

Summary
Background
To investigate the potential mechanisms underlying the protective effects of 18α Glycyrrhizin (GL) on rat hepatic stellate cells (HSCs) and hepatocytes in vivo and in vitro.
Material/Methods
Sprague-Dawley (SD) rats were randomly divided into 5 groups: normal control group, liver fibrosis group, high-dose 18α GL group (25 mg/kg/d), intermediate-dose 18α GL group (12.5 mg/kg/d) and low-dose 18α GL group (6.25 mg/kg/d). The rat liver fibrosis model was induced by carbon tetrachloride (CCl4). The expressions of α-smooth muscle actin (αSMA) and NF-κB were determined by real-time PCR and immunohistochemistry.
Results
18αGL dose-dependently inhibited the CCl4-induced liver fibrosis. There were significant differences in the mRNA and protein expressions of αSMA between the fibrosis group and 18α-GL treatment groups, suggesting that 18α GL can suppress the proliferation and activation of HSCs. Few HSCs were apoptotic in the portal area and fibrous septum in the liver fibrosis group. However, the double-color staining of a-SMA and TUNEL showed that 18α-GL treatment groups increased HSC apoptosis. NF-κB was mainly found in the nucleus in the fibrosis group, while cytoplasmic expression of NF-κB was noted in the 18αGL groups. In the in vitro experiments, 18α GL promoted the proliferation of hepatocytes, but inhibited that of HSCs. HSCs were arrested in the G2/M phase following 18α GL treatment and were largely apoptotic.
Conclusions
18α-GL can suppress the activation of HSCs and induce the apoptosis of HSCs by blocking the translocation of NF-κB into the nucleus, which plays an important role in the protective effect of 18α-GL on liver fibrosis.
Keywords: 18α-glycyrrhizin, hepatocyte, hepatic stellate cell, proliferation, apoptosis
Background
Hepatic stellate cells (HSCs), which are pericytes found in the space of Disse in the liver, constitute the main storage site for vitamin A (in the form of retinyl ester-containing lipid droplets) in the body and contribute to the production of extracellular matrix (ECM) proteins. In normal liver, HSCs are essentially quiescent, but have the ability to trans-differentiate into myofibroblast-like cells in response to liver injury during a process termed “activation” [1]. The activation of HSCs plays a critical role in the fibrogenesis, which is at present still poorly understood. The imbalance between the proliferation and apoptosis of HSCs is one of the main causes of liver fibrosis [2].
Licorice is one of the most ancient medicinal plants and has been used as a flavoring agent. In traditional Chinese medicine, it has been applied in the treatment of various inflammatory diseases [3]. Glycyrrhizin (GL) is the major bioactive triterpene glycoside of licorice root extract and has various pharmacological effects, such as anti-inflammatory, anti-viral and anti-allergic effects, as well as hepatocyte-proliferation and hepatoprotection [4]. It has 2 isomers: 18α-GL and 18β-GL. Due to the effectiveness and safety of 18α-GL, it is frequently used as a hepato-protective agent in clinical practice, especially in the treatment of liver dysfunction. However, the mechanism underlying the hepato-protective effects of 18α-GL remains unknown. The aim of the present study was to investigate the protective effects of 18α-GL on the carbon tetrachloride (CCl4)-induced liver fibrosis in rats, and to study the role of hepatocytes and HSCs in the protective effects of 18α-GL.
Material and Methods
Animals and grouping
Male Sprague-Dawley (SD) rats weighing 180~200 g were purchased from the Animal Center of the School of Medicine, Shanghai Jiao Tong University. Animals were randomly divided into 5 groups (n=8–12 per group): control group, liver fibrosis group, high dose GL group, intermediate dose GL group and low dose GL group. Rats in the control group received a subcutaneous injection of olive oil and an intraperitoneal injection of normal saline (NS) of the same dose. Rats in the remaining 4 groups received a subcutaneous injection of 0.2 ml/100 g CCl4 in olive oil twice weekly for 8 consecutive weeks (the first dose was doubled). From the day of CCl4 injection, rats in high, intermediate and low dose GL groups were intraperitoneally treated with 18α-GL at 25, 12.5 and 6.25 mg/kg, respectively, once daily for 8 weeks. All rats were anesthetized at the end of 8 weeks and the liver was collected. A part of the liver was fixed in 10% formaldehyde for 24 h and the rest was stored at −80°C. All the procedures were approved by the Animal Study Committee of China Shanghai Jiao Tong University.
Histological examination
Liver samples from all animals were processed for light microscopy. Tissue sections embedded in paraffin were stained with hematoxylin-eosin (H&E) and Masson’s trichrome, and then examined and scored by 2 pathologists blind to the study. Four fields were randomly selected from each section and histopathological evaluation was performed twice.
Hepatic fibrosis is divided into 5 stages according to the criteria developed by Scheuer: S0, none; S1, enlarged, fibrotic portal tracts; S2, periportal or portal-portal septa but intact architecture; S3, fibrosis with architectural distortion but without obvious cirrhosis; and S4, probable or definite cirrhosis [5].
