

Abstract
Cellular senescence both protects multicellular organisms from cancer and contributes to their aging1. The preeminent tumor suppressor p53 plays an important role in the induction and maintenance of senescence, but how p53 carries out this function remains poorly understood1–3. Additionally, while increasing evidence supports the notion that metabolic changes underlie many cell fate decisions and p53-mediated tumor suppression, few connections between metabolic enzymes and senescence have been established. Here we describe a novel mechanism by which p53 links the functions. We show that p53 represses the expression of the tricarboxylic acid cycle (TCA cycle)-associated malic enzyme 1 (ME1) and malic enzyme 2 (ME2). Both MEs are important for NADPH production, lipogenesis, and glutamine metabolism, with ME2 having a more profound effect. Through inhibiting MEs, p53 regulates cell metabolism and proliferation. Down-regulation of ME1 and ME2 reciprocally activates p53 through distinct Mdm2 and AMPK-mediated mechanisms in a feed-forward manner, bolstering this pathway and enhancing p53 activation. Down-regulation of ME1 and ME2 also modulates the outcome of p53 activation leading to strong induction of senescence, but not apoptosis, while enforced expression of either ME suppresses senescence. Our findings define physiological functions of MEs, demonstrate a positive feedback mechanism that sustains p53 activation, and reveal a connection between metabolism and senescence mediated by p53.
We previously found that p53 inhibits the important NADPH producer glucose-6-phosphate dehydrogenase (G6PD)4. As this did not fully explain the effect of p53 on NADPH, we investigated whether p53 controls expression of MEs, which catalyze the oxidative decarboxylation of malate to generate pyruvate and either NADPH or NADH5,6 (Supplementary Fig. 1). In mammalian cells three ME isoforms have been identified: a cytosolic NADP+-dependent isoform (ME1), a mitochondrial NAD(P)+-dependent isoform (ME2), and a mitochondrial NADP+-dependent isoform (ME3), with ME1 and ME2 are the major isoforms (Supplementary Fig. 2a)7. By recycling the TCA cycle intermediate malate into the common TCA cycle carbon source pyruvate, MEs may have a regulatory role in matching TCA flux to cellular demand for energy, reducing equivalents, and biosynthetic precursors (Supplementary Fig. 1).
We knocked down p53 in human osteosarcoma U2OS cells and normal diploid fibroblast IMR90 cells using small hairpin (sh) RNA. This led to a significant increase in mRNA levels of ME1 and ME2 (Fig. 1a, b and Supplementary Fig. 2b), accompanied by elevated protein levels and total enzymatic activity of ME1 and ME2 (Fig. 1a, c and Supplementary Fig. 2c). Likewise, expression of ME1 and ME2 were substantially higher in p53 knockout (p53−/−) mouse embryonic fibroblasts (MEFs) compared to p53-wild-type (p53+/+) MEFs (Fig. 1d). The normally short-lived p53 protein is stabilized by DNA damage signals. Cells treated with the genotoxic agents etoposide (ETO) and doxorubicin (DOX) showed both time- and concentration-dependent reductions in the expression of ME1 and ME2 (Fig. 1e and Supplementary Fig. 2b, d–f). When p53 was knocked down, the expression of ME1 and ME2 no longer responded to DNA damage (Fig. 1e). These results demonstrate that the expression of ME1 and ME2 is controlled by p53 both at basal levels and when p53 is stabilized by DNA damage signals.
Figure 1. p53 represses the expression of MEs.
