

Abstract
Both positively and negatively charged residues play pivotal roles in recruiting precursor ions or ion clusters, and lowering interfacial energy in natural biomineralization process. Synergistic utilization of opposite charges, however, has rarely been implemented in the design of cytocompatible synthetic scaffolds promoting hydroxyapatite (HA)-mineralization and osteointegration. We report the use of cytocompatible zwitterionic sulfobetaine ligands to enable 3-dimensional in vitro mineralization of HA across covalently crosslinked hydrogels. The overall charge-neutral zwitterionic hydrogel effectively recruited oppositely charged precursor ions while overcame excessive swelling exhibited by anionic and cationic hydrogels under physiological conditions, resulting in denser and structurally well-integrated mineralized composites. Further controls over the size, content, and spatial distribution of the mineral domains within the zwitterionic hydrogel are accomplished by facile adjustments of hydrogel crosslinking densities and the supersaturation rate governing heterogeneous mineral nucleation and growth. These findings should inspire many creative uses of zwitterionic polymers and polymer coatings for skeletal tissue repair and regeneration.
1. Introduction
According to the classical descriptions of activation energy of homogenous nucleation, nucleation rate, and mineral growth rate for a spherical nucleus, increasing supersaturation (S) and decreasing interfacial free energy (σ) expedite the rate of the nucleation and growth (Supplementary Equations S1–S3) [1]. These parameters similarly govern heterogeneous nucleation and growth. In nature, heterogeneous mineralization of organic extracellular matrices (ECM) in calcified tissues are accomplished by the tight control over ion transport to achieve appropriate local supersaturation (e.g. within diffusion-limited cellular and/or extracellular spaces) and by complex organic ECM templates that stabilize precursor ions, reduce the interfacial energy, and modulate the heterogeneous nucleation and mineral growth [2–5]. For instance, both collagen fibrils in bone and the acidic non-collagenous proteins (NCPs) attached to the collagen matrix are believed to play important roles in facilitating precursor ion infiltration and creating local supersaturation, spatially confining and stabilizing initial amorphous calcium phosphate deposition (ACP), and templating/modulating subsequent transformation into more stable crystalline phases [3, 4, 6–16]. A recent study revisiting the role of collagen in templating mineralization in the presence of HA nucleation inhibitors suggests that the positive net charge close to the C-terminal end of the collagen molecules promotes the infiltration of ACP, and that the clusters of charged amino acids, both in gap and overlap regions of collagen fibrils, form nucleation sites mediating the transformation of ACP into oriented apatite crystals[17]. Taken together, these findings suggest that implementing opposite charge distributions, possibly in an alternating fashion, within a synthetic scaffold may facilitate adequate precursor ion infiltrations and realizing effective in vitro mineralization (e.g. for improving scaffolds’ osteoconductivity) and in vivo osteointegration for scaffold-assisted skeletal tissue repair.
Many synthetic biomaterials designed for template-driven mineralization utilized anionic residues [18–24], cationic residues [25, 26] or uncharged polar ligands [27–29] including in amphiphilic forms [30] to drive the heterogeneous nucleation. In vitro mineralization of many bulk scaffolds, however, often led to limited heterogeneous nucleation/growth of HA beyond the surface of dense substrates or homogeneous precipitation loosely trapped within porous scaffold. Well-integrated dense mineralization of bulk synthetic scaffold in a truly 3-D fashion has been difficult to achieve.
Zwitterionic materials, well known for excellent anti-fouling ability[31–34], have been extensively explored for applications ranging from blood compatible materials[35, 36] to DNA and protein delivery vehicles[37, 38]. Although calcium phosphate nanocomposite of polyelectrolytes and zwitterionic polymers were previously prepared by direct mixing of the mineral components [39, 40] or solidification of precursor ion solutions with the respective polymers [41], the role of zwitterionic ligands in templating heterogeneous nucleation and growth of HA has not been previously explored. Zwitterionic polymers, possessing equal number of oppositely charged residues in the same molecular side chain yet overall electrically neutral, are known to swell more in salt solution than in water, a behaviour opposite to those exhibited by anionic or cationic polyelectrolytes and driven by salt ion-mediated disruption of the extensive physical (electrostatic) crosslinking of the zwitterionic side chains formed in water (Fig. 1a; note the substantial difference in dipole moment: ~24 Debye for sulfobetaine[42] vs. 2 Debye for water[43]). We hypothesize that such antipolyeletrolyte behaviour[44] exhibited by 3-D zwitterionic scaffolds, coupled with the ability of zwitterioic ligands to recruit and retain both cationic and anionic precursor ions or ion clusters and lower the activation energy for heterogeneous nucleation and growth, can enable more extensive 3-D HA-mineralization (Fig. 1b).
Figure 1.

Schematical illustrations of (a) the swelling behaviour of a crosslinked 3-D zwitterionic polymer scaffold in water vs. in aqueous saline solution, and (b) 3-D HA-mineralization of a zwitterionic polymer scaffold driven by a gradual increase of supersaturation resulting from the thermal decomposition of urea.
To test these hypotheses, we examine and compare the potency of photo-crosslinked polymethacrylate hydrogels containing zwitterionic sulfobetaine, cationic tertiary amine, anionic sulfonic acid or uncharged hydroxyl side chain residues in templating 3-D HA-mineralization. The relative cytocompatibility of zwitterionic, cationic and anionic scaffolds were also investigated by direct encapsulation of bone marrow stromal cells within these hydrogel scaffolds. Further, practical controls over the mineralization outcome, including size, content, and density of the mineral growth within the 3-D zwitterionic scaffold, were systematically investigated by altering crosslinker contents of the hydrogel and the external experimental parameters governing the heterogeneous mineralization process.
2. Experimental
2.1. Preparation of chemically crosslinked hydrogels
To investigate the effect of ionic states of mineral nucleating ligands on templated in vitro HA-mineralization, four types of hydrogels, zwitterionic poly[2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (pSBMA), cationic poly[2-(methacryloyloxy)ethyl]trimethylammonium (pTMAEMA), anioic poly(3-sulfopropyl methyacrylate) (pSPMA), and uncharged poly(2-hydroxyethyl methacrylate) (pHEMA), were prepared (Supplementary Table S1, No. 1–4). Monomers SBMA, TMAEMA, SPMA and HEMA and crosslinker ethylene glycol dimethacrylate (EGDMA) were purchased from Aldrich (St. Louis, MO), with the radical inhibitors being removed by distillation (for HEMA), aluminum oxide column followed by freeze-drying (for PTMAEMA) or molecular sieves (for EGDMA) prior to use. In a typical procedure, 2 mmol respective monomer was combined with 50 μL of ethanol solution of EGDMA (10%, v/v), 100 μL PBS solution of 2,2′-Azobis[2-methyl-N-(2-hydroxyethyl)propionamide] (VA-086, 2 %, w/v), and 1850 μL of Milli-Q water. The mixture was bath-sonicated to a clear solution and transferred to a custom-made Teflon mold with cylindrical (6 mm in diameter) and square prism (5 mm × 5 mm) wells (50 μL/well or 100 μL/well) and solidified under irradiation (365 nm, 10 min). The crosslinked hydrogels were equilibrated in Milli-Q water for 2 days at 37 °C with exchange of water every 12 h to ensure the removal of residue initiators or any unreacted monomers. The hydrogels were stored in Milli-Q water until mineralization or further characterizations.
To further study the effect of crosslinker contents on the HA-mineralization outcome of zwitterionic hydrogels, eight pSBMA hydrogels with different crosslinker contents, 2.651 mol% (# 1), 1.326 mol% (#2), 0.663 mol% (#3), 0.265 mol% (#4), 0.133 mol% (#5), 0.027 mol% (#6), 0.020 mol% (#7), and 0.013 mol% (#8), as detailed in Supplementary Table S2, were also prepared using the procedure described above.
2.2 Swelling ratios of the hydrogel
The swelling ratios of the hydrogel by weight (Sw) and by volume (Sv) were determined in Milli-Q water or in PBS (pH=7.4) at room temperature according to Equations 1 and 2:
| Eq-1 |
where Wh and Wd are the weight of a hydrogel in fully hydrated state and dry state, respectively.
| Eq-2 |
where Vh and Vd are the volume of a hydrogel in fully hydrated state and dry state, respectively.
