the pharmaceutical inhibitor over time. Thus chromatin can link central/peripheral compartments by the common biology of histone modifications and serve as an organizing principle or the required mapping function.
Psychiatric epigenetic studies using peripheral blood chromatin levels
■ Clinical studies in schizophrenia patients
The first chromatin studies in schizophrenia were reported in 1975, when Issidorides et al. noted a difference in the nucleohistone staining patterns of peripheral blood neutrophils from patients, suggestive of decondensed heterochromatin [45]. Furthermore, pimozide, a dopamine antagonist, was effective in normalization of this abnormal heterochromatin pattern [45,46]. Later, Kosower et al. observed a smaller heterochromatic C-band on chromosome 1 in schizophrenia patients compared with normal controls, suggestive of condensed chromatin in regions of the genome [47]. In vitro treatment of cultured peripheral blood cells obtained from patients with the epigenetic modifying drug 5-azacytidine (a DNA methyltransferase inhibitor causing reduced methylation of DNA strand) caused a decondensation of heterochromatin and a reduction in the spread of the 1qH heterochromatic C-band, but to a lesser degree in schizophrenia subjects [47]. The author’s laboratory has noted that levels of site-specific histone modifications measured in patient blood samples can distinguish between schizophrenia and bipolar patients, as well as monitor differences in pharmaceutical HDAC inhibitor (valproic acid) response across 4 weeks of clinical treatment [2–5]. Measurements of H3K9ac revealed that schizophrenia subjects are less efficient in ‘opening up’ their chromatin given the same blood levels of the HDAC inhibitor valproic acid [5]. Another recent study has measured the extent of decondensation of the genome in PMBCs from living schizophrenia patients as measured by the level of exposed arginine residues in the core histones [48]. This investigation reports a higher ratio of arginines:lysines, indicating a more reactive genome in patients with a new onset of schizophrenia.
■ Differences in epigenetic parameters between monozygotic twins
Fraga et al. have demonstrated that chromatin parameters measured in PBMCs will distinguish identical twins across their lifespan [49]. Numerous epigenetic studies have demonstrated differences in DNA-methylation profiling of PBMCs between normal monozygotic twins [49,50], as well as monozygotic twins discordant for bipolar disorder [51], or discordant for schizophrenia [52]. Mill et al. demonstrate differences in the methylation of the COMT gene in healthy monozygotic twins measured at 5 years of age, which predates the common age of onset in schizophrenia [50]. Differences in DNA methylation were also reported in a single pair of discordant monozygotic twins using a restriction enzyme method for genome scanning [53].
Chromatin studies in peripheral blood cells from living patients
Circulating peripheral blood cells from a standard venipuncture can be analyzed for total nuclear chromatin, promoter chromatin occupancy and mRNA expression, or examined in nonactivated in vitro cultures. Because a PBMC carries the full complement of epigenetic enzymes and machinery found in all tissues, including neurons [54,55], a reasonable hypothesis is that both tissue types are utilizing similar proteins to execute similar modifications. In a clinical trial where a therapeutic enzyme inhibitor (such as an HDAC inhibitor) is being administered, the chromatin structure in freshly extracted PBMCs will be a first-order function of this pharmaceutical intervention. This dominant effect will diminish any cell-specific differences in epigenetic regulation, allowing the PBMC to be a better estimate of the effect of the pharmaceutical [2,5]. When multiple pharmaceuticals are being administered, as is common in a clinical situation, PBMC chromatin will integrate all possible epigenetic effects from all circulating pharmaceuticals and other modifiers and inform the investigator about the state-of-chromatin in real time. Advantages of in vitro PBMC culture models include:
■ Patient-derived cultures with unique genomic background and enzymatic activity;
■ Isolation of the target cell in a well-defined media that is comparable across experimental and diagnostic conditions;
■ Exposure to equimolar concentrations of experimental HDAC inhibitor;
■ Absence of variable absorption and distribution of a drug;
■ Ability to isolate and exclude epigenetic effects of metabolic, dietary, hormonal and other milieu factors;
■ Ability to segregate a homogeneous population of T-cell PBMCs after 24–120 h of in vitro cultures;
■ Dissect chromatin assemblies using experimental drugs/dosages impossible in a clinical populations; these include MS275, suberoylanilide hydroxamic acid or the highly toxic trichostatin A (TSA).
