

Abstract
Hydroxyapatite (HA) is the principal constituent of bone mineral, and synthetic HA is widely used as a biomaterial for bone repair. Previous work has shown that polyglutamate domains bind selectively to HA and that these domains can be utilized to couple bioactive peptides onto many different HA-containing materials. In the current study we have adapted this technology to engineer polyglutamate domains into cargo-loaded nanocage structures derived from the P22 bacteriophage. P22 nanocages have demonstrated significant potential as a drug delivery system due to their stability, large capacity for loading with a diversity of proteins and other types of cargo, and ability to resist degradation by proteases. Site-directed mutagenesis was used to modify the primary coding sequence of the P22 coat protein to incorporate glutamate-rich regions. Relative to wild-type P22, the polyglutamate-modified nanocages (E2-P22) exhibited increased binding to ceramic HA disks, particulate HA and allograft bone. Furthermore, E2-P22 binding was HA selective, as evidenced by negligible binding of the nanocages to non-HA materials including polystyrene, agarose, and polycaprolactone (PCL). Taken together these results establish a new mechanism for the directed coupling of nanocage drug delivery systems to a variety of HA-containing materials commonly used in diverse bone therapies.
INTRODUCTION
Each year more than 2 million bone grafting procedures are performed to stimulate bone repair or regeneration in orthopedic, neurological and dental applications [1]. Autogenous bone is the gold standard graft material as it contains the patient’s own bone-forming cells and osteogenic proteins, and also provides a scaffold to support bone growth. However, there are limitations associated with autogenous grafts including the restricted amount of donor bone available and the considerable risk of postoperative pain and morbidity at the donor site [1]. Accordingly, allograft bone and synthetic bone-mimetic biomaterials are commonly used as alternatives to autograft [2]. Good clinical outcomes are achieved with allografted bone, but allograft has diminished osteoinductivity due to processing steps that eliminate cells and also denature or destroy osteoregenerative proteins [1, 2]. Synthetic graft materials, such as those comprised of the calcium phosphate hydroxyapatite (HA), also lack osteoinductivity [3]. For these reasons, the development of methods that enable functionalization of allograft or synthetic biomaterials with osteoinductive molecules holds potential for creating graft substrates that have clinical efficacy comparable to that of autografted bone.
In this study we investigated a method for functionalization of graft materials including cadaveric-derived bone allograft, bulk HA, and HA-containing composite tissue engineering scaffolds. The strategy utilized is based on the mechanism by which native bone-binding proteins associate with the biologic HA present in bone mineral. Bone binding proteins such as bone sialoprotein and osteocalcin contain poly-acidic amino acid domains consisting of contiguous aspartate (D) or glutamate (E) residues that interact with the calcium within HA [4–6]. Prior studies from our group and others have shown that polyaspartate or polyglutamate domains can be used to anchor multiple bioactive peptides onto HA including the integrin-binding peptide, RGD [7–9], the proteoglycan-binding peptides, FHRRIKA and KRSR [10], and an osteoinductive collagen-derived peptide, DGEA [11]. For example, the addition of a heptaglutamate domain (E7) to the DGEA peptide markedly increased the amount of peptide loaded onto synthetic HA [11] as well as cortical bone allograft [12], and E7-DGEA peptides were retained on these substrates for at least 2 months in vivo. Improved coupling of DGEA to HA via the E7 domain was shown to have a significant biologic effect, in that E7-DGEA stimulated greater osteoblastic differentiation of mesenchymal stem cells (MSCs) and more robust in vivo bone formation than unmodified DGEA [11].
While osteoinductive peptides are being widely investigated as a tool to enhance graft integration, most of the peptides employed have a limited number of amino acids, encoding a finite amount of biologic information. The goal of the current study was to adapt the polyglutamate approach to achieve anchoring of other types of biomodifiers, specifically protein nanocage structures derived from the P22 bacteriophage. The benefit associated with nanocages is that these structures can be loaded with a variety of cargo including full-length proteins [13], imaging agents [14], and small therapeutic molecules [15]. The protein shell of bacteriophage P22 represents an attractive nano-scale drug delivery system due to the large cargo capacity, resistance to proteolytic cleavage, and stability under extreme pH and temperature [16–19]. Co-expression of the P22 scaffolding and coat proteins in a pET vector/BL21 (DE3) expression system results in the assembly of 60 nm diameter P22 virus like particles made up of 420 identical subunits of coat protein surrounding approximately 300 molecules of scaffolding protein (FIG 1) [20]. Fusion of cargo molecules such as green fluorescent protein (GFP) to the scaffolding protein results in their incorporation into the closed shell (FIG 1) [13]. An insertion tolerant, externally exposed loop on the coat protein has been identified [17, 18]. In the assembled protein shell, as a result of local pentameric and hexameric symmetry, the loops are clustered into twelve patches each containing five loops and sixty patches each containing six loops. To engineer nanocages with HA-selective binding sites, we used site directed mutagenesis to insert a diglutamate (E2) sequence into the loop region. As a result of local clustering, each P22 protein shell presented seventy-two high density negative charge patches to the exterior of each shell. These E2-P22 capsids were tested for binding to allograft and synthetic HA, with the expectation that the E2 modification would increase the binding of the nanocages to HA, providing a mechanism for enhanced delivery of cargo to sites of bone regeneration.