2.3 Mineralization of the hydrogel
Mineralization was carried out by controlled heating of the hydrogels in a urea-containing, acidic solution of hydroxyapatite from 37 °C to 95 °C using a protocol modified over a previous report[27]. Mineralization stock solution was prepared by suspending hydroxyapatite (7.37 g, 34–40% Calcium content, Alfa Aesar) in 500-mL aqueous solution of urea (2 M), followed by the addition of concentrated hydrochloric acid under constant stirring until a clear soluble solution was obtained (final pH 2.5–3.0). Six to ten hydrogels were placed in an Erlenmeyer flask filled with 30 mL of mineralization solution and covered with a perforated aluminum foil. The flask was placed in a high-temperature silicone oil bath with the mineralization solution completely submerged under the oil and heated using a 100-watt immersion heater (Glo-Quartz LHP-IAH4) equipped with a programmable temperature controller (Eurotherm 2408). Controlled heating from 37 °C to 95 °C was carried out at a heating rate of 0.2 °C/min. Mineralized hydrogels were bath-sonicated for 5 min in Milli-Q water to ensure removal of loosely bound minerals, followed by further incubation in fresh Milli-Q water at 37 °C for two days with regular water change of every 12 h to ensure removal of the residual mineralization precursor ions trapped within the mineralized hydrogel. All mineralized specimens were able to recover from their swollen state in high salt mineralization solution to similar dimensions in water at the end of the 2-day post-mineralization equilibration.
To examine the effect of repeated mineralization, pSBMA hydrogels with a crosslinker content of 0.265 mol% were repeatedly mineralized in fresh mineralization solution at a heating rate of 0.2 °C/min up to four times.
2.4 Temporal pH changes of the mineralization solution with different heating rates
To monitor the changes of pH of the mineralization solution over time with different heating rates, six groups of Erlenmeyer flasks (three flasks per group with each flask containing 45 mL of mineralization solution and one pSBMA hydrogel with a crosslinker content of 0.265 mol%) were covered with perforated aluminum foils and heated from 37 °C to 95 °C at a heating rate of 0.02 0.05, 0.1, 0.2, or 1.0 °C/min, respectively. At pre-determined sampling intervals, 2 mL of mineralization solution was retrieved from the flask, cooled to rt before its pH was measured by a VWR Symphony pH meter. Averaged pH values along with the standard deviations for each of the 15 time points at each heating rate were plotted over mineralization time.
2.5 Quantitation of calcium content of mineralized hydrogels
Total calcium content as a function of mineralization conditions was determined by quantifying the Ca2+ ions released from each mineralized hydrogel in a hydrochloric acid solution with a Thermo Scientific calcium ion selective electrode attached to a VWR Symphony pH/ISE meter. In a typical procedure, the mineralized hydrogel was placed in 10 mL of hydrochloric acid solution (pH 3) in a 20-mL glass vial and the pH was adjusted by concentrated hydrochloric acid to around 2.1. The mineral was allowed to be fully released from the hydrogel under constant shaking of the acidic solution on an orbital shaker, as indicated by reaching a transparent appearance of the gel. Ionic Strength Adjustment buffer (ISA, 4 M KCl solution, VWR, 200 μL) was added to the acidic solution containing the released calcium prior to measurement by the calcium ion selective electrode. The total calcium content of each type of mineralized hydrogel (N = 3) was determined using a standard curve generated by a series of acidic (pH 2.1) aqueous Ca2+ ion standard solutions containing 0.1, 0.01, 0.001, and 0.0001 M CaCl2.
2.6 Real-time monitoring of the mineral release from the hydrogels
The progress of the mineral release from the mineralized hydrogel in hydrochloric acid (pH 1.5, 4.5 mL per specimen) was monitored by digital photographs of the specimen taken at 0, 10, 30, 60, 120, 180, 240, and 300 min of the incubation using a digital camera (Canon, VIXIA HF200).
2.7 Zeta potential measurement
To examine how the ionic status of the zwitteironic pSBMA varied as a function of significant pH changes during the mineralization, the zeta potentials of linear pSBMA polymer (synthesized in our lab using the well-controlled atom transfer radical polymerization process, Mn = 12424, PDI = 1.09) under varied solution pH were measured on a Zetasizer (Zen3600, 633 nm laser, Malvern). Linear pSBMA solutions (3 mg/mL in 0.015 M NaCl aq. solution) were adjusted with a final pH of 3.01, 3.99, 5.30, 6.45, 7.03, 7.43 or 9.17 by aqueous HCl or NaOH, and placed in disposable capillary cells (DTS1061, Malvern) for zeta potential measurement at 25 °C.
2.8 Mechanical testing
It is known that increasing mineral content in a poorly integrated polymer-mineral composite can result in the deterioration of the mechanical properties of the composite. Thus, it is important to examine the macroscopic mechanical properties of the mineralized pSBMA hydrogels under the context of increasing mineral contents. The compressive behavior of fully hydrated mineralized zwitterionic hydrogel (# 4 in Supplementary Table S2, with a crosslinker content of 0.265 mol%) as a function of the heating rate, along with the un-mineralized control, was evaluated on a Q800 dynamic mechanical analyzer (DMA, TA Instruments) equipped with a submersion compression fixture. The instrument has an 18-N load cell, a force resolution of 10 μN and a displacement resolution of 1.0 nm. Each specimen fully equilibrated in water was compressed in a force-controlled mode at 25 °C, ramping from 0.02 to 6.0 N at a rate of 1.0 N/min then back to 0.02 N at the same rate. A pre-load of 0.02 N was applied. The averaged compressive stresses at 50% strain (N = 3) of the composites were plotted as a function of the heating rate.
2.9 Microcomputed tomography (μ-CT)
Mineralized hydrogel specimens (N = 3) were scanned on a Scanco μ–CT40 scanner. The effective voxel size of the reconstructed images was 8×8×8 μm3. Data were globally thresholded and reconstructed to quantify the mineral volume (MV, mm3), mineral density (MD, mg HA/ccm), mineral size (diameter or thickness, mm) distribution, mineral density (1/mm) distribution and anisotropy of the composites. MV and MD were determined by a direct analysis. MS/MV and anisotropy were determined by a TRI (plate mode) analysis. Direct thickness distributions of the 3-D mineralized structures were determined with a volume-based thickness definition. It defines the local thickness at a given point in the mineralized structure as the diameter of the largest sphere including the point that can be fitted within the structure. Mineral density (1/mm) distributions within the hydrogels were visualized by reconstructing the respective AIM file with a colored density gradient range of 0.7–1.5 (1/mm). An un-mineralized hydrogel was also scanned as a negative control to ensure proper setting of the threshold for analyses.
2.10 Cell encapsulation in hydrogels
The cyto-compatibility of three types of hydrogels with different overall charges, zwitterionic pSBMA, cationic pTMAEMA, and anioic pSPMA were investigated by the viability of rat bone marrow stromal cells (rMSCs) encapsulated in these hydrogels. In a typical procedure, 2 mmol of the respective hydrogel monomer was dissolved in 2 mL of expansion medium (α-MEM supplemented with 20% FBS, 1% penicillin, 1% streptomycin, and 2% glutamine) mixed with 100 μL PBS solution of 2 wt% VA-086 (Wako) and a predetermined amount of water-soluble crosslinker polyethylene glycol dimethacrylate (PEGDMA, Mn=750, Sigma-Aldrich) as described listed in Table S1 (No. 5–7). Given the drastically different swelling behavior of cationic, anionic and zwitterionic hydrogels, the PEGDMA crosslinker contents applied to the respective hydrogels were adjusted to achieve comparable equilibrium gel volumes in culture media, thus comparable initial cell encapsulation density within the various hydrogels. The mixture was sterile-filtered through a 0.2-μm sterile polyethersulfone syringe filter. Passage 1 rMSC, isolated from the femoral marrow canal of skeletally mature SASCO SD rats (Charles River Lab, 8–10 week old) and enriched by adherent culture [45], were plated in expansion media for 24 h before they were trypsinized, counted, and suspended into the respective monomer/crosslinker/initiator mixture solutions (106 cells/mL). The rMSC-containing solution was then transferred to a custom-made Teflon mold of square prism wells (5 mm × 5 mm; 50 μL/well) and solidified under the irradiation of 365-nm light for 10 min in a sterile hood. All cell–hydrogel constructs were cultured for up to 4 days in humidified incubation (5% CO2, 37 °C) before being subjected to live/dead cell staining.
2.11 Live/dead cell staining and confocal microscopy
The hydrogel–cell constructs were stained using a LIVE/DEAD viability/cytotoxicity kit (Molecular Probes) following the vendor’s protocol. Live cells were stained with green fluorescence by intracellular esterase-catalyzed hydrolysis of Calcein AM, and dead cells were stained red by ethidium homodimer-1 penetrated through the damaged membranes and bound with nucleic acids. After being stained for 30 – 45 min at 37 °C, the stained hydrogel–cell construct was retrieved and mounted in a Cellview culture dish (Greiner) and imaged on a Leica TCS SP2 confocal microscope. Calcein was excited at 488 nm and observed with the FITC filter (518–542 nm) while ethidium homodimer-1 was excited at 543 nm and observed with the TEXAS RED filter (625–665 nm). Confocal Z-stack images of encapsulated rMSC over the depth of 80 μm (8 consecutive 10 μm slices) were overlaid.