Immunohistochemical analysis
Briefly, the sections (5 μm) were deparaffinized and then incubated in phosphate buffered saline solution (PBS) containing 3% H2O2 for 10 min to block the endogenous peroxidase activity. Subsequently, antigen retrieval was carried out in 0.01 mol/l citric acid buffer solution. The sections were then rinsed 3 times with PBS and blocked with Power Block™ Universal Blocking reagent (Biogenex, HK085-5KE, USA) for 10 min and incubated overnight with primary antibodies (α-SMA [1:500], Abcam, ab18460, USA); NF-κB [1:50], Cell Signaling Technology, Inc. cst-“#4764, USA). They were subsequently incubated for 30 min with corresponding secondary antibodies using the Super Sensitive™ Polymer-HRP Two-step Histostaining Reagent (Biogenex, HK518/9-YAK, USA), and visualization was performed with Biogenex stable DAB (3,3′-diaminobenzidine tetrahydrochloride). As a negative control, the primary antibody was replaced with PBS. Sections were counterstained, mounted, and examined by microscopy.
Brown-yellow granules represent positive expression. Five fields were randomly selected from each section, and the color image analysis system (Image-ProPlus(IPP) 6.0 software) was used to determine the protein expressions. The α-SMA labeling index was calculated by the ratio of positive expression area to the total field.
Detection of apoptosis by TUNEL assay
The in situ DNA fragmentation was visualized by the TUNEL method [6]. Briefly, deparaffinized sections were boiled in 0.01 mol/l citric acid buffer solution for 8.5 min and incubated in PBS containing 3% H2O2 for 10 min to block the endogenous peroxidase activity. The sections were incubated with the TUNEL reaction mixture, fluorescein-dUTP (in situ Cell Death Detection, AP kit, Roche, Germany) for 60 min at 37°C. The sections were then rinsed 3 times with PBS and incubated with anti-fluorescein antibody-AP for 30 min at 37°C. After washing 3 times in PBS, 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BICP/NBT, Maxim Biotechnology Development Co., Ltd NBT-2200, China) was added and counterstaining was performed with Nuclear Fast Red (Maxim Biotechnology Development Co., Ltd CTS-3099, China). As a negative control, the TUNEL reaction mixture was replaced with nucleotide mixture. Dual staining for a-SMA and TUNEL was undertaken in representative liver sections to localize apoptotic HSCs. After BICP/NBT was added, sections were washed 3 times with PBS and blocked for 10 min and incubated overnight with a-SMA. They were subsequently incubated for 30 min with corresponding secondary antibodies, and counterstaining was performed with Nuclear Fast Red. After the reaction was terminated by distilled water, the sections were stained with hematoxylin for 3 min. The number of apoptotic cells was counted under a microscope. The percentage of apoptotic cells was calculated from randomly selected fields. At least 1000 cells were counted in 5 random fields and the percentage of TUNEL-positive cells was then calculated (apoptotic index (AI – apoptosis cells/total cells) and HSC AI (apoptosis and a-SMA(+) cells/a-SMA(+) cells).
RNA isolation and real-time PCR
Total RNA was extracted from the liver using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and subjected to RT reaction by PrimeScript® RT reagent Kit (TAKARA, DRR037S, Japan). Real-time PCR was performed according to the manufacturer’s instructions using SYBR® Premix Ex Taq™ Kit (TAKARA, DRR041A, Japan) on the ABI-Prism 7700. Each experiment was performed in triplicate. GAPDH was used as an internal control. The primer sequences are listed in Table 1. The fold-change in the mRNA of target gene relative to that of GAPDH was calculated according to the previously reported method [7].
Table 1.
Primers used in qRT-PCR.
| Gene | Annealing temperature (°C) | Accession No. | Product size (bp) | Number gene primer |
|---|---|---|---|---|
| α-SMA | 58 | NM_031004.2 | 120 | F: 5-AGAAGCCCAGCCAGTCGCCATCA-3 |
| R: 5-AGCAAAGCCCGCCTTACAGAGCC-3 | ||||
| NF-κB | 58 | NM_199267.2 | 101 | F: 5-GACCTGGAGCAAGCCATTAGCC-3 |
| R: 5-CGGACCGCATTCAAGTCATAGTC-3 | ||||
| GAPDH | 58 | NM_017008.3 | 118 | F: 5-AGTTCAACGGCACAGTCAAG-3 |
| R: 5-TACTCAGCACCAGCATCACC-3 |
Growth curve of hepatocytes and HSCs
Hepatocytes (Chang liver cell lines) were purchased from the Cell Resource Center of CAS Shanghai Institute of Life Sciences, and HSCs (rat hepatic stellate cell line, cFSC) were kindly provided by the Laboratory Diagnostics Division of Shanghai Changzheng Hospital. Hepatocytes were maintained in PRMI-1640 (purchased from Austria PAA’s) containing 10% fetal bovine serum (FBS) and HSCs were grown in DMEM containing 10% FBS. Cells in logarithmic phase were digested with 0.25% trypsin and re-suspended at a density of 5×104/ml. Then, these cells were seeded into 24-well plates (1 ml/well) and incubated at 37°C in an atmosphere with 5% CO2 for 24 h. Different concentrations of 18α GL are divided into 5 groups (each plate as a group). 18α-GL of different concentrations was added and the experiment was performed in quadruplicates. The viable cells were counted every day and the growth curve was delineated.
Detection of proliferation by MTT assay
The hepatocytes and HSCs were seeded in 96-well plates at a density of 5×104/ml and incubated in an atmosphere with 5% CO2 at 37°C overnight, followed by observation of cell morphology. One day later, the supernatant was removed and 18α-GL of different concentrations was added, followed by incubation for 24 h. Then, 10 μl of MTT (Sigma) were added into each well, followed by incubation at 37°C in an atmosphere with 5% CO2 for 4 h. Subsequently, the supernatant was removed and 100 μl of DMSO were added into each well, followed by incubation for 10 min under continuous oscillation. Absorbance (A) was measured at 550 nm with a microplate reader (Thermo Labsystems, Finland).