FIG 1. P22 Capsid Expression System.
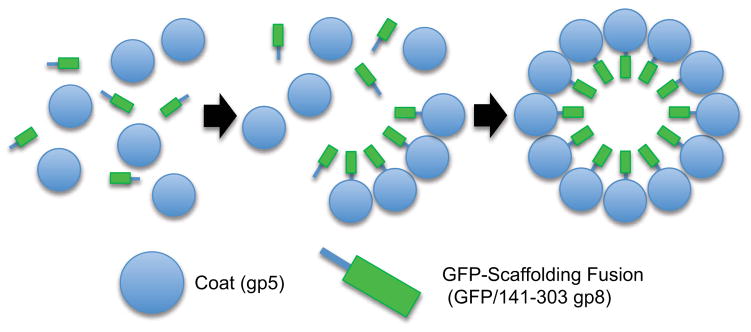
Co-expression of P22 coat (gp5) and GFP-scaffolding fusion (GFP/141–303 gp8) in BL21(DE3) results in the assembly of 60 nm diameter VLPs. Assembly is mediated by the C-terminus of the scaffolding protein and results in the incorporation of GFP into the closed VLP shell at an approximate ratio of 420 coat subunits to 300 scaffolding subunits. As previously described, the GFP-scaffolding fusion remains incorporated in the closed shell and demonstrates a mechanism of packaging non-native proteins into the P22 VLPs [13].
MATERIALS AND METHODS
Capsid Expression System
Co-expression of wild-type P22 scaffolding and coat proteins using the P22 assembler plasmid generates P22 virus-like particles (VLPs) in a BL21 DE3 expression system (FIG 1). The P22 assembler plasmid has been previously manipulated to express a fusion of GFP and truncated P22 scaffolding (GFP/141–303 gp8) that fully encapsulates and retains the GFP molecule at the same packaging efficiency as wildtype P22 scaffolding (gp8) during self-assembly [13]. Site-directed mutagenesis was performed on the GFP expressing P22 assembler plasmid in order to replace codon 183 (T) of P22 coat protein with the desired coding sequences. Plasmids from successful colonies were purified and the presence of the intended mutagenesis was confirmed by sequencing.
Purification of Capsids
BL21 DE3 containing the P22 assembler plasmid was grown to ~0.6 OD, induced with 1mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and incubated for an additional 4 hours. Cells were then pelleted, lysed by french press, and the lysate debris cleared by centrifugation. Capsids were purified from the clarified lysate by pelleting through 20% (w/v) sucrose at 40K (Beckman type 42.1) for 2 hours followed by sedimentation through a 5–20% (w/v) sucrose gradient at 38K (Beckman SW41) for 30 minutes. Sucrose gradient fractions were isolated and analyzed by SDS-PAGE to identify the VLP fractions. Pooled VLP fractions were then characterized by electrospray ionization mass spectrometry (ESI-MS) and transmission electron microscopy (TEM) to confirm proper coat protein expression, capsid morphology, and packaged GFP/141–303 gp8. Molecular graphics were performed with the UCSF Chimera package. Chimera was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS 9P41GM103311).
Material substrates
HA powder (MP Biomedicals, Solon, Ohio) was measured (5–50mg) into eppendorf tubes or pressed into disks using a 15.875 mm die under 3000 psi. All disks were sintered at 1000°C and allowed to cool in the furnace at decreasing intervals before being stored under sterile conditions. Electrospun scaffolds ± nano-HA were fabricated as described previously [21]. Briefly, two types of scaffolds were produced by electrospinning: (1) 100% polycaprolactone (PCL) and (2) 80wt% PCL +20wt% HA (PCL/HA). 15kV voltages were applied using a high-voltage power supply (Gamma High Voltage Research, Ormond Beach, FL). A syringe pump was used to feed polymer solution into the needle at a feed rate of 2 mL/h and scaffolds were collected onto an aluminum collecting plate. The allograft used for these studies was commercial-grade, mineralized cortical particulate bone graft (OraGRAFT®), obtained from LifeNet Health (Virginia Beach, Virginia). For the assays of binding selectivity, polystyrene dishes or tissue culture dishes coated with poly-D-lysine were purchased from Corning (Corning, New York).
Capsid coating onto materials
GFP loaded P22 or E2-P22 nanocages were used at equimolar concentrations based on protein quantification values. Working concentrations of 0.1–1.0 μM were used for coating materials, and for every experiment, solutions were checked to ensure that wild-type P22 and E2-P22 had equivalent starting fluorescence values.
Imaging of nanocage binding to substrates
P22 or E2-P22 capsid solutions at 1μM were coated onto bulk HA disks, electrospun scaffolds (100%PCL and PCL/HA), and agarose for 2 hours, or particulate HA for 2 minutes. Following coating, substrates were washed three times for 1 minute with agitation to remove unbound capsids. Particulate HA, bulk HA disks, electrospun scaffolds (100%PCL and PCL/HA), and agarose were then fluorescently imaged using a Nikon dissecting scope to evaluate GFP signal, indicating relative capsid binding.