2.12 MTT Cell viability assay
The viability of the rMSCs encapsulated in the zwitterionic pSBMA and the uncharged PEGMA control with established cytocompatibility (prepared by crosslinking PEGMA monomer from Sigma-Aldrich, Mn = 360, at the same crosslinker content of 1.33 mol% applied to pSBMA) were evaluated by an MTT cell proliferation kit (Roche) at 24 and 96 h after the initial cell encapsulation. The gel-cell constructs were cultured in expansion media (α-MEM supplemented with 20% FBS, 1% penicillin, 1% streptomycin, and 2% glutamine). MTT labeling reagent (50 μL) was added to each well containing culture media & cell–hydrogel construct and incubated for 11 h in humidified incubation (5% CO2, 37 °C). Solubilization solution (500 μL) was then added and incubated for an additional 12 h to release the purple formazan from the 3-D hydrogels. The absorbance of the collected supernatant (N=3) was read at 570 nm (with background correction read at 690 nm) on a MULTISCANFC spectrophotometer (Thermo Scientific).
2.13 Scanning electron microscopy (SEM)
The mineral morphologies at both the surface and the cross-section of air-dried mineralized hydrogels, sputter coated with 4-nm gold, were observed by SEM on a Quanta 200 FEG MKII microscope equipped with an Oxford-Link Inca 350 X-ray spectrometer (Oxford Instruments) under an accelerating voltage of 10 kV and a spot size of 3.0 μm. For the cross-section observation, air-dried mineralized hydrogels were bisected by a razor blade.
2.14 Focused ion beam (FIB) processing and transmission electron microscopy (TEM)
Air-dried cubic mineralized specimens were first bisected with a razor blade to expose the mineral domains at the cross-section and sputter coated with 8 nm of gold. Selected mineral domains at the cross-section were further sectioned with a Zeiss NVision 40 dual-beam Focused Ion Beam (FIB) using 30 kV Ga+ ions. A U-cut (1.5 nA) was performed, followed by further ion polishing (5 kV, 40 pA) to thin the regions of interest to achieve TEM transparency (< 100 nm in thickness). The thinned FIB specimens were then examined on an FEI transmission electron microscope (Tecnai 12 Spirit) at 80kV and on a high-resolution transmission electron microscope (JEOL 2000FX) at 200 kV. The relative intensities of the 002, 004, and 211 diffraction arcs or spots in selected area electron diffraction (SAED) were analyzed by ImageJ. The chemical compositions of the thinned mineral specimens (FIB samples) were examined on a JEOL 2010 TEM/STEM equipped with Energy Dispersive X-ray Spectroscopy (EDS). Three random points on each specimen were scanned.
3. Results and Discussion
3.1. Zwitterionic sulfobetaine as a potent mineral nucleating ligand
To test the hypothesis that zwitterionic ligands are effective mineral nucleating ligands and cytocompatible matrix for potential tissue engineering applications, comparisons between the zwitterionic (pSBMA) with other charged or uncharged ligands, similar polymethacrylate hydrogels bearing cationic (pTMAEMA), anionic (pSPMA), and polar uncharged (pHEMA) side chains that photo-crosslinked with identical crosslinker content (Supplementary Table S1) were investigated. The photo-crosslinked polymethacrylate hydrogels were placed in the mineralization solution and subjected to heterogeneous HA-mineralization driven by gradual pH increases resulting from controlled thermal decomposition of urea upon heating from 37 °C to 95 °C at a rate of 0.2 °C/min[27, 46].
SEM and μ–CT analyses revealed that although substantial mineralization occurred on all hydrogel surfaces with varying surface mineral morphologies (Supplementary Fig. S1), the degrees of mineralization within the interior of these 3-D hydrogel scaffolds varied significantly (Fig. 2a). Consistent with previous finding[27], the mineralization of the uncharged polar pHEMA hydrogel was limited to the surface of the hydrogel. By contrast, the interior of cationic and anionic hydrogels were mineralized with spherical mineral nodules (Fig. 2b), likely benefiting from a combination of their exceptionally high swelling ratios that facilitated precursor ion infiltrations, and the ability of the charged residues to stabilize initial ACP and decrease the interfacial energy for subsequent nucleation and growth of mineral crystals[17]. The most striking observation is that the zwitterionic hydrogel not only was substantially mineralized, but also exhibited much higher mineral content than its anionic and cationic counterparts mineralized under identical conditions (nearly 3 times higher in mineral content when normalized against their respective equilibrium volumes in PBS, Fig. 2c). This finding supports our hypothesis that zwitterionic hydrogels are more advantageous than both cationic/anionic and uncharged hydrogels in templating extensive 3-D mineralization. The sufficient (unlike pHEMA) yet not excessive (unlike pTMAEMA or pSPMA) swelling behavior of pSBMA in aqueous salt solutions as well as the ability of the zwitterionic ligands to recruit and stabilize oppositely charged precursor ions or ion clusters have likely made the difference in achieving 3-D mineralization with much higher mineral density. It is worth noting that within the range of pH increases during the urea thermal decomposition-mediated mineralization process (Fig. 2d), there is no significant fluctuation in zeta potential of the zwitterionic ligands as shown by the measurements taken with a linear pSBMA model polymer (Fig. 2e). This observation is consistent with previous reports [47] and suggests that the ability of the zwitterionic sulfobetaine ligands within the crosslinked pSBMA hydrogel to facilitate active infiltration of oppositely charged precursor ions or ion clusters is maintained throughout the mineralization process.
Figure 2.

(a) Micro-CT sagittal cross-section views and (b) mineral content (n=3, determined by μ-CT) of chemically crosslinked polymethacrylate hydrogels with varied side chain chemistries and ionic charges upon mineralization. All hydrogels were mineralized at a heating rate of 0.2 °C/min. The mineral content was normalized against equilibrium volume (EV) of the respective hydrogels in PBS. (c) Swelling ratio of the un-mineralized hydrogels (n=3) by weight (Sw) in pure water and PBS, respectively. (d) Temporal pH changes of the mineralization solution (n=3). (e) Zeta potential of linear pSBMA (Mn = 12424, PDI = 1.09, 3 mg/mL in 0.015 M NaCl aqueous solution) as a function of pH.
The mineral nodules formed within the interior of pSBMA were structurally well-integrated with the templating hydrogel matrix, as supported by SEM micrographs of the cross-sections of the composite (Fig. 3a). No delamination of the spherical mineral nodules from the hydrogel matrix was observed when the air-dried composites were fractured to expose the cross-sections. To examine the crystallinity and alignment of these mineral nodules at the nano-scale, the exposed cross-section was processed by focused ion beam to obtain an ultir-thin section of a typical spherical mineral nodule (FIB, Fig. 3a) and subjected to TEM, HR-TEM, SAED, and EDS analyses. The mineral nodule was predominantly crystalline in nature (Supplementary Fig. S2), and the corresponding SAED of the TEM micrograph (Fig. 3b) exhibited more intense arcs corresponding to the (002) and (004) faces of HA than that corresponding to the (211) face (Fig. 3c), suggesting a preferential alignment of HA crystals along the c axis. The lattice spacings of 0.28 nm and 0.34 nm measured from the HR-TEM micrographs of randomly selected areas (Fig. 3d) correspond to the (002) and (211) faces of HA[48, 49], agreeing with the intense diffraction ring patterns revealed by the SAED over the same areas (Fig. 3d inset). Finally, S signal associated with the pSBMA hydrogel matrix (Fig. 3e) was consistently detected along with the Ca and P signals from the EDS spectra recorded on the ultra-thin FIB specimens (multiple randomly chosen areas were analysed for each specimen), further supporting structural integration of the mineral nodules with the templating organic matrix.
Figure 3.

(a) SEM and TEM micrographs illustrating FIB processing and thinning of a mineral noduled formed within the interior of a mineralized pSBMA specimen. (b) TEM micrograph and (c) the corresponding SAED pattern & lattice plane intensities of the FIB-thinned mineralized specimen. (d) HR-TEM micrograph and SAED (inset) of a randomly chosen area shown in b). (e) Representative EDS spectrum acquired on the ultra-thin FIB specimen.