Detection of cell cycle of hepatocytes and HSCs by flow cytometry
Hepatocytes were treated with TGF-β1 (Sigma, 8 ng/ml) and then with 18α-GL (0~1 mg/ml) in the medium, while HSCs were maintained in medium supplemented with 18α-GL alone. One day later, these cells were digested by trypsin and then collected by centrifugation. After washing with PBS, these cells were fixed in 70% cold alcohol at −20°C overnight. Cell cycle was analyzed by MCYCLE software with flow cytometry.
Apoptosis of hepatocytes and HSCs
Hepatocytes were maintained in the medium containing 18α-GL (0~1 mg/ml) and TGF-β1 (8 ng/ml), while HSC were maintained in medium supplemented with 18α-GL alone. One day later, these cells were digested by trypsin and then collected by centrifugation. After washing with PBS, the apoptosis of these cells was determined with Annexin V Kit (Roche) by flow cytometry.
Statistical analysis
Statistical analysis was performed with SPSS Version 11.0 statistic software package. Data were expressed as means ± standard deviation (SD). Comparisons between groups were performed with analysis of variance (ANOVA), Student’s T test or Kruskal-Wallis test. A value of P<0.05 was considered statistically significant.
Results
Histopathological findings
In the control group, the structure of the liver was clear, and the size of hepatocytes was constant. The hepatic lobule was intact, without denaturation or necrosis (Figure 1A). There were a few thin and short blue collagen fibers around the blood vessels (Figure 1D). In the liver fibrosis group, fatty degeneration was apparent and ballooning degeneration of hepatocytes was found around the limiting plate (Figure 1B). In the fibrosis group, the number of blue collagen fibers was significantly increased. These fibers were distributed from the portal area and central vein to the hepatic lobules, and collagenous fibers formed a thick textile fiber gap and pseudolobules (Figure 1E). In the 18α-GL group, the proliferation of fibrous tissues was absent (Figure 1C, 1F). The grades of liver fibrosis in each group are shown in Table 2.
Figure 1.
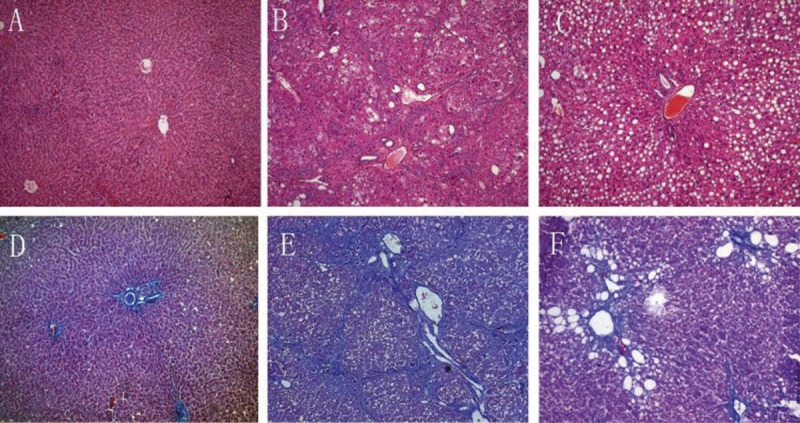
Representative Light Photomicrographs of liver Tissue Showing Effects of 18αGL on Histological Changes (H&E and Masson Staining, Original Magnification 100). (A–C) represented H&E staining of the control group, liver fibrosis group, and high-dose 18αGL groups, respectively. (D,E) represented Masson staining of control group, liver fibrosis group, and high-dose 18αGL groups, respectively.
Table 2.
Grades of liver fibrosis in each group.
| n | Mean ± SD | MR (mean rank) | |
|---|---|---|---|
| Control group | 8 | 0 | 4.5 |
| Fibrosis group | 12 | 3.75±0.45 | 39.13 |
| High dose GL group | 12 | 2.91±0.9 | 26.83* |
| Intermediate dose GL group | 12 | 3.00±0.85 | 27.83* |
| Low dose GL group | 10 | 3.3±0.82 | 32.35* |
P<0.05 vs. fibrosis group.
The mean rank of fibrosis in the 3 18α-GL groups was significantly lower than that in the fibrosis group (H=27.153, P<0.05). The histopathological changes in the intermediate and low dose 18α-GL groups were between those in the fibrosis group and those in the high dose 18α-GL group. These results show 18α-GL may prevent and improve CCL4-induced liver fibrosis.
Effect of 18α-GL on the activation of HSCs
The activated HSCs were detected by immunohistochemistry for α-SMA. Results showed α-SMA was mainly expressed in the vascular walls in the portal area, and rarely found in the perisinusoidal space of the liver parenchyma in the control group (Figure 2A, 2D). However, liver tissues were strongly positive for α-SMA in the fibrosis group (Figure 2B, 2E). In the 3 18α-GL treatment groups, α-SMA was less noted in the liver (Figure 2C). RT-PCR revealed there was a significant difference in the mRNA expression of α-SMA between the fibrosis group and the 3 18α-GL treatment groups. The ratio of positive protein and mRNA expression of α-SMA are shown in Figure 2F, 2G.
Figure 2.