Time course for capsid binding
Sintered HA disks were coated with 0.1μM of P22 or E2-P22 capsids. Samples were taken from the coating solutions at 30 minutes, 2 hours, 6 hours, or 24 hours and fluorescence read using a Biorad VersaFluor fluorometer. These readings (indicating depletion of fluorescence) were subtracted from fluorescence of the original coating solution to calculate the percent of capsids bound to HA disks over a 24-hour time course.
Dose dependent binding of capsids to HA and allograft
Particulate HA and allograft bone chips were measured into 5, 10, 25, and 50 mg aliquots and coated with GFP-loaded P22 or E2-P22 for either 2 minutes (HA) or 2 hours (allograft). HA or allograft particles were then precipitated by centrifugation, and samples of the supernatant were analyzed for fluorescence (representing the unbound nanocages). Percent of capsids bound was calculated by subtracting the fluorescence values of the supernatants from the starting fluorescence of the coating solution.
Western blot for coat protein
HA or allograft particles (5–50mg) were coated with GFP-loaded P22 or E2-P22 as described above. After spinning the particles down from the solution, we removed the supernatants and denatured the capsids in sodium dodecyl sulfate (SDS) buffer (50 mm Tris, 2% SDS, 5% β-mercaptoethanol) at 95°C for 5 minutes. Proteins were resolved by SDS-PAGE and transferred to polyvinyldifluoride (PVDF) membranes. Membranes were blocked in 5% dried nonfat milk buffer (in Tris-buffered saline containing 0.01% Tween 20) for 1 hour at room temperature. The membranes were then subjected to a primary polyclonal antibody against the P22 capsid protein (generated in the Prevelige lab) followed by an HRP-conjugated secondary antibody (Amersham Life Sciences, Buckinghamshire, United Kingdom) diluted in blocking buffer. Capsid protein was detected using chemiluminescence reagents (Amersham Life Sciences, Buckinghamshire, United Kingdom).
Statistics
All experiments were performed with at least three independent runs, and each independent experiment was performed in triplicate. Data sets were graphed to show averages and standard error, and statistical significance was assessed using Student’s t-test parametric analysis. A confidence level of at least 95% (p < 0.05) was considered significant and denoted by (“*”). Additionally, (“#”) indicated a p<0.01 and (“❖”) indicated a p<0.001.
RESULTS
Generation of E2-modified P22 nano-cages The P22 coat protein A-domain (FIG 2A, right) is permissive of peptide primary sequence inserts without affecting folding of the monomeric protein. Many inserted sequences have shown a minimal effect on the assembly or the morphology of the resulting virus like particles (VLPs) when compared to the wild-type sequence (data not shown). We hypothesized that the region would also be permissive of a 7-mer glutamate insert (E7: GGG(E)7GGG), resulting in P22 capsids that display the E7 peptide on the surface. Although the resulting protein was expressed and properly folded, as assayed by circular dichroism, we were unable to detect any fully assembled particles (data not shown). We speculate that this is because the local symmetry of 5-fold and 6-fold capsomers caused each E7 A-domain loop to localize near neighboring E7 A-domain loops in the quaternary structure, and the resulting charge density of 35 glutamates per pentamer or 42 glutamates per hexamer destabilized the intersubunit coat-coat interactions required for assembly.
FIG 2. Engineering polyglutamate-modified P22 capsid.

Using the UCSF Chimera package molecular graphics were performed. (A) In a 3IYI surface (rendered to 8 angstrom resolution) an A-domain flexible loop at residues 180–185 is highlighted in red to indicate the overall distribution of inserted sequences in the fully assembled capsid. The P22 virus-like particle forms a T=7 icosahedral structure of 60nm in diameter consisting of 12 pentameric capsomers and 60 hexameric capsomers. (B) Atomic surface rendering of the 3IYI hexameric capsomer structural model with all acidic residues indicated in red. The unaltered, P22 capsomer is shown on the left and a presumed model with the E2 sequence (GGGEEGGG) is shown on the right. (C) Negative-stain TEM images of P22 assembler products indicate that both P22 and E2-P22 assembler plasmids produce capsid structures with similar morphology.
As an alternative approach, we engineered E2 sequences (GGG(E)2GGG) into each coat protein, using the close proximity and localization of A domains during assembly to our advantage. We hypothesized this would decrease the localized charge-charge repulsion of the A-domain loops during assembly, while simultaneously achieving a high enough charge density (E10 for each pentamer and E12 for each hexamer) in each capsomer to facilitate enhanced loading of P22 particles to HA (FIG 2B). Engineering the E2 sequence into the coat protein in the P22 assembler plasmid encoding a GFP-scaffolding fusion protein (GFP/141–303 gp8) resulted in the assembly of particles similar in morphology to unmodified P22 capsids (FIG 2C). GFP was used as an encapsulated reporter molecule to facilitate binding assays, and under the conditions used in this study, the small nanocage pore size did not allow GFP release.
E2-P22 binding to synthetic HA materials
To assess whether insertion of E2 into the coat protein facilitated greater binding of P22 particles to HA materials, we coated particulate HA and ceramic HA disks with equimolar solutions of GFP-loaded wild-type P22 or E2-P22 and used fluorescent imaging to visualize bound capsids (FIG 3A). On both particulate HA and HA disks, E2-P22 cages were present at a greater density than unmodified P22, as shown by greater fluorescent signal (FIG 3A).