3.2. Cytocompatibility of zwitterionic pSBMA hydrogel vs. cationic and anionic counterparts
We further test the hypothesis that the overall charge-neutral zwitterionic sulfobetaine ligands exhibit further advantage over polyelectrolyte mineral nucleating ligands in terms of cytocompatibility. To compare the cytotoxicity of these ligands imposed on skeletal progenitor cells, rat bone marrow stromal cells (rMSC) were encapsulated within pSBMA, pTMAEMA and pSPMA hydrogels crosslinked by poly(ethylene glycol) dimethacrylate (PEGDMA, Mn = 750). The PEGDMA crosslinker was chosen for its aqueous solubility and well established cytocompatibility, which would help narrow down contributing factors to any observed cytotoxicity to the anionic, cationic or zwitterionic ligands. Given the dramatically different swelling behavior of these hydrogels, the PEGDMA crosslinker content was adjusted for each gel type (Supplementary Table S1) to ensure comparable equilibrated cell-hydrogel construct volume in culture media (thus comparable 3-D cell encapsulation density). Live/dead cell straining performed after 96 h culture of the cell-hydrogel constructs revealed a significant number of dead cells in the cationic pTMAEMA hydrogel, consistent with previous reports on the cytotoxicity associated with polycationic scaffolds.[50, 51] By contrast, MSCs encapsulated within the zwitterionic pSBMA hydrogel remained mostly viable after the 4-day culture (Fig. 4). Indeed, the viability and early proliferation of rMSCs encapsulated within the pSBMA hydrogel, determined by MTT cell viability assay, was comparable to those encapsulated within the cytocompatible PEGMA hydrogel control (Supplementary Fig. S3). This observation is consistent with previous reports on the cytotoxicity associated with highly charged polyelectrolyte, particularly polycationic, scaffolds [50, 51]. These observations, combined with the ability of sulfobetaine ligands to template more extensive/higher-density, well-integrated 3-D mineralization of synthetic scaffolds than its charged or uncharged counterparts, suggests a promising role of this zwitterionic ligand in enabling improved osteointegration of scaffold-assisted skeletal tissue repair. Motivated by such exciting potentials, we further investigate feasible regulations of the HA mineralization outcome templated by the zwitterionic hydrogel.
Figure 4.
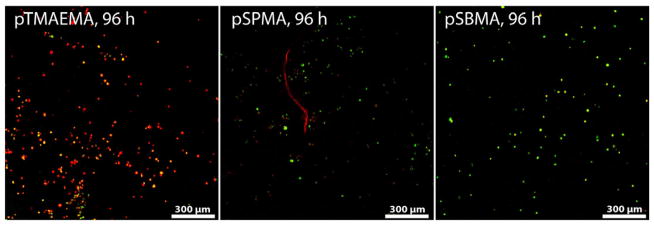
Confocal Z-stack (80 μm) images of live (green)/dead (red) rat bone marrow stromal cells (rMSC, 106 cells/mL) encapsulated in the cationic (pTMAEMA), anionic (pSPMA), and zwitterionic (pSBMA) hydrogels after culturing the cell-hydrogel constructs for 96 h.
3.3. Regulating the HA-mineralization outcome of zwitterionic pSBMA hydrogels
To better understand the HA-mineralization mediated by the zwitterionic ligand and to optimize the mineralization outcome, we further investigated whether and how the size, spatial distribution and total mineral content of the mineral domains within the pSBMA hydrogel can be modulated. We hypothesize that by altering the controlled heating rate of the urea decomposition-mediated heterogeneous mineralization process (thus the rate/degree of supersaturation) and the crosslinker content (thus the swelling ratio of the pSBMA hydrogel in high ionic-strength mineralization solution and the diffusibility of precursor ions), fine control over the in vitro mineralization outcome can be accomplished (type I, II, and III, Fig. 5). Finally, we hypothesize that by repeating an optimized mineralization process, the extent of the 3-D mineralization can be further improved.
Figure 5.

Modulating mineral morphology and spatial distribution of the mineralized pSBMA as a function of the chemical crosslinker content and the heating rate of the urea thermal decomposition-mediated mineralization process. Type I: isolated small spherical mineral nodules resulting from faster heating rates (1.0 and 0.5 °C/min) or higher crosslinker contents (2.651 and 1.326 mol%); Type II: large spherical mineral nodules resulting from medium heating rates (0.2 and 0.1 °C/min) or medium crosslinker contents (0.663, 0.265, and 0.133 mol%); Type III: stripe-like bicontinuous mineral aggregates of small spherical nodules resulting from slower heating rates (0.02 °C/min) or lower crosslinker contents (<0.027 mol%).
3.3.1 Regulating HA mineralization outcome by heating rate
We first demonstrated that by altering the heating rate (0.02–1.0 °C/min; Supplementary Table S2), thus the rate of pH changes governing the supersaturation[52] and the duration of the urea thermal decomposition-mediated mineralization process, the extent of mineralization within pSBMA hydrogel at a given crosslinker content (e.g. 0.265 mol% EGDMA) can be drastically altered (Figs. 6a–g). At the fast heating rates of ≥0.5 °C/min, the hydrogel was minimally mineralized (Figs. 6a & 6b, Supplementary Figs. S4 & S5). This is likely due to the insufficient time (as a result of the fast pH increases revealed by pH-time plot in Supplementary Fig. S6) for the rapidly supersaturated precursor ions or pre-nucleation ion clusters to diffuse into the hydrogel for heterogeneous nucleation and growth into stable micrometer-sized minerals identifiable by SEM and μ-CT. At heating rates of 0.1–0.5 °C/min, however, the hydrogels could be substantially mineralized (Figs. 6a & 6b, Supplementary Figs. S4 & S5), with the mineral volume and total calcium content (determined by μ-CT analysis in Fig. 6c and calcium release assays in Fig. 6d, respectively) increased with decreasing heating rates. More sufficient time for sustained mineral growth and more gradual pH increases resulting from the slower heating rates (Supplementary Fig. S6) are primary contributing factors to the more robust mineralization throughout the 3-D scaffold. However, such an effect plateaued with further decreases of the heating rate, which did not lead to substantial continued increase in mineral content (Figs. 6c & d).
Figure 6.

Effect of heating rate (overall mineralization time) on the pSBMA hydrogel mineralization outcomes. (a) μ-CT views (sagittal and coronal) of the mineralized hydrogels and the respective mineral size distribution as a function of the heating rate of the urea thermal decomposition. (b) SEM micrographs revealing the mineral nodule morphologies at the cross-sections of mineralized hydrogels as a function of the heating rate. (c) Mineral volume of the mineralized hydrogels (determined by μ-CT, n=3) as a function of the heating rate and the overall mineralization time. (d) Total calcium content of mineralized hydrogel (determined by selective ion electrode upon acid treatment, n=3) as a function of heating rate and overall mineralization time. (e) Mineral density of mineralized hydrogels (determined by μ-CT) as a function of heating rate. (f) Anisotropy of the mineralized hydrogels (determined by μ-CT, n=3) as a function of heating rate and overall mineralization time. (g) Mineral density distribution across the mineralized hydrogels as visualized by color mapping of sagittal cross-sections (by μ-CT) as a function of heating rate. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison. Pairwise comparisons of each parameter with those obtained at the fastest heating rate are statistically significant (P < 0.05) unless denoted as NS (not significant). Complete pairwise comparisons are shown in Supplementary Tables S4–S7.
Interestingly, the size and spatial distribution of the mineral domains within the hydrogel also dramatically altered with the heating rate (Figs. 6a & 6b, Supplementary Figs. S4 & S5). At the faster heating rates of 0.5–1.0 °C/min, isolated small spherical mineral nodules (Type I, Fig. 5), characterized with a narrow mineral size distribution (8–24 μm; Fig. 6a), relatively low mineral density (Figs. 6e & g) and low anisotropy (Fig. 6f), were formed across the hydrogel as supported by SEM micrographs (Supplementary Figs. S4, S7a &e). Although the higher supersaturation resulting from the faster heating (e.g. 0.5 °C/min, Supplementary Fig. S6) could expedite the heterogeneous nucleation and growth[1], the limited time (Supplementary Fig. S6, Supplementary Table S3) may have prevented adequate diffusion of precursor ions into the hydrogel to compensate the locally depleted precursor ion (due to mineral growth), resulting in the limited size of the mineral nodules. When slower heating rates (0.1–0.2 °C/min) were employed, larger spherical mineral nodules (Type II, Fig. 5) with narrow Gaussian mineral size distribution (diameters 30–60 μm; Figs. 6a & 6b, Supplementary Figs. S4, S7b & f) resulting from more sufficient time for ion diffusions and mineral growth were indeed obtained. The increase in size of the mineral nodules was accompanied by a relative increase in anisotropy (Fig. 6f). At the heating rate of 0.05 °C/min, even larger spherical mineral domains containing multiple nucleation centers, with their sizes deviating from the typical Gaussian distributions, were formed (Figs. 6a & 6b, Supplementary Figs. S7c & g). Such morphologies represent the departure from the typical type II mineral morphology (Fig. 5). When the heating rate was further reduced to 0.02 °C/min, bicontinuous mineral aggregates of small, Liesegang ring-like,[53] spherical nodules were formed in high anisotropy across the hydrogel (Figs. 6a, b & f, Supplementary Figs. S4, S5, S7d & h) We hypothesize that the relative lower level of supersaturation and the more sufficient time resulting from the slower increase in pH with the much reduced heating rate would allow the slowly grown minerals to aggregate by interparticle forces[54]. The heating rate-dependent density, volume and spatial distribution of the minerals within the mineralized composites were also confirmed by the temporal changes in appearance of the composites as they were demineralized by acid treatment (Supplementary Fig. S8).