Experimental considerations
■ How reliable (or stable) are blood chromatin parameters on repeated sampling in human subjects?
Fraga et al. have measured chromatin modifications over the lifespan [49]. Histone modifications are ‘stable’ in repeated sampling over a period of 12 weeks performed in different age cohorts of identical monozygotic twins. Replicate blood chromatin samples from eight identical twin pairs demonstrated no significant fluctuations in the acetylated H3 fraction over 12 weeks [49].
■ Saturable response of HDAC inhibitors in primary PBMC cultures
PBMCs from living subjects can be cultured without activation and treated with HDAC inhibitors.Figure 3 shows a typical response to varying concentrations of valproic acid spanning the blood levels seen in clinical psychiatry. We have reported similar dose–response effects with the HDAC inhibitor TSA [2]. The advantage of primary PBMC cultures is that experimental drugs such as TSA (toxic) – a high-affinity specific HDAC inhibitor – can be tested on patient cell tissue. Other experimental drugs, such as antagonists of histone methyltransferases (BIX-01294), can be applied to measure changes to H3K9me2. A blunted response to TSA in PBMC cultures from blood obtained from schizophrenia patients has been demonstrated. This finding indicated an obdurate restrictive chromatin in these subjects [4]. However, it should be noted that these findings would require replication in specific cell types, since a blunted response could also indicate a shift in differential cell count.
Figure 3. Peripheral blood mononuclear cells extracted from human peripheral blood, plated at 1 million cells per ml and treated with valproic acid for 48 h in vitro.

The range for clinical therapy is between 0.3–0.9 mM blood levels for bipolar disorder and seizure disorders.
■ ChIP in cultured PBMCs from patients
The analysis of the ‘chromatin plasticity’ of a particular promoter can be measured in treatment-resistant patients to monitor the effectiveness of pharmaceuticals targeting chromatin. GAD67 is the protoptyic schizophrenia candidate gene that is widely reported as downregulated in the brains of schizophrenia patients and is an important theoretical component in our understanding of the cortical GABAergic inter neuron. We have hypothesized that this reduced expression could be due to a restrictive chromatin state. This hypothesis cannot be tested in brain tissue from living patients, but can be tested in peripheral tissue. As a preamble to such an investigation, we have reported on the occupancy of the GAD67 promoter by H3K9ac in PBMCs, and demonstrate that this indicator of ‘open’ transcriptionally facilitative chromatin is enhanced upon treatment of the cultured PBMC cells with an HDAC inhibitor, such as valproic acid or TSA [2,3]. A future investigation could sample the response of this promoter in cultured cells from living schizophrenia patients to a predetermined dose of a chromatin-modifying pharmaceutical.
Limitations of peripheral tissue to approximate brain function
Peripheral blood chromatin will not provide information specific to a brain region or neuronal phenotype. There are a vast number of neuronal phenotypes (not currently numerable based on neuroanatomy, morphology, gene-expression profile, firing patterns, circuitry and so on) [56]. Attempts to match a particular peripheral blood cell (such as a CD4− helper T lymphocyte) to a neuronal type (such as a GABA interneuron in layer IV of the frontal cortex) will be a recapitulation of earlier attempts to simulate neuronal function using peripheral tissue and may be difficult to interpret. In addition, peripheral blood chromatin may not be useful to monitor neuroanatomical, synaptic signal transduction or neuronal circuitry which cannot be supported given the lack of synaptic connections on peripheral blood cells.
Conclusion & future perspective
The analysis of gene regulation in living patients undergoing the full evolution of their illness including the effects of the environment and prescribed pharmaceuticals should be the natural progression of clinical research from single-point post-mortem brain studies. Peripheral blood chromatin provides a protein biosensor capable of integrating the totality of epigenetic influences operative in a given patient. Pharmaceuticals are being developed for the more than 50 catalytic enzymes involved in this system of epigenetic regulation. Translational investigations exploring the role of the epigenome in patients, particularly those that are treatment resistant, will require monitoring of epigenetic response, even at a given histone residue. These living cells can be measured fresh from a patient, or grown in vitro as primary cultures for experimentation. This approach can be validated by establishing shared epigenetic enzymes/modifiers in the proxy PBMC versus the target neuronal tissue of interest. Readouts can include either genome-wide or promoter-specific histone-protein modifications, as well as mRNA output from gene networks or individual genes. In as much as the brain shares the internal epigenetic milieu, these blood levels will approximate changes at carefully selected nonsynaptic networks and could be used to monitor epigenetic correlates of pharmaceuticals directed at the epigenome. Indeed, these approaches could lead to methods of screening patients prior to the implementation of chromatin altering treatments. In other words, sampling a patient’s PBMCs could provide a mechanism for individualizing treatments prior to an actual clinical trial.