FIG 3. Enhanced loading of P22 capsids facilitated by E2 modification.

(A) Particulate HA or sintered HA disks were coated with 1μM GFP-loaded P22 or E2-P22. Following a brief wash to remove unbound capsids, substrates were imaged using fluorescent microscopy to qualitatively evaluate capsid binding. (B) Sintered HA disks were coated for up to 24 hours with 0.1μM concentrations of GFP-loaded P22 or E2-P22. Time course for capsid binding from solution was evaluated by measuring depletion of fluorescence from the coating solution. Residual solution fluorescence was subtracted from the starting fluorescence to calculate the amount of capsids bound. (C) 5–50mg of particulate HA were incubated with 0.1μM solutions of P22 or E2-P22 (loaded with GFP). After centrifugation to precipitate the HA particles and bound nanocages, supernatants were monitored for fluorescence in order to determine the amount of nanocages bound. (D) 5–50mg of particulate HA were incubated with 0.1μM of P22 or E2-P22, and after centrifugation, the supernatants (containing unbound nanocages) were immunoblotted for the P22 coat protein. Blots were subjected to densitometry to quantify the relative amount of unbound nanocages.
To quantitatively measure the effect of the E2-modification on the rate of capsid binding, the coating solution was monitored for depletion of fluorescence over 24 hours (with depletion from solution correlating to binding of the GFP-containing particles to HA substrates). These studies showed that the E2 domains directed greater loading of E2-P22 onto HA, when compared with wild-type P22, at all time points (FIG 3B). Additionally, we evaluated the binding of wild-type P22 or E2-P22 to increasing quantities of particulate HA (5–50 mg). We found that as the quantity of HA increased, binding of both P22 and E2-P22 increased; however, at all quantities E2-P22 was bound at significantly greater amounts than unmodified P22 (FIG 3C).
As an independent approach to assess the HA-binding capacity of E2-P22, we performed immunoblotting experiments to track the amount of nanocages remaining in solution following incubation with HA. Wild-type P22 and E2-P22 containing solutions were mixed with HA particles, and at the end of the incubation, samples were centrifuged to precipitate the HA (with bound nanocages) and the supernatants were immunoblotted for the P22 coat protein (FIG 3D). Similar to studies of solution fluorescence, we found that both P22 and E2-P22 were depleted from solution with increasing amounts of HA, however E2-P22 was depleted from the coating solution at greater quantities than P22, indicating better binding of E2-P22 to HA. These results were confirmed by quantifying relative density of protein bands using image analysis software (Image J) (FIG 3D).
Binding of E2-P22 to allograft bone
Having established greater binding of E2-P22 to synthetic HA, we next evaluated the potential of the E2 modification to enhance P22 binding to allograft bone. Equimolar concentrations of P22 and E2-P22 were incubated with increasing quantities of allograft bone particles (5–50mg), and binding was quantified by measuring depletion of fluorescence from the coating solution. Similar to studies of synthetic HA, greater binding of E2-P22 compared with wild-type P22 was observed for all concentrations of allograft bone tested (FIG 4A). Additionally, we monitored P22 and E2-P22 binding to allograft by immunoblotting the coating solutions for the amount of residual P22 coat protein (representing unbound viral particles) following precipitation of the particulate allograft. In line with fluorescence studies, we observed that as the quantity of allograft increased, both wild-type P22 and E2-P22 were depleted from solution; however, E2-P22 was depleted in greater amounts than wild-type P22, reflecting enhanced binding (FIG 4B).
FIG 4. E2 modification enhanced loading of P22 onto allograft bone.

5–50mg of particulate allograft bone were incubated with 0.1μM P22 or E2-P22 for 2 hours. Following centrifugation to precipitate the bone particles with bound nanocages, supernatants were evaluated by (A) measuring depletion of fluorescence from solution and (B) immunoblotting for depletion of the P22 coat protein.
Selectivity of E2-directed binding
To further assess whether modification of viral capsids with polyglutamate domains affects capsid selectivity for substrates, we evaluated P22 binding to various test surfaces. Wild-type P22 or E2-P22 particles were coated onto polystyrene dishes, which present a neutral, relatively hydrophobic, surface, or alternatively, onto standard tissue culture dishes coated with the positively-charged amino acid, poly-D-lysine. Low, but equivalent amounts of the wild-type P22 and E2-P22 particles were found to associate with polystyrene (FIG 5A). In contrast, significantly greater binding of E2-P22 compared with wild-type P22 was observed on the poly-D-lysine surfaces (FIG 5A), suggesting that the E2 domain functions through an ionic mechanism to alter the binding affinity of P22 for substrates. Additionally, we monitored wild-type P22 and E2-P22 binding to either agarose particles, or to two types of electrospun tissue engineering scaffolds: (i) 100% polycaprolactone (PCL) or (ii) polycaprolactone containing HA nanoparticles (80%PCL/20%HA “PCL/HA”). Wild-type P22 or E2-P22 particles were coated onto these substrates for 2 hours and imaged. As shown in FIG 5B, agarose coated with P22 or E2-P22 exhibited minimal fluorescence, indicating poor binding of both capsids (as compared with the positive control: HA particles coated with E2-P22). Similarly, very low amounts of wild-type P22 and E2-P22 were bound to electrospun scaffolds composed of 100% PCL (FIG 5C). However, when nanoparticles of HA were incorporated into the electrospun PCL scaffolds (PCL/HA), E2-P22 exhibited strong binding to the scaffold surface (FIG 5D). Thus, the binding of E2-P22 appears to be selective for HA-containing materials, with negligible binding observed on agarose, polystyrene, and PCL. Taken together, these results provide proof of concept that the polyglutamate/HA interaction offers an effective mechanism for: (1) anchoring distinct particles onto multiple HA graft materials, and (2) altering the binding selectivity of viral capsids.