The mineral nodules in all composites examined were structurally well-integrated with the hydrogel matrix, as evidenced by SEM micrographs of the cross-sections of the composites (Supplementary Fig. S7). No delamination of the spherical mineral nodules from the hydrogel matrix was observed when the air-dried composites were fractured to expose the cross-sections. In addition, we observed steady increases in stiffness and compressive modulus of the hydrated composites as the total mineral contents increased with slower heating rates (Supplementary Fig. S9), further supporting the good structural integration between the organic and inorganic components. It is likely that the increased connectivity of the mineral domains (higher anisotropy) within the composite mineralized with the slowest heating rate examined (0.02 °C/min) also contributed to further improvements in its compressive modulus (e.g. modulus at 50% strain, Supplementary Fig. S9b) as there was no significant difference between its total mineral content and that mineralized with the heating rate of 0.05 °C/min.
3.3.2 Regulating HA mineralization outcome by crosslinking content of pSBMA
We next tested the hypothesis that the chemical crosslinker content can significantly influence the solute diffusibility as a result of the unique swelling behavior of the zwitterionic hydrogel in a high-salt environment (Fig. 7c) such as the supersaturated mineralization solution, thereby influencing the mineralization outcome. Six hydrogels with EGDMA crosslinker contents ranging from 0.027 to 2.651 mol% were mineralized with a fixed heating rate of 0.2 °C/min (Supplementary Table S2). The mineral volume, mineral size distribution, and total mineral content of these composites, determined by μ-CT analyses and calcium release assays, inversely correlated with the crosslinker content (Figs. 7a, b, d & e), with substantial mineralization across the 3-D scaffolds (Fig. 7a, Supplementary Figs. S10 & S11). SEM micrographs (Fig. 7b, Supplementary Fig. S12) revealed that pSBMA with higher crosslinker contents (2.651 and 1.326 mol%) were mineralized with smaller spherical mineral nodules (type I, Fig. 5) while those with medium crosslinker contents (0.663, 0.265, and 0.133 mol%) were mineralized with larger spherical mineral nodules (type II, Fig. 5) within the hydrogel interior. The increase in size of the mineral domains (Fig. 7a) with decreasing crosslinker content at the given heating rate could be attributed to the significantly higher swelling ratio for the hydrogels with lower crosslinker content (e.g. volume swelling ratios in PBS: ~22 for the 0.027 mol% gel vs. ~2 for the 2.651 mol% gel, Fig. 7c). The significant increase in volume of the swollen hydrogel may have further facilitated the zwitterionic ligand-mediated active infiltration of precursor ions and more sustained growth of mineral domains. Closer examination of the composites mineralized from the hydrogel with very low crosslinker content (0.027 mol%) by SEM (Fig. S12) revealed large spherical mineral domains comprising of multiple nucleation centers, signaling a departure from the typical type II mineral morphology and the transitioning into the type III mineral morphology where smaller mineral nodules aggregate. Interestingly, unlike mineral volume and total calcium content, the mineral density of these composites steadily increased with increasing crosslinker contents from 0.027 to 0.663 mol%, before reaching a plateau at the crosslinker content beyond 1.326 mol% (Supplementary Fig. S13, note that no statistically significant difference in mineral density was observed from the composites with 1.326 and 2.651 mol% crosslinker contents). The loosely crosslinked hydrogels have likely allowed better diffusion of precursor ions, thereby replenishing precursor ions due to mineral nucleation and growth in a timely manner to sustain a relative high supersaturation (S) within the hydrogel microenvironment, resulting in a fast mineral growth rate JG[1].
Figure 7.

Effect of chemical crosslinker content on the pSBMA hydrogel mineralization outcome. (a) μ-CT views (sagittal and coronal) of the mineralized hydrogels and respective mineral size distribution in diameters (determined by μ-CT) as a function of the crosslinker content. (b) SEM micrographs of the cross-sections of the mineralized hydrogels as a function of the crosslinker content. (c) Swelling ratios of pSBMA hydrogels by volume (Sv) in water (red) and in PBS (green) as a function of the chemical crosslinker content (n=3). The swelling ratios of pSBMA hydrogels in water by weight (Sw, black) and by volume (Sv, red) are plotted in the inset. (d) Mineral volume of the mineralized hydrogels (determined by μ-CT, n=3) as a function of crosslinker content. (e) Total calcium content of the mineralized hydrogels (determined by selective ion electrode upon acid treatment, n=3) as a function of the crosslinker content. All hydrogels were mineralized with a heating rate of 0.2 °C/min. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison. Pairwise comparisons of each parameter with those obtained with lowest crosslinker content (0.027 mol%) are statistically significant (P < 0.05) unless denoted as NS (not significant). Complete pairwise comparisons are shown in Supplementary Tables S9 & S10.
3.3.3 A model of synergistic modulation of mineralization outcome by heating rates and crosslinker contents
The above investigation of the roles of heating rate (mineralization time) and covalent crosslinker content on the mineralization outcome of the zwitterionic pSBMA hydrogel supported that three predominant types of mineral domains can be obtained by tuning these experimental parameters (Fig. 5). We found that lower contents of isolated small spherical mineral nodules (Type I) tended to form with either faster heating rates (1.0 and 0.5 °C/min) or higher pSBMA crosslinker contents (2.651 and 1.326 mol%). These conditions limited either the time for sufficient precursor ion diffusions across the hydrogel and/or space for continued mineral growth within the hydrogel. Higher mineral content and larger spherical mineral nodules within the pSBMA hydrogels (type II) could be obtained with medium heating rates (0.2 and 0.1 °C/min) or medium crosslinking densities (0.663, 0.265 and 0.133 mol%) as these conditions allowed more sufficient time and space for sufficient ion diffusion, maintaining the local supersaturation for sustained mineral growth. Formation of stripe-like bicontinuous mineral aggregates of small spherical mineral nodules within the hydrogel (Type III) could be accomplished with either much slower heating rate (0.02 °C/min) or very low crosslinking densities (<0.027 mol%). These conditions may have allowed sufficient time, space or polymer chain mobility (e.g. higher polymer chain motilities are expected within the less crosslinked hydrogel network) for the slow-growing mineral domains to interact and aggregate. Borderline crosslinker content such as 0.027 mol% and borderline heating rate such as 0.05 °C/min led to mineral morphologies departing from Type II and transitioning to Type III. To validate this proposed model, we demonstrated that lowering the mineralization heating rate of the pSBMA hydrogel with borderline crosslinker content of 0.027 mol% from 0.2 to 0.05 °C/min (increasing mineralization time from 4.8 to 19.3 h) indeed resulted in the transitioning from Type II spherical mineral morphology to Type III stripe-like morphology with increasing mineral volume (Figs. 8a & b). Likewise, lowering the crosslinker content from a medium 0.265 mol% to the lower 0.027 mol% at a given borderline heating rate of 0.05 °C/min also led to the transitioning from the Type II morphology to the higher mineral content, stripe-like type III morphologies (Figs. 8a & c).
Figure 8.

Effects of prolonged mineralization (slower heating rate) vs. crosslinker content, as well as repeating mineralization cycles, on the mineralization outcome of pSBMA hydrogel. (a) SEM micrographs and μ-CT views of the sagittal cross-sections of the mineralized hydrogels with lower crosslinker content and shorter mineralization time (faster heating rate, left), lower crosslinker content and longer mineralization time (slower heating rate, middle), and higher crosslinker content and longer mineralization time (slower heating rate, right). (b) Mineral volume (determined by μ-CT, n=3) as a function of mineralization time (heating rate) at a low hydrogel crosslinker content of 0.027 mol%. (c) Mineral volume (determined by μ-CT, n=3) as a function of hydrogel crosslinker content with slower heating rate of 0.05 °C/min (mineralization time of 19.3 h).
3.3.4. Effect of repeated mineralization cycles on mineralization outcome
Finally, we demonstrated that by repeating the urea thermal decomposition-mediated HA-mineralization process to pSBMA (e.g. with the borderline 0.027 mol% crosslinker content and a heating rate of 0.2 °C/min), the overall mineral content and the number of the mineral modules were steady increased (Fig. 9). This finding offers a practical approach to further modulating the mineralization outcome, achieving even more extensively mineralized zwitterionic hydrogel-HA composites if desired.
Figure 9.