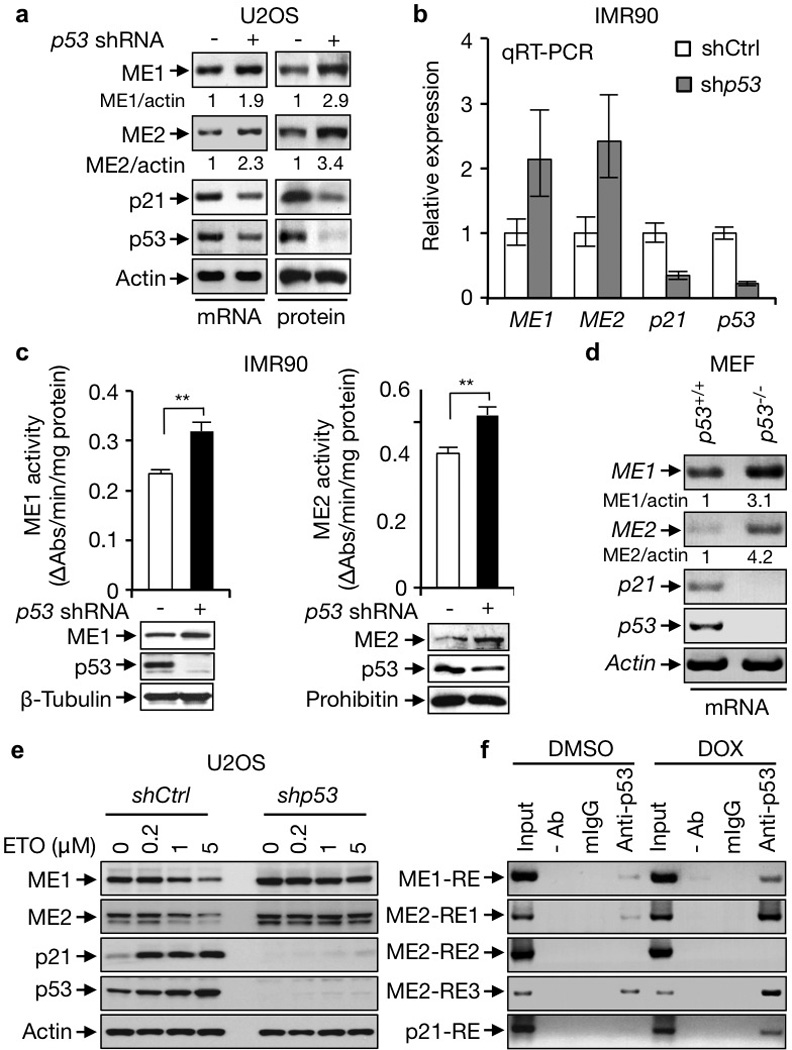
a, ME mRNA and protein expression in U2OS cells stably expressing p53 shRNA or control shRNA. Relative ME/actin ratios are given. b, c, mRNA expression (b), total activity, and protein levels (c) of MEs in p53-depleted and control IMR90 cells. Data shown are mean ± SD, n=3. d, ME expression in p53+/+ and p53−/− MEFs. e, p53-depleted and control U2OS cells were treated with increasing amounts of ETO and assayed for ME expression. f, p53+/+ HCT116 cells treated with or without DOX (1 µg/ml) were subjected to ChIP assay with anti-p53 (DO-1), a control mouse IgG, or no antibody (−). *, p<0.05; **, p<0.01; ***, p<0.001.
By analyzing the ME gene sequences, we identified a putative p53 response element (RE)8 in the first intron of the ME1 gene (ME1-RE) and three putative REs in the first intron of the ME2 gene (ME2-RE1 to ME2-RE3) (Supplementary Fig. 3a). Chromatin immunoprecipitation (ChIP) assays revealed that p53 bound to the genomic regions of the ME1-RE, ME2-RE1, and ME2-RE3, but not ME2-RE2. This binding was further enhanced when p53 was stabilized by treatment with DOX (Fig. 1f). p53 repressed the expression of a luciferase gene driven by the genomic fragments containing ME1-RE, ME2-RE1, or ME2-RE3, but not ME2-RE2 (Supplementary Fig. 3b). p53-mediated repression of certain target genes involves histone deacetylases (HDACs)9. Treatment with trichostatin A (TSA), an inhibitor of HDACs, abrogated p53-mediated repression of ME1 and ME2 genes (Supplementary Fig. 2g).
p53 deficiency also led to a strong increase in ME3 transcript (Supplementary Fig. 4a). A putative p53 RE is present in the first intron of the ME3 gene (ME3-RE) (Supplementary Fig. 4b). p53 bound to the genomic region of ME3-RE in cells (Supplementary Fig. 4c) and reduced the expression of a luciferase reporter driven by this RE (Supplementary Fig. 4d). Given the low abundance of ME3 expression in cell lines that have been tested (Supplementary Fig. 2a)7, we focused on ME1 and ME2 in subsequent analyses.