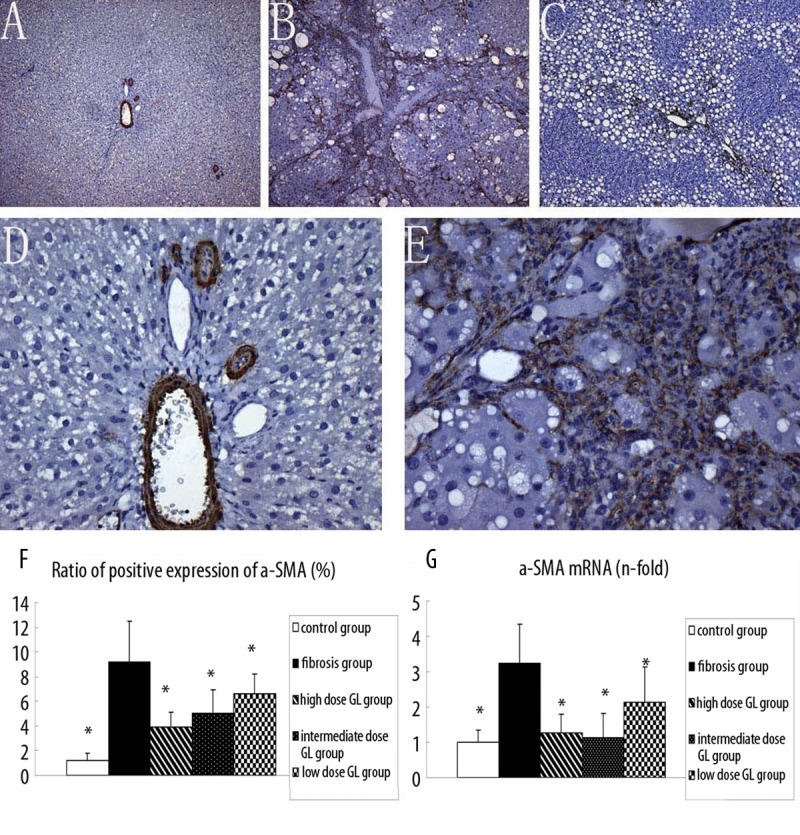
Effects of 18αGL on α-SMA Protein Expression in Rats liver Tissues (Positive as Brown, Original Magnification 100 (A–C) 400 (D,E), and mRNA level of a-SMA in five group. (A–C) represented the α-SMA deposition in control group, liver fibrosis group, and high-dose 18αGL groups, respectively. (D and E) are magnified image of (A and B). (F) bargraph showed the ratio of positive expression of a-SMAs. (G) bargraph showed mRNA level of a-SMA in five groups by qPCR quantity. Values are mean ± S.D * p<0.05 vs. liver fibrosis group.
Apoptosis of HSCs and hepatocytes
Only a small amount of apoptotic cells was found in the normal liver (Figure 3A, 3D, 3G), while the apoptotic cells were markedly increased in the fibrosis group (Figure 3B). In the 3 18α-GL treatment groups, the number of apoptotic cells in the liver was markedly larger than that in the control group (Figure 3C). Furthermore, the majority of apoptotic cells in the liver parenchyma were hepatocytes and only a few HSCs were apoptotic in the portal area and fibrous septum in the fibrosis group (Figure 3E, 3H). However, in the 18α-GL group, the apoptotic HSCs in the portal area increased, suggesting that 18α-GL may induce the HSC apoptosis (Figure 3F, 3I). The apoptosis index of HSCs and hepatocytes are shown in Table 3. Thus, in the following experiments, the effects of 18α-GL on the apoptosis of hepatocytes and HSCs were investigated in vitro independently.
Figure 3.
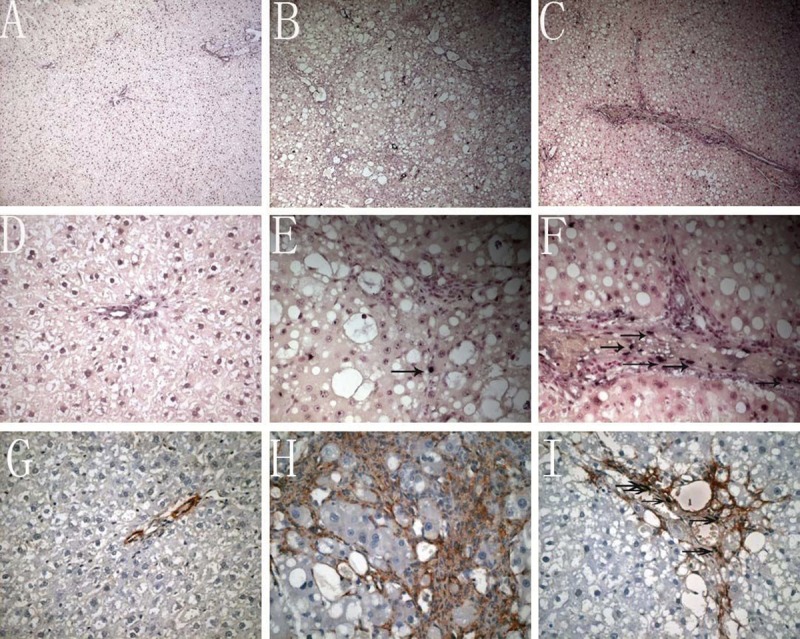
Effects of 18αGL on apoptosis in Rats liver Tissues by TUNEL stained (the nucleus dark blue is positive Original Magnification 100 (A–C) 400 (D–F). Dual staining for a-SMA and TUNEL was undertaken in representative liver sections to localize apoptotic HSCs (Dark brownish stained positive apoptotic bodies with TUNEL reaction. Red stain signified the area of HSC. Co-localization of both stain represented apoptotic HSCs (original magnification 400 (G–I)). (A–C) represented TUNEL staining of control group, liver fibrosis group, and high-dose 18αGL groups, respectively. (D–F) are magnified image of (A–C), respectively (G–I). Dual staining for a-SMA and TUNEL of control group, liver fibrosis group, and high-dose 18αGL groups, respectively. Single arrow indicates apoptosis of HSCs.