Executive summary.
Histone protein modifications in peripheral blood mononuclear cells
■ Epigenetic modifications of histone proteins measured in circulating blood cells can provide an estimate of the internal epigenetic milieu in a living subject.
■ The biochemical nature of epigenetic modifiers predisposes them to distribute in both central (brain) and peripheral (blood) tissue compartments.
■ Levels of a histone modification in circulating blood will significantly capture this milieu, especially if there is a powerful pharmaceutical inhibitor capable of overriding pathophysiological signals.
Chromatin regulation of transcription measured in peripheral blood mononuclear cells
■ The transcriptome has many points of similarity across all tissues. Consequently, a mapping function such as a specific chromatin signature deposited along nodes of a commensurate gene network is a practical starting point to monitor chromatin status.
■ mRNA readouts of chromatin assemblies can be limited to predetermined nonsynaptic cellular gene networks similar in their chromatin mapping in central and peripheral tissue.
In vitro cultures from living subjects
■ Peripheral blood cells from individual patients will possess full genomic variability, and can be cultured to dissect the chromatin status of a single promoter of interest using standard culture methodologies.
■ Candidate genes identified from genetic linkage studies or other theoretically relevant targets can be examined using an increasing number of experimental enzyme inhibitors not yet approved for clinical trials.
Financial & competing interests disclosure
This work was partly funded by NIH 1RO1MH09435801A1 (principal investigator: RP Sharma). The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Sims RJ, 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19(11):629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Gavin DP, Kartan S, Chase KA, Jayaraman S, Sharma RP. Histone deacetylase inhibitors and candidate gene expression: an in vivo and in vitro approach to studying chromatin remodeling in a clinical population. J. Psychiatr. Res. 2009;43(9):870–876. doi: 10.1016/j.jpsychires.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Gavin DP, Rosen C, Chase KA, Grayson DR, Sharma RP. Dimethylated lysine 9 of histone 3 is elevated in schizophrenia and exhibits a divergent response to histone deacetylase inhibitors in lymphocyte cultures. J. Psychiatr. Neurosci. 2009;34(3):232–237. [PMC free article] [PubMed] [Google Scholar]
- 4.Gavin DP, Kartan S, Chase KA, Grayson DR, Sharma RP. Reduced baseline acetylated histone 3 levels, and a blunted response to HDAC inhibition in lymphocyte cultures from schizophrenia subjects. Schizophr. Res. 2008;103(1–3):330–332. doi: 10.1016/j.schres.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma RP, Rosen C, Kartan S, et al. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: preliminary results from a clinical population. Schizophr. Res. 2006;88(1–3):227–231. doi: 10.1016/j.schres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40(7):897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Maze I, Covington HE, 3rd, Dietz DM, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327(5962):213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer A, Sampath SC, Intrator A, et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron. 2009;64(5):678–691. doi: 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma RP, Gavin DP, Chase KA. Heterochromatin as an incubator for pathology and treatment non-response: implication for neuropsychiatric illness. Pharmacogenomics J. 2012 doi: 10.1038/tpj.2011.64. doi:10.1038/tpj.2011.64. Epub ahead of print.■ Discusses the possible role of restrictive chromatin in the therapeutic response of psychiatric patients.
- 11.Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004;68(6):1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 12.Takizawa N, Watanabe K, Nouno K, Kobayashi N, Nagata K. Association of functional influenza viral proteins and RNAs with nuclear chromatin and sub-chromatin structure. Microbes Infect. 2006;8(3):823–833. doi: 10.1016/j.micinf.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Jones DR, Divecha N. Linking lipids to chromatin. Curr. Opin Genet. Dev. 2004;14(2):196–202. doi: 10.1016/j.gde.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007;8(5):355–367. doi: 10.1038/nrn2132.Good overview of epigenetic mechanisms in psychiatric disorders.