Although ME1 and ME2 have been extensively characterized in vitro, there is a paucity of information on their functions in cells. Silencing the expression of ME1 or particularly ME2 with small interfering RNA (siRNA) reduced cellular NADPH levels (Fig. 2a and Supplementary Fig. 5a). This effect was also observed when a separate set of ME siRNAs, as well as ME shRNAs, were used (Supplementary Fig. 5b, c). In contrast, forced expression of ME1 or especially ME2, or the addition of an ME substrate (dimethyl L-malate), elevated cellular NADPH levels (Fig. 2b, lanes 1–3; Supplementary Fig. 5d). To determine whether the effect of MEs is due to their enzymatic activity, we generated two ME1 mutations (ME1mut1 and ME1mut2) and three ME2 mutations (ME2mut1 to ME2mut3), each of which exhibited little or no enzymatic activity in vitro as well as in vivo (Supplementary Fig. 6). None of these mutants were able to increase cellular NADPH levels (Fig. 2b, lanes 4–10). Thus, both ME1 and ME2 are required for maintaining cellular NADPH levels through their enzymatic activity, with ME2 having a more profound effect. As previously observed4, knockdown of p53 led to a significant increase in NADPH levels. This increase was partially reversed through the silencing of ME1 and was near completely reversed through silencing of ME2 (Fig. 2a and Supplementary Fig. 5a, b). These results indicate that p53 regulates NADPH metabolism through the suppression of both MEs, particularly ME2.
Figure 2. ME1 and ME2 influence NADPH production, lipid production, and glutaminolysis.
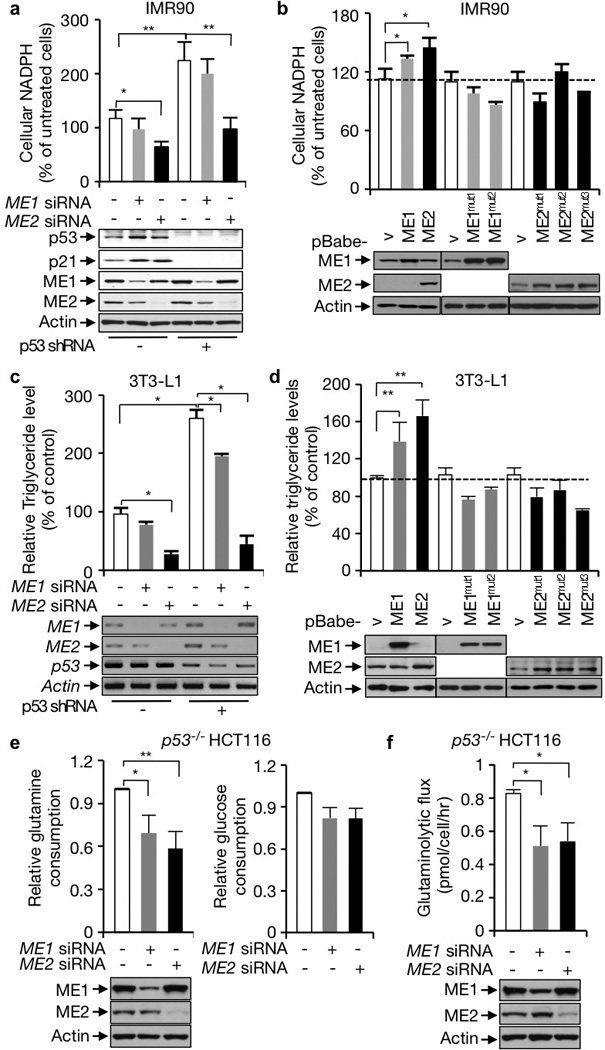
a, b, NADPH levels (means ± SD, n=3) in p53-depleted and control IMR90 cells transfected with control, ME1, or ME2 siRNA (a), or in IMR90 cells stably over-expressing wild-type MEs, mutant MEs, or vector control. Protein expression is shown below. c, d, Triglyceride contents (means ± SD, n=3) in p53-depleted and control 3T3-L1 cells transfected with control, ME1, or ME2 siRNA (c), or 3T3-L1 cells stably expressing wild-type MEs, mutant MEs, or vector control (d). mRNA expression is shown below. e, f, Effect of ME1 and ME2 knockdown in p53−/− HCT116 cells on glucose and glutamine consumption (means ± SD, n=3) (e), and glutaminolytic flux (f).