Table 3.
Apoptosis Index of HSCs and hepatocytes.
| n | AI% | HSC AI% | |
|---|---|---|---|
| Control group | 8 | 0.21±0.18* | 0.11±0.10 |
| Fibrosis group | 12 | 3.97±1.44 | 0.37±0.43 |
| High dose GL group | 12 | 3.70±1.20 | 3.03±1.11* |
| Intermediate dose GL group | 12 | 3.75±1.22 | 2.75±1.37* |
| Low dose GL group | 10 | 3.69±1.26 | 1.98±1.09* |
P<0.05 vs. fibrosis group.
NF-κB activation
NF-κB activation is closely related to the apoptosis of HSCs. Immunohistochemistry and PCR were employed to detect the levels of NF-κB in the liver. Under normal condition, NF-κB locates in the cytoplasm with a small amount of expression in the liver (Figure 4A). As a response to injury, NF-κB transfers from the cytoplasm to the nucleus, and then plays a critical role in the regulation of gene transcription. The immunohistochemistry showed that in the fibrosis group NF-κB was mainly found in the nucleus (Figure 4B), while in the 3 18α-GL groups, NF-κB was predominantly noted in the cytoplasm (Figure 4C). However, there was no significant difference in the mRNA expression of NF-κB between the 18α-GL treatment groups and the liver fibrosis group (Figure 4D), which was significantly increased when compared with that in the control group. We speculate that 18α-GL may block the translocation of NF-κB into the nucleus and inhibit its activation.
Figure 4.

Effects of 18αGL on NF-κB protein expression in rats liver tissues by immunohistochemistry staining (Positive as Brown, Original Magnification 400 (A–C), and mRNA level of NF-κB in five groups. (A–C) represented NF-κB staining of control group, liver fibrosis group, and high-dose 18αGL groups, respectively. Red arrows indicate nuclear-positive, and yellow arrows indicate the plasma positive. (D) The bargraph showed the mRNA level of NF-κB in five groups by qPCR quantity. Values are mean ± S.D; * p<0.05 vs. liver fibrosis group.
Effect of 18α-GL on the proliferation of hepatocytes and HSCs
The 18α-GL of different concentrations had distinct effects on the proliferation of hepatocytes. After treatment with 18α-GL at 0.001 mg/ml, 0.01 mg/ml and 0.1 mg/ml, the number of viable cells was not different from that in the control group (0 mg/ml 18α-GL) (P>0.05). However, following the treatment with 18α-GL at 1 mg/ml, the proliferation of viable hepatocytes was significantly promoted when compared with that in the control group (P<0.05), especially at 48 h after treatment (Figure 5A).
The 18α-GL of different concentrations had distinct effects on the proliferation of HSCs. The number of HSCs followed the treatment with 18α-GL at 0.001 mg/ml, 0.01 mg/ml and 0.1 mg/ml was similar to that in the control group (P>0.05). Nevertheless, the treatment with 18α-GL at 1 mg/ml markedly inhibited the proliferation of HSCs when compared with the control group (P<0.05), and this effect was in a time- and concentration-dependent manner (Figure 5B).
Figure 5.
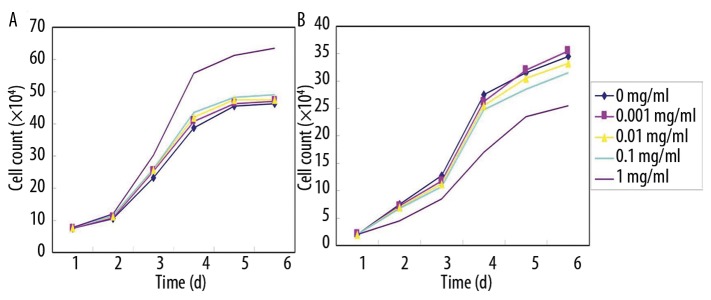
Effects of 18α GL at different concentrations (0–1 mg/ml) on the growth of hepatocytes and HSCs. (A) hepatocytes; (B) HSCs.
The promotive effect and suppressive effects of 18α-GL at designed concentrations on hepatocytes and HSCs, respectively, were further confirmed by the MTT assay (Tables 4 and 5).
Table 4.
Effects of 18α-GL on the proliferation of hepatocytes (x±s).
| Group | Concentration of 18α-GL (mg/ml) | OD value (x±s) |
|---|---|---|
| Control | 0 | 1.694±0.058 |
| 18α-GL group | 0.001 | 1.717±0.072 |
| 0.01 | 1.789±0.126 | |
| 0.1 | 1.751±0.110 | |
| 1 | 2.045±0.170* |
P<0.001 vs. control group.
Table 5.
Effects of 18α GL on the proliferation of HSCs.
| Group | Concentration of 18α-GL (mg/ml) | OD value (x±s) |
|---|---|---|
| Control | 0 | 1.401±0.145 |
| 18α-GL group | 0.001 | 1.434±0.139 |
| 0.01 | 1.320±0.050 | |
| 0.1 | 1.378±0.101 | |
| 1 | 0.568±0.065* |
P<0.001 vs. control group.
Changes in the cell cycle of hepatocytes and HSCs following 18α-GL treatment
TGF-β1 can negatively regulate the cell growth and arrest cells in G1 phase. In the present study, our results also confirmed the hepatocytes were arrested in G1 phase after TGF-β1 treatment. After 18α–GL (1 mg/ml) treatment, flow cytometry showed the TGF-β1-treated hepatocytes in G0/G1 phase were significantly decreased when compared with the control group with TGF-β1 treatment alone (P<0.05), but the cells in S phase were dramatically increased (P <0.05) (Table 6).