- 15.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J. Neurosci. 2008;28(14):3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine A, Huang Y, Drisaldi B, et al. Molecular mechanism for a gateway drug: epigenetic changes initiated by nicotine prime gene expression by cocaine. Sci. Transl Med. 2011;3(107):107–109. doi: 10.1126/scitranslmed.3003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aoyagi S, Archer TK. Dynamic histone acetylation/deacetylation with progesterone receptor-mediated transcription. Mol. Endocrinol. 2007;21(4):843–856. doi: 10.1210/me.2006-0244. [DOI] [PubMed] [Google Scholar]
- 18.Sun JM, Chen HY, Davie JR. Effect of estradiol on histone acetylation dynamics in human breast cancer cells. J. Biol. Chem. 2001;276(52):49435–49442. doi: 10.1074/jbc.M108364200. [DOI] [PubMed] [Google Scholar]
- 19.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108(4):465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 20.Sharma RP. Schizophrenia, epigenetics and ligand-activated nuclear receptors: a framework for chromatin therapeutics. Schizophr. Res. 2005;72(2–3):79–90. doi: 10.1016/j.schres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 21.Kirillova GP, Hrutkay RJ, Shurin MR, et al. Dopamine receptors in human lymphocytes: radioligand binding and quantitative RT-PCR assays. J. Neurosci. Methods. 2008;174(2):272–280. doi: 10.1016/j.jneumeth.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vile JM, Strange PG. High-affinity binding sites for neuroleptic drugs in human peripheral blood lymphocytes and their relation to dopamine receptors. A long-standing controversy. Biochem. Pharmacol. 1995;49(6):747–753. doi: 10.1016/0006-2952(94)00426-m. [DOI] [PubMed] [Google Scholar]
- 23.Vetoshkin AV, Fomenko AM, Zozulia AA. Lymphocyte serotonin receptors: a radioreceptor study. Biull. Eksp Biol. Med. 1982;94(7):52–53. [PubMed] [Google Scholar]
- 24.Kohm AP, Sanders VM. Norepinephrine: a messenger from the brain to the immune system. Immunol. Today. 2000;21(11):539–542. doi: 10.1016/s0167-5699(00)01747-3. [DOI] [PubMed] [Google Scholar]
- 25.Marazziti D, Catena Dell’osso M, et al. Alterations of the dopamine transporter in resting lymphocytes of patients with different psychotic disorders. Psychiatry Res. 2010;175(1–2):54–57. doi: 10.1016/j.psychres.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Fujii T, Takada-Takatori Y, Kawashima K. Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J. Pharmacol. Sci. 2008;106(2):186–192. doi: 10.1254/jphs.fm0070109. [DOI] [PubMed] [Google Scholar]
- 27.Mehrishi JN, Mills IH. Opiate receptors on lymphocytes and platelets in man. Clin. Immunol. Immunopathol. 1983;27(2):240–249. doi: 10.1016/0090-1229(83)90074-0. [DOI] [PubMed] [Google Scholar]
- 28.Guidotti A, Dong E, Kundakovic M, et al. Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling. Trends Pharmacol. Sci. 2009;30(2):55–60. doi: 10.1016/j.tips.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447(7141):178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 30.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42(6):961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh J, Gage FH. Epigenetic control of neural stem cell fate. Curr. Opin Genet. Dev. 2004;14(5):461–469. doi: 10.1016/j.gde.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 33.Caron H, van Schaik B, van der Mee M, et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291(5507):1289–1292. doi: 10.1126/science.1056794.■■ Survey of the trancriptome across 12 different tissues.
- 34.Versteeg R, van Schaik BD, van Batenburg MF, et al. The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Res. 2003;13(9):1998–2004. doi: 10.1101/gr.1649303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Middleton FA, Pato CN, Gentile KL, et al. Gene expression analysis of peripheral blood leukocytes from discordant sib-pairs with schizophrenia and bipolar disorder reveals points of convergence between genetic and functional genomic approaches. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005;136B(1):12–25. doi: 10.1002/ajmg.b.30171.■ Transcriptome analysis from peripheral blood mononuclear cells (PBMCs) of psychiatric patients with gene locations mapped to genetic linkage findings.