FIG 5. E2-enhanced loading is selective for HA-containing materials.

(A) Polystyrene or poly-D-lysine-coated surfaces were incubated with equimolar concentrations of GFP loaded P22 or E2-P22. After two hours, fluorescence of the coating solution was monitored, and these values (representing unbound capsids) were subtracted from the starting solution values (total capsids) to obtain the percent capsid binding. (B) P22 or E2-P22 (loaded with GFP) was coated onto agarose particles or HA particles (positive control) and evaluated by fluorescence microscopy. (C) P22 or E2-P22 was coated onto 100% PCL electrospun scaffolds or HA particles. (D) E2-P22 was coated onto electrospun scaffolds composed of 100% PCL, or PCL scaffolds containing HA nanoparticles (E2-P22 binding to HA particles served as a positive control).
DISCUSSION
Synthetic HA biomaterials and allograft bone are commonly used in the medical and dental fields as substitutes for autograft bone, however these materials lack osteoinductivity [1–3]. To address this deficiency, various recombinant proteins (e.g. Bone Morphogenic Proteins) or bioactive peptides have been passively adsorbed onto the surface of these substrates [22–25]; however passive adsorption provides little control over the dose or kinetics of protein/peptide delivery [24]. Alternatively, some investigators have evaluated encapsulating molecules within cage or vesicular structures to enhance bone regeneration (reviewed in [26]). Encapsulation of bioactive cargo may be beneficial because it confers protection of cargo from proteolytic degradation, and also protects the body from non-targeted effects resulting from broad dissemination of osteogenic molecules [27, 28]. Additionally, the use of vesicles or nanocages provides a mechanism for greater control of drug release, and in particular, may facilitate sustained drug delivery [16]. A multitude of vesicular structures including micelles, liposomes, and poly(lactic-co-glycolic acid) (PLGA) nanospheres have been used to deliver bioactive molecules from synthetic HA or native bone graft carriers (reviewed in [27]). Some of these vehicles have been chemically modified to enhance affinity for HA or bone surfaces [29–33]. Wang et. al. used bisphosphonates (BP) to decorate liposomes and micelles, and showed that BP on the surface of these particles increased particle binding to synthetic HA and a composite HA/collagen matrix [29]. As well, BP-coupled PLGA nanoparticles loaded with selected chemotherapeutics exhibited accumulation in bone following intravenous injection, resulting in higher local concentrations of drug [34]. Targeting to bone has also been observed when BP domains were coupled directly to proteins or other molecules such as estradiol, prostaglandin, and the cancer therapeutic, methotrexate [35–38]. These collective results support the fundamental concept of using HA-binding domains to enhance both the systemic delivery of therapeutics to bone and the coupling of osteoinductive molecules onto the surface of graft materials.
The use of BP as a mechanism to target bone is efficient and warranted in some applications where its pharmacological effect could be beneficial, however one concern is the potential for unintended effects introduced by the use of a pharmacologically active agent as a bone targeting moiety. In some instances, BPs can cause apoptosis, inhibit cell migration, and reduce angiogenesis, potentially negatively affecting bone healing and regeneration [39]. Given these issues, we have developed a second modality to deliver cargo on HA. Specifically, we investigated the use of polyglutamate domains to anchor cargo-loaded P22 nanocages onto HA. Although polyacidic amino acid sequences have been employed to couple synthetic peptides on HA [7–12, 40], the current study is the first to exploit these domains to anchor nanocages onto HA. To engineer P22 nanocages with polyglutamate, site-directed mutagenesis was used to insert diglutamate (E2) domains into the P22 capsid protein. It was found that E2-modified P22 capsids exhibited significantly better binding to HA particles, sintered HA disks, and allograft bone. In contrast, negligible binding of E2-P22 was observed to agarose, polystyrene, and 100% PCL electrospun scaffolds, suggesting that the E2 domains conferred selectivity for HA. One anticipates that insertional mutagenesis could be employed to modify many other types of protein cages, including other VLPs, providing a relatively straightforward alternative to some of the complex chemical coupling technologies required for modifying particles with BP or other bone-binding domains.