As NADPH provides reducing equivalents for reductive biosynthesis, we examined the role of MEs in lipid production. MEF cells and 3T3-L1 preadipocytes were cultured with a cocktail that stimulated their differentiation into adipocytes4,10. Triglycerides and total lipid levels in these cells declined when ME1 or particularly ME2 was depleted in these cells (Fig. 2c and Supplementary Fig. 7). In contrast, over-expression of ME1 or especially ME2, but none of the ME1 and ME2 mutants, increased lipid abundance (Fig. 2d). Concordant with previously published data4, we observed a marked increase in lipid levels in p53-deficient cells compared to p53-proficient cells. ME1 knockdown partially reversed this increase, while ME2 knockdown prevented it entirely correlating with its greater influence on cellular NADPH levels (Fig. 2c and Supplementary Fig. 7). These results suggest that the enhanced lipid accumulation in p53 deficient cells is dependent on the MEs, especially ME2.
Silencing of ME2, as well as ME1, did not significantly alter NADH levels or the NAD+/NADH ratio in IMR90 cells (Supplementary Fig. 8a, b), despite that ME2 is characterized as either NADP+- or NAD+-dependent5,6. NADH can be oxidized by the electron transport chain for ATP production. Silencing of each ME did not significantly alter the abundance of cellular ATP or ADP in IMR90 cells (Supplementary Fig. 8c). In U2OS cells silencing of ME1, but not ME2, reduced NADH levels and increased the NAD+/NADH ratio (Supplementary Fig. 8d, e). These results are consistent with a cell-type specific role of ME1 and a minimal role of ME2 in maintaining cellular NADH and ATP levels.
Next we investigated the role of ME1 and ME2 in the metabolism of glucose and glutamine. In p53−/− HCT116 cells silencing of either ME, especially ME2, strongly reduced glutamine consumption (Fig. 2e), while silencing of either ME had a moderate effect on glucose consumption. We extended this analysis by evaluating the rate of glutaminolysis. Depletion of either ME1 or ME2 noticeably slowed down glutaminolytic flux (Fig. 2f). These results suggest that both ME1 and ME2 have a major role in glutamine metabolism but a relative minor role in glucose metabolism.
p53 plays a critical role in the induction and maintenance of senescence1–3. We noticed that in IMR90 cells, a well-established senescence model, silencing of each ME by either siRNA or shRNA caused a profound increase in cells expressing senescence-associated β-galactosidase (SA-β-gal) and stopping growth (Fig. 3a, b and Supplementary Fig. 9a–f). The induction of senescence in ME-knockdown cells was also indicated by the dramatic accumulation the promyelocytic leukemia protein nuclear bodies (PML-NBs)11,12 (Fig. 3c and Supplementary Fig. 9g). Remarkably, even a moderate reduction (20–30%) in either ME1 or ME2 strongly elicited senescence (Supplementary Fig. 10a). ME loss-induced senescence also occurred in tumor cell lines (Supplementary Fig. 10b, c). In p53-deficient primary and tumor cell lines, senescence decreased markedly, and ME depletion lost its ability to induce this phenotype (Fig. 3b, c and Supplementary Figs. 9b, c, f, g, 10c). In contrast, ME depletion did not cause cell death (Supplementary Fig. 11a), and it induced the expression of p53-target genes implicated in senescence2,13 but not apoptosis (Supplementary Fig. 11b). These data suggest that down-regulation of ME induces senescence via p53.