- 36.Sullivan PF, Fan C, Perou CM. Evaluating the comparability of gene expression in blood and brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006;141B(3):261–268. doi: 10.1002/ajmg.b.30272. [DOI] [PubMed] [Google Scholar]
- 37.Guidotti A, Auta J, Chen Y, et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology. 2011;60(7–8):1007–1016. doi: 10.1016/j.neuropharm.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung I, Shulha HP, Jiang Y, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl Acad. Sci. USA. 2010;107(19):8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009;41(2):246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SA, Tsao TT, Yang KC, et al. Construction and analysis of the protein–protein interaction networks for schizophrenia, bipolar disorder, and major depression. BMC Bioinformatics. 2011;12(Suppl. 13):S20. doi: 10.1186/1471-2105-12-S13-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, et al. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol. Psychiatry. 2011;16(7):751–762. doi: 10.1038/mp.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garbett K, Gal-Chis R, Gaszner G, Lewis DA, Mirnics K. Transcriptome alterations in the prefrontal cortex of subjects with schizophrenia who committed suicide. Neuropsychopharmacol. Hung. 2008;10(1):9–14. [PubMed] [Google Scholar]
- 43.Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol. Psychiatry. 2007;62(7):711–721. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang B, Capitao C, Dean B, Thomas EA. Differential age- and disease-related effects on the expression of genes related to the arachidonic acid signaling pathway in schizophrenia. Psychiatry Res. 2012;196(2–3):201–206. doi: 10.1016/j.psychres.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Issidorides MR, Stefanis CN, Varsou E, Katsorchis T. Altered chromatin ultrastructure in neutrophils of schizophrenics. Nature. 1975;258(5536):612–614. doi: 10.1038/258612a0.■■ First chromatin study to report differences in PBMCs in schizophrenia patients.
- 46.Stefanis CN, Issidorides MR. Histochemical changes in the blood cells of schizophrenic patients under pimozide treatment. Biol. Psychiatry. 1976;11(1):53–68. [PubMed] [Google Scholar]
- 47.Kosower NS, Gerad L, Goldstein M, et al. Constitutive heterochromatin of chromosome 1 and Duffy blood group alleles in schizophrenia. Am. J. Med. Genet. 1995;60(2):133–138. doi: 10.1002/ajmg.1320600209. [DOI] [PubMed] [Google Scholar]
- 48.Kloukina-Pantazidou I, Havaki S, Chrysanthou-Piterou M, et al. Chromatin alterations in leukocytes of first-episode schizophrenic patients. Ultrastruct. Pathol. 2010;34(3):106–116. doi: 10.3109/01913121003644781. [DOI] [PubMed] [Google Scholar]
- 49.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl Acad. Sci. USA. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102.■■ Monozygotic twin study examing chromatin and other epigenetic parameters in PBMCs across patient lifespan.
- 50.Mill J, Dempster E, Caspi A, et al. Evidence for monozygotic twin (MZ) discordance in methylation level at two CpG sites in the promoter region of the catechol-O-methyltransferase (COMT) gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006;141B(4):421–425. doi: 10.1002/ajmg.b.30316. [DOI] [PubMed] [Google Scholar]
- 51.Kuratomi G, Iwamoto K, Bundo M, et al. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol. Psychiatry. 2008;13(4):429–441. doi: 10.1038/sj.mp.4002001. [DOI] [PubMed] [Google Scholar]
- 52.Petronis A, Gottesman II, Kan P, et al. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophr. Bull. 2003;29(1):169–178. doi: 10.1093/oxfordjournals.schbul.a006988.■ Elegant argument for the epigenetic basis of psychiatric disorders.
- 53.Tsujita T, Niikawa N, Yamashita H, et al. Genomic discordance between monozygotic twins discordant for schizophrenia. Am. J. Psychiatry. 1998;155(3):422–424. doi: 10.1176/ajp.155.3.422. [DOI] [PubMed] [Google Scholar]
- 54.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003;370(3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dangond F, Gullans SR. Differential expression of human histone deacetylase mRNAs in response to immune cell apoptosis induction by trichostatin A and butyrate. Biochem. Biophys. Res. Commun. 1998;247(3):833–837. doi: 10.1006/bbrc.1998.8891. [DOI] [PubMed] [Google Scholar]
- 56.Migliore M, Shepherd GM. Opinion: an integrated approach to classifying neuronal phenotypes. Nat. Rev. Neurosci. 2005;6(10):810–818. doi: 10.1038/nrn1769. [DOI] [PubMed] [Google Scholar]
- 57.Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care. 2002;25(2):275–278. doi: 10.2337/diacare.25.2.275. [DOI] [PubMed] [Google Scholar]