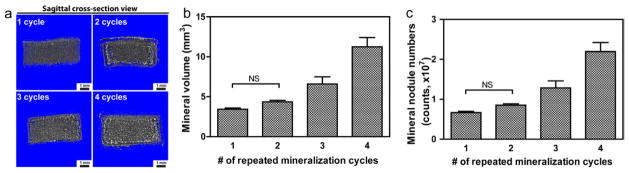
Effect of repeated mineralization cycles on the mineralization outcome of pSBMA. (a) Sagittal μ-CT cross-section views, (b) mineral content, and (c) mineral nodule numbers (both by μ-CT, n=3) as a function of the number of repeated mineralization cycles. Statistical significance are determined by one-way ANOVA with Tukey’s multiple comparison. All pairwise comparisons are significant (P < 0.05) unless denoted as NS (not significant).
4. Conclusions
Inspired by the synergistic roles of oppositely charged residues in natural biomineralization processes, here we explore the strategic use of zwitterionic ligands to more effectively template the HA-mineralization of synthetic scaffolds. We showed that zwitterionic sulfobetaine is effective in templating extensive heterogeneous HA-mineralization across 3-D hydrogel scaffolds. The uniquely positioned opposite charges have likely played a critical role in facilitating active infiltration of oppositely charged precursor ions or pre-nucleation ion clusters[17, 26] and lowering the interfacial energy for heterogeneous nucleation and growth. Although 3-D mineralization of HA can also be achieved in anionic or cationic hydrogels, the significantly more cytocompatible pSBMA, along with its appropriate (not excessive) swelling behavior under physiological conditions, make the zwitterionic hydrogel uniquely suited for potential in vivo applications and for the preparation of higher density composites. The end-stage the mineralization outcome of the zwitterionoic hydrogel, including size, density, and the spatial distribution of the templated mineral growth, can be readily controlled through facile adjustments of the heating rate of the urea thermal decomposition-mediated mineralization process and the crosslinker content of the zwitterionic hydrogels. The inexpensive sulfobetaine can be incorporated into a wide variety of synthetic scaffolds, offering greater versatility than natural ECM such as collagen for scaffold-assisted skeletal tissue repair. Other zwitterionic molecules such as the phosphobetaine and carboxybetaine may also be explored as cytocompatible mineral nucleating ligands, and, collectively, could usher in creative utilization of zwitterionic polymers or polymer coatings for a wide range of orthopedic and dental applications.
Supplementary Material
Figure S1. SEM micrographs revealing surface and cross-section mineral domain morphologies of the mineralized hydrogels containing identical crosslinker content (1.326 mol%) but varied side chain charges. All hydrogels were subjected to heterogeneous mineralization by controlled heating in an acidic solution of HA containing 2-M urea from 37 °C to 95 °C at a heating rate of 0.2 °C/min.
Figure S2. HR-TEM micrograph of the mineralized composite formed at the heating rate of 0.2 °C/min, suggesting predominantly crystalline phase of the mineral component.
Figure S3. Cell viability of rMSC encapsulated by pSBMA and the PEGMA control (with identical crosslinker content of 1.33 mol%) as determined by MTT assay (n=3). No significant difference (p > 0.05) was observed between the cells encapsulated by the 2 hydrogel formulations at a given time, while significant increases in viable cells were detected over time in each hydrogel formulation Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison.
Figure S4. SEM micrographs of the surfaces and cross-sections of mineralized pSBMA hydrogels as a function of the heating rate. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol%.
Figure S5. Reconstructed μ-CT images of mineralized pSBMA hydrogels obtained with various heating rates. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol%.
Figure S6. Temporal pH changes of the mineralization solution as a function of the heating rate and overall mineralization time (n=3). The first pH plateaus (from pH 3.8 to 4.0, indicated by the green box) observed in all six pH-mineralization time curves could be attributed to the deprotonation of H2PO4-ions. All specimens contained the same hydrogel crosslinker content of 0.265 mol%.
Figure S7. SEM micrographs revealing the different mineral nodule sizes and morphologies as a function of the heating rate. a & e, Specimen mineralized at 0.5 °C/min where small spherical mineral nodules were formed (type I). b & f, Specimen mineralized at 0.2 °C/min where large spherical mineral nodules were formed (type II). c & g, Specimen mineralized at 0.05 °C/min where larger spherical mineral domains containing multiple smaller nodules were formed, indicating a departure from a typical type II mineral morphology. d & h, Specimen mineralized at 0.02 °C/min where bicontinuous stripe-like mineral aggregates were formed (type III). All mineralized specimens contained the same crosslinker content of 0.265 mol%.
Figure S8. Digital photograph images of the mineralized pSBMA hydrogels at various time points of the acid treatment. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol%.
Figure S9. Compressive mechanical properties of the mineralized pSBMA hydrogels as a function of the heating rate. a, Compressive stress-strain curves of hydrated specimens as a function of the heating rate of their mineralizations. The data were recorded on a Q800 DMA equipped with a submersion compression fixture at 25 °C. b, Stress at 50 % compressive strain of the mineralized specimens as a function of the heating rate and the respective mineralization time. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison tests. Pairwise comparisons with unmineralized pSBMA hydrogel are statistically significant (P < 0.05) unless denoted as NS (not significant). Complete pairwise comparisons are in shown in Supplementary Tables S8. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol %.
Figure S10. Digital photograph images of the mineralized hydrogels with different crosslinker contents at various time points of the acid treatment. All the hydrogel specimens were mineralized with a heating rate of 0.2 °C/min.
Figure S11. Reconstructed μ-CT images of mineralized pSBMA hydrogels containing different crosslinker content. All specimens were mineralized with a heating rate of 0.2 °C/min.
Figure S12. SEM micrographs of the surfaces and cross-sections of the mineralized pSBMA hydrogels containing different crosslinker contents. All hydrogel specimens were mineralized with a heating rate of 0.2 °C/min.
Figure S13. Mineral density (a) distribution colored mapping (b) of pSBMA hydrogels as a function of crosslinking content (determined by μ-CT). The mineral density increased gradually with the increase of crosslinker content (obviously from 0.027 to 0.265 mol%, but not so clear from 0.265 to 2.651 mol% due to the mineral size). All specimens were mineralized with a heating rate of 0.2 °C/min. Complete pairwise comparisons of mineral density are shown in Supplementary Table S11.
Table S1. Preparation of hydrogels with different side chain charges for mineralization (No.1–4) and cyto-compatibility (No. 5–7) studies.
Table S2. Preparation and mineralization conditions of zwitterionic pSBMA hydrogels
Table S3. Sampling intervals for the monitoring of pH changes of the mineralization solution with different heating rates.