As the 18α-GL could exert inhibitory effect on the proliferation of HSCs, the following experiment was done to investigate the effect of 18α-GL on the cell cycle of HSCs. After 18α-GL (1 mg/ml) treatment, the HSCs in G2/M phase were remarkably elevated when compared with the control group (P<0.001) (Table 7).
Table 6.
DNA content in each phase of Hepatocytes after treatment with 18α-GL and TGF-β1 (8 ng/ml) (x±s, n=3).
| 18α-GL (mg/ml) | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|
| 0 (control) | 59.9±0.6 | 33.9±0.7 | 6.2±0.2 |
| 0.1 | 62.6±1.2 | 34.5±0.3 | 2.8±1.0# |
| 1 | 54.7±1.1** | 42.4±1.1* | 2.8±1.1## |
P<0.05 vs. control group;
P<0.01,
P<0.05,
P<0.05 vs. control group.
Table 7.
DNA content in each phase of HSCs after 18α-GL treatment (x±s, n=3).
| 18α-GL (mg/ml) | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|
| 0 (control) | 29.9±3.5 | 65.7±3.3 | 4.5±0.5 |
| 0.1 | 28.3±1.7 | 66.7±3.1 | 5.0±1.4 |
| 1 | 29.8±1.7 | 58.8±1.1 | 11.4±0.6* |
P<0.001 vs. control group.
Effect of 18α-GL on the apoptosis of hepatocytes and HSCs
The effect of 18α-GL on the TGF-β1-induced apoptosis of hepatocytes was further investigated. In TGF-β1-treated hepatocytes, after treatment with 18α-GL at 0.1 mg/ml and 1 mg/ml, the apoptosis rate was markedly lower than that in the control group (P<0.05) and the higher the 18α-GL concentration, the lower the apoptosis rate of hepatocytes, showing a dose-dependent manner (Table 8).
According to the results above, we hypothesized that 18α GL might inhibit the proliferation of HSCs by enhancing cell apoptosis. In the following experiment, we investigated the effect of 18α-GL on the apoptosis of HSCs. After treatment with 18α-GL at 1 mg/ml, the apoptotic rate of HSCs was significantly higher than that in the control group (P<0.01) (Table 9), which was consistent with our findings in vivo.
Table 8.
Apoptosis rate of hepatocytes after treatment with 18α-GL and TGF-β1 (x±s, n=3).
| 18α-GL (mg/ml) | Apoptosis (%) | F | P value |
|---|---|---|---|
| 0 (control) | 23.27±1.86 | ||
| 0.1 | 16.83±0.84 | 9.976 | <0.05 |
| 1 | 8.64±0.22 | 61.366 | <0.01 |
Table 9.
Apoptosis rate of HSCs after 18α-GL treatment (x±s, n=3).
| 18α-GL (mg/ml) | Apoptosis (%) | F | P value |
|---|---|---|---|
| 0 (control) | 2.93±0.29 | ||
| 0.1 | 3.36±0.50 | 1.621 | >0.05 |
| 1 | 4.81±0.16 | 31.591 | <0.01 |
Discussion
Glycyrrhetinic acid is an active metabolite of glycyrrhizin extracted from licorice root (Glycyrrhiza spp.), and has been applied as a herbal drug in the prevention of tumors as well as treatment of viral hepatitis and liver injury in China and Japan [8,9]. Animal experiments have demonstrated that glycyrrhizin and its metabolite can be used to treat liver fibrosis due to their anti-fibrotic or anti-hepatotoxic properties [10,11]. Randomized controlled trials also confirm that glycyrrhizin and its derivatives can improve the hepatocellular injury in chronic hepatitis B or C patients, and reduce the risk for hepatocellular carcinoma in patients with hepatitis C virus-induced cirrhosis [12]. However, the mechanism by which glycyrrhizin delays the progression of liver diseases is still unknown. Some research has revealed the hepatoprotective activity of licorice [13], and anti-viral capacity and immune modulatory activity of glycyrrhizin [14,15]. As mentioned above, GL has 2 isomers: 18α-GL and 18β-GL. In recent years, 18β-GL has been extensively investigated, and, in our lab, the protective effect of 18α-GL on CCL4-induced liver fibrosis and its impact on hepatocytes and HSCs were explored in vitro.
The present study showed 18α-GL could dose-dependently inhibit CCL4 -induced liver fibrosis, and the effects could be attributed to significant suppression of the proliferation and activation of HSCs and induction of apoptosis of HSCs following 18α-GL treatment, which may be related to the blocking of NF-κB translocation into the nucleus. In addition, in vitro experiments, 18α-GL could promote the proliferation of hepatocytes in a dose-dependent manner, which was contrary to the effect of 18α-GL on HSCs. Moreover, 18α-GL could also regulate cell cycle by arresting cells in G2/M phase and induce the apoptosis of HSCs.