Currently there is substantial interest in developing nanocage technologies. As examples, synthetic inorganic nanocages, liposomes, and VLPs, have been employed for in vivo imaging and drug delivery [41, 42]. VLPs in particular have emerged as versatile architectures for the development of functional hybrid nanostructures. VLPs self-assemble into capsid structures of consistent size and shape, establishing a limit to the quantity of cargo that can be contained, and inherently providing a means for dose control that is absent from nanocages that are either more difficult to manufacture or do not produce homologous products. VLPs derived from P22 offer many beneficial features as a carrier system. They are physically stable and can be readily produced in quantity. As demonstrated here, the external surface can be modified to promote selective binding to target materials and tissues. Additionally, a unique characteristic of P22 compared with other VLP systems is the ability to modulate the overall volume and porosity of the capsid structure, providing tunable control of the degree and kinetics of cargo release [43, 44]. Another advantage is that a variety of approaches to incorporate cargo into P22 have been developed, which enables loading of multiple types of molecules. For example, our group has shown that incorporation of cargo can be accomplished by covalent modification of the interior surface to present nickel for binding 6X-His tagged proteins [45], or to initiate controlled internal polymer formation using atom transfer radical polymerization [46]. The latter method has been adapted to use the encapsulated polymer as a scaffold for the attachment of small functional molecules, such as MRI contrast agents [46]. The approach utilized in this investigation, fusion of large proteins to the scaffolding, results in incorporation and entrapment of the cargo. However the use of mutant scaffolding proteins with weaker capsid binding affinity [47] should enhance the release of small fusion proteins. Alternatively, a larger fusion protein could be released by pre-treatment of the particles with heat. This treatment results in the formation of stable shells with pores sufficiently large to allow protease to access and cleave the trapped protein, thus providing a mechanism for controlled release of small proteins and peptides [13]. The considerable adaptability of P22 nanocages with regard to both the engineering of the capsid sequence and the capacity to carry diverse cargo molecules highlights the potential utility of this vehicle for many types of clinical applications.
CONCLUSION
P22-derived nanocage structures were engineered with polyglutamate domains to facilitate enhanced binding to synthetic HA and allograft bone. Glutamate residues were incorporated into a specific region of the P22 coat protein and were found to cluster following capsid assembly, resulting in focal domains with concentrated negative charge on the surface of the capsid. The addition of these distinct high-density glutamate regions enabled loading of increased numbers of nanocages onto the surface of HA-containing substrates, suggesting that polyglutamate-modified nanocages have enhanced capacity for local delivery of osteoinductive molecules. Future studies will be aimed at engineering the polyglutamate-modified particles with bone inductive cargo such as bone morphogenic proteins. However, clinical applicability of the polyglutamate-modified P22 nanocages extends beyond the delivery of therapeutic proteins, because in addition to proteins, the P22 vehicle has been successfully engineered with cargo such as metal ions for imaging, and nonproteinaceous small molecules. Finally, it is anticipated that the fundamental polyglutamate coupling strategy can be adapted to endow many other types of particles or cargo-loaded vehicles with selectivity for HA.
Acknowledgments
This research was supported by NIH grant R01AR51539 (SLB), as well as grants from the Osseointegration Foundation (SLB), and the UAB BioMatrix Engineering and Regenerative Medicine Center (PEP). BKC was supported by NIH/NIDCR predoctoral fellowship 1F31DE021613 (BKC). We are grateful for technical support provided by Kuni J. Scissum.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Giannoudis PV, Dinopoulos H, Tsiridis E. Bone substitutes: An update. Injury. 2005;36 (Suppl 3):S20–7. doi: 10.1016/j.injury.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 2.Bostrom MP, Seigerman DA. The clinical use of allografts, demineralized bone matrices, synthetic bone graft substitutes and osteoinductive growth factors: A survey study. HSS J. 2005;1:9–18. doi: 10.1007/s11420-005-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nandi SK, Roy S, Mukherjee P, Kundu B, De DK, Basu D. Orthopaedic applications of bone graft & graft substitutes: A review. Indian J Med Res. 2010;132:15–30. [PubMed] [Google Scholar]
- 4.Ganss B, Kim RH, Sodek J. Bone sialoprotein. Crit Rev Oral Biol Med. 1999;10:79–98. doi: 10.1177/10454411990100010401. [DOI] [PubMed] [Google Scholar]
- 5.Hoang QQ, Sicheri F, Howard AJ, Yang DS. Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature. 2003;425:977–80. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