Table S4 Complete statistical significance data (p values) of the mineral volume pairwise comparisons as a function of heating rate (corresponding to Fig. 5c) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S5. Complete statistical significance data (p values) of the calcium content pairwise comparisons as a function of heating rate (corresponding to Fig. 5d) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S6. Complete statistical significance data (p values) of the mineral density pairwise comparisons as a function of heating rate (corresponding to Supplementary Fig. 5e) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S7. Complete statistical significance data (p values) of the mineral anisotropy pairwise comparisons as a function of heating rate (corresponding to Fig. 5f) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S8. Complete statistical significance data (p values) of the mechanical property pairwise comparisons as a function of heating rate (corresponding to Supplementary Figure S8b) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S9. Complete statistical significance data (p values) of the mineral volume pairwise comparisons as a function of crosslinker content (corresponding to Fig. 6d) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S10. Complete statistical significance data (p values) of the calcium content pairwise comparisons as a function of crosslinker content (corresponding to Fig. 6e) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S11. Complete statistical significance data (p values) of the mineral density pairwise comparisons as a function of crosslinker content (corresponding to Supplementary Fig. S13a) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Acknowledgments
The work was supported by the National Institutes of Health Grant R01AR055615. Core resources supported by the National Center for Research Resources Grants S10RR027082 and S10RR021043 were used. FIB was performed at the Center for Nanoscale Systems (CNS) at Harvard University supported by the National Science Foundation award ECS-0335765. The authors thank Emily Domingue for technical assistance on mineral release studies.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mann S. Biomineralization: Principles and concepts in bioinorganic materials chemistry. Oxford: Oxford University Press; 2001. [Google Scholar]
- 2.Mann S. Molecular recognition in biomineralization. Nature. 1988;332:119–24. [Google Scholar]
- 3.Weiner S, Wagner HD. The material bone: Structure mechanical function relations. Annu Rev Mater Sci. 1998;28:271–98. [Google Scholar]
- 4.Traub W, Arad T, Weiner S. 3-Dimensional ordered distribution of crystals in turkey tendon collagen-fibers. Proc Natl Acad Sci USA. 1989;86:9822–6. doi: 10.1073/pnas.86.24.9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George A, Veis A. Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition. Chem Rev. 2008;108:4670–93. doi: 10.1021/cr0782729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter GK, Goldberg HA. Modulation of crystal formation by bone phosphoproteins: role of glutamic acid-rich sequences in the nucleation of hydroxyapatite by bone sialoprotein. Biochem J. 1994;302:175–9. doi: 10.1042/bj3020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tye CE, Rattray KR, Warner KJ, Gordon JAR, Sodek J, Hunter GK, et al. Delineation of the hydroxyapatite-nucleating domains of bone sialoprotein. J Biol Chem. 2003;278:7949–55. doi: 10.1074/jbc.M211915200. [DOI] [PubMed] [Google Scholar]
- 8.He G, Dahl T, Veis A, George A. Nucleation of apatite crystals in vitro by self-assembled dentin matrix protein, 1. Nat Mater. 2003;2:552–8. doi: 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- 9.Veis A, Perry A. The phosphoprotein of the dentin matrix. Biochemistry. 1967;6:2409–16. doi: 10.1021/bi00860a017. [DOI] [PubMed] [Google Scholar]
- 10.George A, Bannon L, Sabsay B, Dillon JW, Malone J, Veis A, et al. The carboxyl-terminal domain of phosphophoryn contains unique extended triplet amino acid repeat sequences forming ordered carboxyl-phosphate interaction ridges that may be essential in the biomineralization process. J Biol Chem. 1996;271:32869–73. doi: 10.1074/jbc.271.51.32869. [DOI] [PubMed] [Google Scholar]
- 11.Olszta MJ, Cheng XG, Jee SS, Kumar R, Kim YY, Kaufman MJ, et al. Bone structure and formation: A new perspective. Mat Sci Eng R. 2007;58:77–116. [Google Scholar]
- 12.Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res. 2011;108:1494–509. doi: 10.1161/CIRCRESAHA.110.234260. [DOI] [PubMed] [Google Scholar]
- 13.Boskey AL, Spevak L, Paschalis E, Doty SB, McKee MD. Osteopontin deficiency increases mineral content and mineral crystallinity in mouse bone. Calcif Tissue Int. 2002;71:145–54. doi: 10.1007/s00223-001-1121-z. [DOI] [PubMed] [Google Scholar]
- 14.Fantner G, Hassenkam T, Kindt J, Weaver J, Birkedal H, Pechenik L, et al. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat Mater. 2005;4:612–6. doi: 10.1038/nmat1428. [DOI] [PubMed] [Google Scholar]
- 15.Gebauer D, Colfen H. Prenucleation clusters and non-classical nucleation. Nano Today. 2011;6:564–84. [Google Scholar]
- 16.Mahamid J, Aichmayer B, Shimoni E, Ziblat R, Li C, Siegel S, et al. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc Natl Acad Sci USA. 2010;107:6316–21. doi: 10.1073/pnas.0914218107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nudelman F, Pieterse K, George A, Bomans PHH, Friedrich H, Brylka LJ, et al. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat Mater. 2010;9:1004–9. doi: 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer LC, Newcomb CJ, Kaltz SR, Spoerke ED, Stupp SI. Biomimetic systems for hydroxyapatite mineralization inspired by bone and enamel. Chem Rev. 2008;108:4754–83. doi: 10.1021/cr8004422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartgerink JD, Beniash E, Stupp SI. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science. 2001;294:1684–8. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 20.Cha C, Kim ES, Kim IW, Kong H. Integrative deign of a poly(ethylene glycol)-poly(propylene glycol)-alginate hydrogel to control three dimensional biomineralization. Biomaterials. 2011;32:2695–703. doi: 10.1016/j.biomaterials.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 21.Chao Z, Chu CC. Biomimetic mineralization of acid polysaccharide-based hydrogels: towards porous 3-dimensional bone-like biocomposites. J Mater Chem. 2012;22:6080–6087. [Google Scholar]
- 22.Phadke A, Zhang C, Hwang Y, Vecchio K, Varghese S. Templated mineralization of synthetic hydrogels for bone-like composite materials: role of matrix hydrophobicity. Biomacromolecules. 2010;11:2060–8. doi: 10.1021/bm100425p. [DOI] [PubMed] [Google Scholar]
- 23.Murphy WL, Mooney DJ. Bioinspired growth of crystalline carbonate apatite on biodegradable polymer substrata. J Am Chem Soc. 2002;124:1910–7. doi: 10.1021/ja012433n. [DOI] [PubMed] [Google Scholar]
- 24.Stephenson A, DeYoreo J, Wu L, Wu K, Hoyer J, Dove P. Peptides enhance magnesium signature in calcite: insights into origins of vital effects. Science. 2008;322:724–7. doi: 10.1126/science.1159417. [DOI] [PubMed] [Google Scholar]
- 25.Taubert A, Junginger M, Kita-Tokarczyk K, Schuster T, Reiche J, Schacher F, et al. Calcium phosphate mineralization beneath a polycationic monolayer at the air-water interface. Macromol Biosci. 2010;10:1084–92. doi: 10.1002/mabi.201000093. [DOI] [PubMed] [Google Scholar]
- 26.Koumoto K, Zhu PX, Masuda Y. The effect of surface charge on hydroxyapatite nucleation. Biomaterials. 2004;25:3915–21. doi: 10.1016/j.biomaterials.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 27.Song J, Saiz E, Bertozzi CR. A new approach to mineralization of biocompatible hydrogel scaffolds: An efficient process toward 3-dimensional bonelike composites. J Am Chem Soc. 2003;125:1236–43. doi: 10.1021/ja028559h. [DOI] [PubMed] [Google Scholar]
- 28.Hutchens SA, Benson RS, Evans BR, O’Neill HM, Rawn CJ. Biomimetic synthesis of calcium-deficient hydroxyapatite in a natural hydrogel. Biomaterials. 2006;27:4661–70. doi: 10.1016/j.biomaterials.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 29.Ma PX. Biomimetic materials for tissue engineering. Adv Drug Deliver Rev. 2008;60:184–98. doi: 10.1016/j.addr.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CL, Qi JH, Zuckermann RN, DeYoreo JJ. Engineered biomimetic polymers as tunable agents for controlling CaCO3 mineralization. J Am Chem Soc. 2011;133:5214–7. doi: 10.1021/ja200595f. [DOI] [PubMed] [Google Scholar]
- 31.Banerjee I, Pangule RC, Kane RS. Antifouling Coatings: Recent Developments in the Design of Surfaces That Prevent Fouling by Proteins, Bacteria, and Marine Organisms. Adv Mater. 2011;23:690–718. doi: 10.1002/adma.201001215. [DOI] [PubMed] [Google Scholar]
- 32.Chen SF, Li LY, Zhao C, Zheng J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer. 2010;51:5283–93. [Google Scholar]
- 33.Ishihara K, Nomura H, Mihara T, Kurita K, Iwasaki Y, Nakabayashi N. Why do phospholipid polymers reduce protein adsorption? J Biomed Mater Res. 1998;39:323–30. doi: 10.1002/(sici)1097-4636(199802)39:2<323::aid-jbm21>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 34.Yan H, Zhu HM, Shen J. Molecular dynamics simulation study on zwitterionic structure to maintain the normal conformations of Glutathione. Sci China Ser B. 2007;50:660–4. [Google Scholar]
- 35.Nakaya T, Li YJ. Phospholipid polymers. Prog Polym Sci. 1999;24:143–81. [Google Scholar]
- 36.Liu PS, Chen Q, Wu SS, Shen J, Lin SC. Surface modification of cellulose membranes with zwitterionic polymers for resistance to protein adsorption and platelet adhesion. J Membrane Sci. 2010;350:387–94. [Google Scholar]
- 37.Jiang S, Cao Z. Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications. Adv Mater. 2010;22:920–52. doi: 10.1002/adma.200901407. [DOI] [PubMed] [Google Scholar]
- 38.Andrew JK, Shaoyi J. Poly(zwitterionic)protein conjugates offer increased stability without sacrificing binding affinity or bioactivity. Nat Chem. 2012;4:59–63. doi: 10.1038/nchem.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stupp SI, Ciegler GW. Organoapatites: Materials for artificial bone. I. Synthesis and microstructure. J Biomed Mater Res. 1992;26:169–83. doi: 10.1002/jbm.820260204. [DOI] [PubMed] [Google Scholar]