Hepatic fibrosis is a scarring process associated with an increase and altered deposition of ECM in the liver. At present, fibrosis is considered to be a reversible process [16]. At the cellular and molecular levels, this process is mainly characterized by the “activation” of HSCs [17,18]. HSC activation consists of discrete phenotype responses, retinoid loss, proliferation, contractility, chemotaxis fibrogenesis and matrix degradation. Several types of cells and cytokines play important roles in the regulation of HSC activation. Currently, anti-fibrotic therapeutic strategies include inhibition of HSC proliferation or stimulation of HSC apoptosis, down-regulation of collagen production or promotion of its degradation [19,20]. The present study demonstrated that 18α-GL not only inhibited the activation or proliferation of HSCs, but also promoted the apoptosis of HSCs. These effects may be responsible for the significant improvement of liver fibrosis. In addition, in vitro experiments revealed that 18α-GL could regulate the cell cycle by arresting cells in G2/M phase and induce the apoptosis of HSCs. However, the exact signaling pathways underlying the effects of 18α-GL on the proliferation and apoptosis are largely unclear. The NF-κB is a key component in the cellular response to a variety of extracellular stimuli. The activation of NF-κB is associated with the phosphorylation of IκB, followed by its degradation by the proteasome and subsequent nuclear translocation. In the nucleus, NF-κB can bind to the DNA elements of target genes and positively regulate the transcription of genes involved in immune and inflammatory responses, cell growth and apoptosis [21]. As NF-κB is a potent pro-survival transcription factor in activated HSC [22,23], its inhibition could cause apoptosis of activated HSCs. There is indirect evidence showing the anti-apoptotic effect of NF-κB in HSCs. Jiang’s study showed that in phagocytosing HSCs there are different anti-apoptotic pathways induced, including a NADPH (nicotinamide adenine dinucleotide phosphate reduced) oxidase-dependent PI3K/Akt/ NF-κB induction pathway. NF-κB activation and subsequent upregulation of anti-apoptotic protein A1 promotes HSC survival [24]. Another study has shown that some nerve growth factors are expressed during fibrotic liver injury and may regulate the number of activated HSCs via inducing apoptosis, which is related to the increase of NF-κB activity, reduction of p50/p65 binding and decrease of NF-κB CAT reporter activities [25]. There is evidence showing that competitive antagonism of NF-κB can inhibit the inflammatory response and prevent CCL4-induced hepatic injury and fibrosis [26]. siRNA targeting NF-κB p65 can effectively enhance the HSC apoptosis and attenuate the ECM production [27]. Therefore, the anti-fibrotic effect of 18α-GL shown in the present study may be associated with the inhibition of NF-κB and the subsequent inflammatory response.
Furthermore, our result also revealed 18α-GL could promote the proliferation of hepatocytes, the hepatocytes in G0/G1 phase were significantly decreased after treatment with TGF-β1 and 18α-GL, and those in S phase markedly increased. Our results suggest that 18α-GL might be able to antagonize the TGF-β1-induced apoptosis of hepatocytes in vitro. We speculate that the proliferation and activation of HSCs are key participants in fibrogenesis. The apoptosis of hepatocytes through the activation of death receptors is also common in cholestasis, chronic alcoholic liver fibrosis, Wilson’s disease and viral hepatitis [28–30]. Liver tissue repair, inflammation, regeneration, and fibrosis may all be triggered by apoptosis [31,32]. Although both the apoptosis of hepatocytes and fibrosis are the features of chronic liver diseases, the potential relationship between these 2 processes remains unclear. Canbay et al. [33] revealed that hepatocyte apoptosis in the BDL mouse was, in part, mediated by Fas. Their observations also suggested Fas-mediated liver injury during extrahepatic cholestasis could result in fibrogenesis and collagen deposition in the liver. Thus, they conclude that Fas-mediated hepatocyte injury is mechanistically linked to liver fibrogenesis. In the future we will investigate the mechanism underlying the effect of 18α-GL on hepatocyte apoptosis. Taken together, we speculate that 18α-GL may become a promising anti-fibrogenic drug for the treatment of chronic liver diseases.
Abbreviations
- 18α-GL
18α-glycyrrhizin
- HSC
hepatic stellate cell
- NF-κB
nuclear factor-κ;B
- α-SMA
alpha-sooth muscle actin
- TGF-β1
transforming growth factor-β1
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
Source of support: This study was supported by National Natural Science Foundation of China (No: 30871162), the National Key Technologies Research and Development Program of China during the 11th Five-year Plan Period (2008ZX10002-006), the National High Technology Research and Development Program of China (863 Program, No: 2006AA 02A 411), Science and Technology Commission of Shanghai Municipality (No: 09XD1403200 and No: 10411955300)
References
- 1.Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38:S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 2.Gressner AM. The cell biology of liver fibrogenesis – an imbalance of proliferation, growth arrest and apoptosis of myofibroblasts. Cell Tissue Res. 1998;292:447–52. doi: 10.1007/s004410051073. [DOI] [PubMed] [Google Scholar]
- 3.Eisenbrand G. Glycyrrhizin. Mol Nutr Food Res. 2006;50:1087–88. doi: 10.1002/mnfr.200500278. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, Zhang L, Gurley E, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47(6):1905–15. doi: 10.1002/hep.22239. [DOI] [PubMed] [Google Scholar]