- 6.Stayton PS, Drobny GP, Shaw WJ, Long JR, Gilbert M. Molecular recognition at the protein-hydroxyapatite interface. Crit Rev Oral Biol Med. 2003;14:370–6. doi: 10.1177/154411130301400507. [DOI] [PubMed] [Google Scholar]
- 7.Itoh D, Yoneda S, Kuroda S, Kondo H, Umezawa A, Ohya K, et al. Enhancement of osteogenesis on hydroxyapatite surface coated with synthetic peptide (EEEEEEEPRGDT) in vitro. J Biomed Mater Res. 2002;62:292–8. doi: 10.1002/jbm.10338. [DOI] [PubMed] [Google Scholar]
- 8.Fujisawa R, Mizuno M, Nodasaka Y, Kuboki Y. Attachment of osteoblastic cells to hydroxyapatite crystals by a synthetic peptide (glu7-pro-arg-gly-asp-thr) containing two functional sequences of bone sialoprotein. Matrix Biol. 1997;16:21–8. doi: 10.1016/s0945-053x(97)90113-x. [DOI] [PubMed] [Google Scholar]
- 9.Sawyer AA, Weeks DM, Kelpke SS, McCracken MS, Bellis SL. The effect of the addition of a polyglutamate motif to RGD on peptide tethering to hydroxyapatite and the promotion of mesenchymal stem cell adhesion. Biomaterials. 2005;26:7046–56. doi: 10.1016/j.biomaterials.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Sawyer AA, Hennessy KM, Bellis SL. The effect of adsorbed serum proteins, RGD and proteoglycan-binding peptides on the adhesion of mesenchymal stem cells to hydroxyapatite. Biomaterials. 2007;28:383–92. doi: 10.1016/j.biomaterials.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 11.Culpepper BK, Phipps MC, Bonvallet PP, Bellis SL. Enhancement of peptide coupling to hydroxyapatite and implant osseointegration through collagen mimetic peptide modified with a polyglutamatetamate domain. Biomaterials. 2010;31:9586–94. doi: 10.1016/j.biomaterials.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Culpepper BK, Bonvallet PP, Reddy MS, Ponnazhagan S, Bellis SL. Polyglutamate directed coupling of bioactive peptides for the delivery of osteoinductive signals on allograft bone. Biomaterials. 2013;34:1506–13. doi: 10.1016/j.biomaterials.2012.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neil A, Reichhardt C, Johnson B, Prevelige PE, Douglas T. Genetically programmed in vivo packaging of protein cargo and its controlled release from bacteriophage p22. Angew Chem Int Ed Engl. 2011;50:7425–8. doi: 10.1002/anie.201102036. [DOI] [PubMed] [Google Scholar]
- 14.Qazi S, Liepold LO, Abedin MJ, Johnson B, Prevelige P, Frank JA, et al. P22 viral capsids as nanocomposite high-relaxivity MRI contrast agents. Mol Pharm. 2012 Jul 16; doi: 10.1021/mp300208g. ePub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV-DOX delivers. Mol Pharm. 2012 Aug 6; doi: 10.1021/mp3002057. ePub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang S, Uchida M, O’Neil A, Li R, Prevelige PE, Douglas T. Implementation of p22 viral capsids as nanoplatforms. Biomacromolecules. 2010;11:2804–9. doi: 10.1021/bm100877q. [DOI] [PubMed] [Google Scholar]
- 17.Kang S, Lander GC, Johnson JE, Prevelige PE. Development of bacteriophage p22 as a platform for molecular display: Genetic and chemical modifications of the procapsid exterior surface. Chembiochem. 2008;9:514–8. doi: 10.1002/cbic.200700555. [DOI] [PubMed] [Google Scholar]
- 18.Lanman J, Tuma R, Prevelige PE., Jr Identification and characterization of the domain structure of bacteriophage p22 coat protein. Biochemistry. 1999;38:14614–23. doi: 10.1021/bi9915420. [DOI] [PubMed] [Google Scholar]
- 19.Galisteo ML, Gordon CL, King J. Stability of wild-type and temperature-sensitive protein subunits of the phage p22 capsid. J Biol Chem. 1995;270:16595–601. doi: 10.1074/jbc.270.28.16595. [DOI] [PubMed] [Google Scholar]
- 20.Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, et al. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci U S A. 2011;108:1355–60. doi: 10.1073/pnas.1015739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phipps MC, Clem WC, Catledge SA, Xu Y, Hennessy KM, Thomas V, et al. Mesenchymal stem cell responses to bone-mimetic electrospun matrices composed of polycaprolactone, collagen I and nanoparticulate hydroxyapatite. PLoS One. 2011;6:e16813. doi: 10.1371/journal.pone.0016813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ono I, Gunji H, Kaneko F, Saito T, Kuboki Y. Efficacy of hydroxyapatite ceramic as a carrier for recombinant human bone morphogenetic protein. J Craniofac Surg. 1995;6:238–44. doi: 10.1097/00001665-199505000-00011. [DOI] [PubMed] [Google Scholar]
- 23.Ono I, Gunji H, Suda K, Kaneko F, Murata M, Saito T, et al. Bone induction of hydroxyapatite combined with bone morphogenetic protein and covered with periosteum. Plast Reconstr Surg. 1995;95:1265–72. doi: 10.1097/00006534-199506000-00019. [DOI] [PubMed] [Google Scholar]
- 24.LeBaron RG, Athanasiou KA. Extracellular matrix cell adhesion peptides: Functional applications in orthopedic materials. Tissue Eng. 2000;6:85–103. doi: 10.1089/107632700320720. [DOI] [PubMed] [Google Scholar]