- 40.Stupp SI, Mejicano GC, Hanson JA. Organoapatites: Materials for artificial bone. II. Hardening reactions and properties. J Biomed Mater Res. 1993;27:289–99. doi: 10.1002/jbm.820270303. [DOI] [PubMed] [Google Scholar]
- 41.Kanapathipillai M, Yusufoglu Y, Rawal A, Hu YY, Lo CT, Thiyaigarajan P, et al. Synthesis and characterization of ionic block copolymer templated calcium phosphate nanocomposites. Chem Mater. 2008;20:5922–32. [Google Scholar]
- 42.Galin M, Chapoton A, Galin JC. Dielectric increments, intercharge distances and conformation of quaternary ammonioalkylsulfonates and alkoxydicyanoethenolates in aqueous and tirfluoroethanol solutions. J Chem Soc-Perkin Trans. 1993;2:545–53. [Google Scholar]
- 43.Clough SA, Beers Y, Klein GP, Rothman LS. Dipole-moment of water from stark measument of H2O, HDO, and D2O. J Chem Phys. 1973;59:2254–9. [Google Scholar]
- 44.Lee WF, Chen CF. Poly(2-hydroxyethyl methacrylate-co-sulfobetaine) hydrogels. II. Synthesis and swelling behaviors of the [2-hydroxyethyl methacrylate-co-3-dimethyl(methacryloyloxyethyl)ammonium propane sulfonate] hydrogels. J Appl Polym Sci. 1998;69:2021–34. [Google Scholar]
- 45.Xu JW, Filion TM, Prifti F, Song J. Cytocompatible poly(ethylene glycol)-co-polycarbonate hydrogels cross-linked by copper-free, strain-promoted click chemistry. Chem-Asian J. 2011;6:2730–7. doi: 10.1002/asia.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song J, Malathong V, Bertozzi CR. Mineralization of synthetic polymer scaffolds: A bottom-up approach for the development of artificial bone. J Am Chem Soc. 2005;127:3366–72. doi: 10.1021/ja043776z. [DOI] [PubMed] [Google Scholar]
- 47.Reichmuth DS, Kirby BJ. Effects of ammonioalkyl sulfonate internal salts on electrokinetic micropump performance and reversed-phase high-performance liquid chromatographic separations. J Chromatogr A. 2003;1013:93–101. doi: 10.1016/s0021-9673(03)01468-7. [DOI] [PubMed] [Google Scholar]
- 48.Nelson DGA, McLean JD. High-resolution electron microscopy of octacalcium phosphate and its hydrolysis products. Calcif Tissue Int. 1984;36:219–32. doi: 10.1007/BF02405321. [DOI] [PubMed] [Google Scholar]
- 49.Suvorova EI, Buffat PA. Electron diffraction from micro- and nanoparticles of hydroxyapatite. J Microsc-Oxf. 1999;196:46–58. doi: 10.1046/j.1365-2818.1999.00608.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhu YB, Gao CY, Shen JC. Surface modification of polycaprolactone with poly(methacrylic acid) and gelatin covalent immobilization for promoting its cytocompatibility. Biomaterials. 2002;23:4889–95. doi: 10.1016/s0142-9612(02)00247-8. [DOI] [PubMed] [Google Scholar]
- 51.Hong SP, Leroueil PR, Janus EK, Peters JL, Kober MM, Islam MT, et al. Interaction of polycationic polymers with supported lipid bilayers and cells: Nanoscale hole formation and enhanced membrane permeability. Bioconjug Chem. 2006;17:728–34. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- 52.Nancollas GH, Zhang J. Formation and dissolution mechanisms of calcium phosphates in aqueous systems. In: Brown PW, Constantz B, editors. Hydroxyapatite and Related Materials. Ann Arbor: CRC Press; 1994. pp. 73–81. [Google Scholar]
- 53.Henisch HK, Garciaruiz JM. Crystalgrowth in gels and Liesegangringformation: II. Crystallization criteria and successive precipitation. J Cryst Growth. 1986;75:203–11. [Google Scholar]
- 54.Benning LG, Waychunas GA. Kinetics of Water-Rock Interaction. Springer; 2008. Nucleation, growth, and aggregation of mineral phases Mechanisms and kinetic controls; p. 840. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SEM micrographs revealing surface and cross-section mineral domain morphologies of the mineralized hydrogels containing identical crosslinker content (1.326 mol%) but varied side chain charges. All hydrogels were subjected to heterogeneous mineralization by controlled heating in an acidic solution of HA containing 2-M urea from 37 °C to 95 °C at a heating rate of 0.2 °C/min.
Figure S2. HR-TEM micrograph of the mineralized composite formed at the heating rate of 0.2 °C/min, suggesting predominantly crystalline phase of the mineral component.
Figure S3. Cell viability of rMSC encapsulated by pSBMA and the PEGMA control (with identical crosslinker content of 1.33 mol%) as determined by MTT assay (n=3). No significant difference (p > 0.05) was observed between the cells encapsulated by the 2 hydrogel formulations at a given time, while significant increases in viable cells were detected over time in each hydrogel formulation Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison.
Figure S4. SEM micrographs of the surfaces and cross-sections of mineralized pSBMA hydrogels as a function of the heating rate. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol%.
Figure S5. Reconstructed μ-CT images of mineralized pSBMA hydrogels obtained with various heating rates. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol%.
Figure S6. Temporal pH changes of the mineralization solution as a function of the heating rate and overall mineralization time (n=3). The first pH plateaus (from pH 3.8 to 4.0, indicated by the green box) observed in all six pH-mineralization time curves could be attributed to the deprotonation of H2PO4-ions. All specimens contained the same hydrogel crosslinker content of 0.265 mol%.
Figure S7. SEM micrographs revealing the different mineral nodule sizes and morphologies as a function of the heating rate. a & e, Specimen mineralized at 0.5 °C/min where small spherical mineral nodules were formed (type I). b & f, Specimen mineralized at 0.2 °C/min where large spherical mineral nodules were formed (type II). c & g, Specimen mineralized at 0.05 °C/min where larger spherical mineral domains containing multiple smaller nodules were formed, indicating a departure from a typical type II mineral morphology. d & h, Specimen mineralized at 0.02 °C/min where bicontinuous stripe-like mineral aggregates were formed (type III). All mineralized specimens contained the same crosslinker content of 0.265 mol%.
Figure S8. Digital photograph images of the mineralized pSBMA hydrogels at various time points of the acid treatment. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol%.
Figure S9. Compressive mechanical properties of the mineralized pSBMA hydrogels as a function of the heating rate. a, Compressive stress-strain curves of hydrated specimens as a function of the heating rate of their mineralizations. The data were recorded on a Q800 DMA equipped with a submersion compression fixture at 25 °C. b, Stress at 50 % compressive strain of the mineralized specimens as a function of the heating rate and the respective mineralization time. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparison tests. Pairwise comparisons with unmineralized pSBMA hydrogel are statistically significant (P < 0.05) unless denoted as NS (not significant). Complete pairwise comparisons are in shown in Supplementary Tables S8. All mineralized hydrogel specimens contained the same crosslinker content of 0.265 mol %.
Figure S10. Digital photograph images of the mineralized hydrogels with different crosslinker contents at various time points of the acid treatment. All the hydrogel specimens were mineralized with a heating rate of 0.2 °C/min.
Figure S11. Reconstructed μ-CT images of mineralized pSBMA hydrogels containing different crosslinker content. All specimens were mineralized with a heating rate of 0.2 °C/min.
Figure S12. SEM micrographs of the surfaces and cross-sections of the mineralized pSBMA hydrogels containing different crosslinker contents. All hydrogel specimens were mineralized with a heating rate of 0.2 °C/min.
Figure S13. Mineral density (a) distribution colored mapping (b) of pSBMA hydrogels as a function of crosslinking content (determined by μ-CT). The mineral density increased gradually with the increase of crosslinker content (obviously from 0.027 to 0.265 mol%, but not so clear from 0.265 to 2.651 mol% due to the mineral size). All specimens were mineralized with a heating rate of 0.2 °C/min. Complete pairwise comparisons of mineral density are shown in Supplementary Table S11.
Table S1. Preparation of hydrogels with different side chain charges for mineralization (No.1–4) and cyto-compatibility (No. 5–7) studies.
Table S2. Preparation and mineralization conditions of zwitterionic pSBMA hydrogels
Table S3. Sampling intervals for the monitoring of pH changes of the mineralization solution with different heating rates.
Table S4 Complete statistical significance data (p values) of the mineral volume pairwise comparisons as a function of heating rate (corresponding to Fig. 5c) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S5. Complete statistical significance data (p values) of the calcium content pairwise comparisons as a function of heating rate (corresponding to Fig. 5d) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S6. Complete statistical significance data (p values) of the mineral density pairwise comparisons as a function of heating rate (corresponding to Supplementary Fig. 5e) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S7. Complete statistical significance data (p values) of the mineral anisotropy pairwise comparisons as a function of heating rate (corresponding to Fig. 5f) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S8. Complete statistical significance data (p values) of the mechanical property pairwise comparisons as a function of heating rate (corresponding to Supplementary Figure S8b) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S9. Complete statistical significance data (p values) of the mineral volume pairwise comparisons as a function of crosslinker content (corresponding to Fig. 6d) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S10. Complete statistical significance data (p values) of the calcium content pairwise comparisons as a function of crosslinker content (corresponding to Fig. 6e) as determined by one-way ANOVA with Tukey’s multiple comparison tests.
Table S11. Complete statistical significance data (p values) of the mineral density pairwise comparisons as a function of crosslinker content (corresponding to Supplementary Fig. S13a) as determined by one-way ANOVA with Tukey’s multiple comparison tests.