- 5.Scheuer PJ. Classification of chronic viral hepatitis: a need for reassessment. J Hepatol. 1991;13:372–74. doi: 10.1016/0168-8278(91)90084-o. [DOI] [PubMed] [Google Scholar]
- 6.Huang HF, Linsenmeyer TA, Li MT, et al. Acute effects of spinal cord injury on the pituitary-testicular hormone axis and Sertoli cell functions: a time course study. J Androl. 1995;16:148–57. [PubMed] [Google Scholar]
- 7.Schmittgen TD, Zakrajsek BA, Mills AG, et al. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: Comparison of endpoint and real-time methods. Analytical Biochemistry. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 8.Van Rossum TGJ, Vulto AG, De Man RA, et al. Review article: glycyrrhizin as a potential treatment for chronic hepatitis C. Aliment Pharmacol Ther. 1998;12:199–205. doi: 10.1046/j.1365-2036.1998.00309.x. [DOI] [PubMed] [Google Scholar]
- 9.Lin G, Nnane IP, Cheng TY. The effects of pretreatment with glycyrrhizin and glycyrrhetinic acid on the retrorsine-induced hepatotoxicity in rats. Toxicon. 1999;37:1259–70. doi: 10.1016/s0041-0101(98)00263-3. [DOI] [PubMed] [Google Scholar]
- 10.Luk JM, Zhang QS, Lee NP, et al. Hepatic stellate cell-targeted delivery of M6P-HSA-glycyrrhetinic acid attenuates hepatic fibrogenesis in a bile duct liGLtion rat model. Liver Int. 2007;27:548–57. doi: 10.1111/j.1478-3231.2007.01452.x. [DOI] [PubMed] [Google Scholar]
- 11.Wang JY, Zhang QS, Guo JS, et al. Effects of glycyrrhetinic acid on collagen metabolism of hepatic stellate cells at different stages of liver fibrosis in rats. World J Gastroenterol. 2001;7:115–19. doi: 10.3748/wjg.v7.i1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiore C, Eisenhut M, Krausse R, et al. Antiviral effects of Glycyrrhiza species. Phytother Res. 2008;22:141–48. doi: 10.1002/ptr.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JR, Park SJ, Lee HS, et al. Hepatoprotective Activity of Licorice Water Extract against Cadmium-induced Toxicity in Rats. Evid Based Complement Alternat Med. 2009;6:195–201. doi: 10.1093/ecam/nem078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abe M, Akbar SMF, Hasebe A, et al. Glycyrrhizin enhances interleukin-10 production by liver dendritic cells in mice with hepatitis. J Gastroenterol. 2003;38:962–67. doi: 10.1007/s00535-003-1179-7. [DOI] [PubMed] [Google Scholar]
- 15.Dai JH, Iwatani Y, Ishida T, et al. Glycyrrhizin enhances interleukin-12 production in peritoneal macrophages. Immunology. 2001;103:235–43. doi: 10.1046/j.1365-2567.2001.01224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927–39. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- 17.Reeves HL, Friedman SL. Activation of hepatic stellate cells – A key issue in liver fibrosis. Front Biosci. 2002;7:D808–D826. doi: 10.2741/reeves. [DOI] [PubMed] [Google Scholar]
- 18.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21:S84–87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 19.Li JT, Liao ZX, Ping J, et al. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol. 2008;43:419–28. doi: 10.1007/s00535-008-2180-y. [DOI] [PubMed] [Google Scholar]
- 20.Hagens WI, Beljaars L, Mann DA, et al. Cellular targeting of the apoptosis-inducing compound gliotoxin to fibrotic rat livers. J Pharmacol Exp Ther. 2008;324:902–10. doi: 10.1124/jpet.107.132290. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin AS. The transcription factor NF-kappa B and human disease. J Clin Invest. 2001;107(1):3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oakley F, Meso M, Iredale JP, et al. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128(1):108–20. doi: 10.1053/j.gastro.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Lang A, Schoonhoven R, Tuvia S, et al. Nuclear factor kappaB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J Hepatol. 2000;33(1):49–58. doi: 10.1016/s0168-8278(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 24.Jiang JX, Mikami K, Venugopal S, et al. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51(1):139–48. doi: 10.1016/j.jhep.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oakley F, Trim N, Constandinou CM, et al. Hepatocytes express nerve growth factor during liver injury – Evidence for paracrine regulation of hepatic stellate cell apoptosis. Am J Pathol. 2003;163(5):1849–58. doi: 10.1016/S0002-9440(10)63544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Son G, Iimuro Y, Seki E, et al. Selective inactivation of NF-kappa B in the liver using NF-kappa B decoy suppresses CCl4-induced liver injury and fibrosis. American J Physiol Gastrointest Liver Physiol. 2007;293(3):G631–39. doi: 10.1152/ajpgi.00185.2007. [DOI] [PubMed] [Google Scholar]
- 27.Cui DL, Zhang S, Ma JJ, et al. Short interfering RNA targeting NF-kappa B induces apoptosis of hepatic stellate cells and attenuates extracellular matrix production. Dig Liver Dis. 2010;42(11):813–17. doi: 10.1016/j.dld.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 28.Guicciardi ME, Gores GJ. Apoptosis as a Mechanism for Liver Disease Progression. Semin Liver Dis. 2010;30:402–10. doi: 10.1055/s-0030-1267540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491–508. doi: 10.2174/1566524033479555. [DOI] [PubMed] [Google Scholar]
- 30.GLlle PR, Krammer PH. CD95-induced apoptosis in human liver disease. Semin Liver Dis. 1998;18:141–51. doi: 10.1055/s-2007-1007150. [DOI] [PubMed] [Google Scholar]
- 31.Ghavami S, Hashemi M, Kadkhoda K, et al. Apoptosis in liver diseases – detection and therapeutic applications. Med Sci Monit. 2005;11(11):RA337–45. [PubMed] [Google Scholar]
- 32.Rust C, Gores GJ. Apoptosis and liver disease. Am J Med. 2000;108(7):567–74. doi: 10.1016/s0002-9343(00)00370-3. [DOI] [PubMed] [Google Scholar]
- 33.Canbay A, Higuchi H, Bronk SF, et al. Fas enhances fibrogenesis in the bile duct ligated mouse: A link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–30. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]