- 25.Yasuda H, Yano K, Wakitani S, Matsumoto T, Nakamura H, Takaoka K. Repair of critical long bone defects using frozen bone allografts coated with an rhBMP-2-retaining paste. J Orthop Sci. 2012;17:299–307. doi: 10.1007/s00776-012-0196-x. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Leeuwenburgh SC, Li Y, Jansen JA. The use of micro- and nanospheres as functional components for bone tissue regeneration. Tissue Eng Part B Rev. 2012;18:24–39. doi: 10.1089/ten.teb.2011.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Low SA, Kopecek J. Targeting polymer therapeutics to bone. Adv Drug Deliv Rev. 2012;64:1189–204. doi: 10.1016/j.addr.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Miller SC, Kopeckova P, Kopecek J. Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev. 2005;57:1049–76. doi: 10.1016/j.addr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 29.Wang G, Mostafa NZ, Incani V, Kucharski C, Uludag H. Bisphosphonate-decorated lipid nanoparticles designed as drug carriers for bone diseases. J Biomed Mater Res A. 2012;100:684–93. doi: 10.1002/jbm.a.34002. [DOI] [PubMed] [Google Scholar]
- 30.Hengst V, Oussoren C, Kissel T, Storm G. Bone targeting potential of bisphosphonate-targeted liposomes. Preparation, characterization and hydroxyapatite binding in vitro. Int J Pharm. 2007;331:224–7. doi: 10.1016/j.ijpharm.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 31.Cenni E, Granchi D, Avnet S, Fotia C, Salerno M, Micieli D, et al. Biocompatibility of poly(d,l-lactide-co-glycolide) nanoparticles conjugated with alendronate. Biomaterials. 2008;29:1400–11. doi: 10.1016/j.biomaterials.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 32.Salerno M, Cenni E, Fotia C, Avnet S, Granchi D, Castelli F, et al. Bone-targeted doxorubicin-loaded nanoparticles as a tool for the treatment of skeletal metastases. Curr Cancer Drug Targets. 2010;10:649–59. doi: 10.2174/156800910793605767. [DOI] [PubMed] [Google Scholar]
- 33.Wang G, Kucharski C, Lin X, Uludag H. Bisphosphonate-coated BSA nanoparticles lack bone targeting after systemic administration. J Drug Target. 2010;18:611–26. doi: 10.3109/10611861003622560. [DOI] [PubMed] [Google Scholar]
- 34.Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012;33:7164–73. doi: 10.1016/j.biomaterials.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 35.Gil L, Han Y, Opas EE, Rodan GA, Ruel R, Seedor JG, et al. Prostaglandin e2-bisphosphonate conjugates: Potential agents for treatment of osteoporosis. Bioorg Med Chem. 1999;7:901–19. doi: 10.1016/s0968-0896(99)00045-0. [DOI] [PubMed] [Google Scholar]
- 36.Uludag H, Kousinioris N, Gao T, Kantoci D. Bisphosphonate conjugation to proteins as a means to impart bone affinity. Biotechnol Prog. 2000;16:258–67. doi: 10.1021/bp990154m. [DOI] [PubMed] [Google Scholar]
- 37.Hosain F, Spencer RP, Couthon HM, Sturtz GL. Targeted delivery of antineoplastic agent to bone: Biodistribution studies of technetium-99m-labeled gem-bisphosphonate conjugate of methotrexate. J Nucl Med. 1996;37:105–7. [PubMed] [Google Scholar]
- 38.Morioka M, Kamizono A, Takikawa H, Mori A, Ueno H, Kadowaki S, et al. Design, synthesis, and biological evaluation of novel estradiol-bisphosphonate conjugates as bone-specific estrogens. Bioorg Med Chem. 2010;18:1143–8. doi: 10.1016/j.bmc.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 39.Ziebart T, Pabst A, Klein MO, Kammerer P, Gauss L, Brullmann D, et al. Bisphosphonates: Restrictions for vasculogenesis and angiogenesis: Inhibition of cell function of endothelial progenitor cells and mature endothelial cells in vitro. Clin Oral Investig. 2011;15:105–11. doi: 10.1007/s00784-009-0365-2. [DOI] [PubMed] [Google Scholar]
- 40.Kasugai S, Fujisawa R, Waki Y, Miyamoto K, Ohya K. Selective drug delivery system to bone: Small peptide (asp)6 conjugation. J Bone Miner Res. 2000;15:936–43. doi: 10.1359/jbmr.2000.15.5.936. [DOI] [PubMed] [Google Scholar]
- 41.Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–30. doi: 10.1146/annurev-bioeng-071910-124709. [DOI] [PubMed] [Google Scholar]
- 42.Koudelka KJ, Manchester M. Chemically modified viruses: Principles and applications. Curr Opin Chem Biol. 2010;14:810–7. doi: 10.1016/j.cbpa.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Teschke CM, McGough A, Thuman-Commike PA. Penton release from p22 heat-expanded capsids suggests importance of stabilizing penton-hexon interactions during capsid maturation. Biophys J. 2003;84:2585–92. doi: 10.1016/S0006-3495(03)75063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang W, Li Z, Zhang Z, Baker ML, Prevelige PE, Jr, Chiu W. Coat protein fold and maturation transition of bacteriophage p22 seen at subnanometer resolutions. Nat Struct Biol. 2003;10:131–5. doi: 10.1038/nsb891. [DOI] [PubMed] [Google Scholar]
- 45.Uchida M, Morris DS, Kang S, Jolley CC, Lucon J, Liepold LO, et al. Site-directed coordination chemistry with p22 virus-like particles. Langmuir. 2012;28:1998–2006. doi: 10.1021/la203866c. [DOI] [PubMed] [Google Scholar]
- 46.Lucon J, Qazi S, Uchida M, Bedwell GJ, Lafrance B, Prevelige PE, Jr, et al. Use of the interior cavity of the p22 capsid for site-specific initiation of atom-transfer radical polymerization with high-density cargo loading. Nat Chem. 2012;4:781–8. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cortines JR, Weigele PR, Gilcrease EB, Casjens SR, Teschke CM. Decoding bacteriophage p22 assembly: Identification of two charged residues in scaffolding protein responsible for coat protein interaction. Virology. 2011;421:1–11. doi: 10.1016/j.virol.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]