Figure 3. A role for MEs in suppressing p53-mediated senescence.
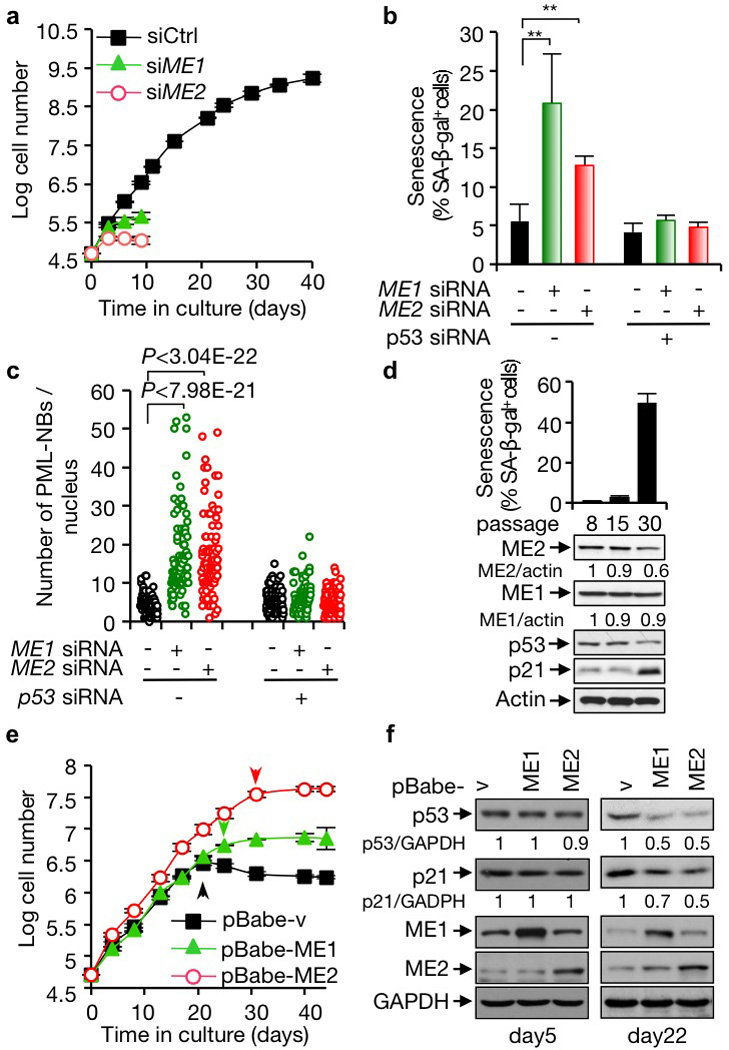
a, The replicative lifespan (means ± SD, n=3) of IMR90 cells transfected with control, ME1, or ME2 siRNA. b, c, IMR90 cells transfected with ME1, ME2, p53, or control siRNA as indicated. Percentages of SA-β-gal-positive cells (means ± SD, n=3) (b) and numbers of PML NBs (c) are shown. See Supplementary Figs. 9f, g for representative images. d, Percentages of SA-β-gal-positive cells (means ± SD, n=3) (top) and protein expression (bottom) in IMR90 cells at different passages. e, f, Replicative lifespan (mean ± SD, n=3) (e), and protein expression (f) of IMR90 cells with and without overexpression of ME1 or ME2. Arrows indicate the onset of senescence (e).
We next examined the role of MEs in replicative senescence of normal human cells, a p53-regulated process1,3. IMR90 cells were serially passaged in culture until a substantial number of them (~50%) entered senescence. The expression of ME1 remained at comparable levels at different passages, while the expression of ME2, which stayed unchanged initially, noticeably declined at the late stage (Fig. 3d). To test whether the decline in ME2 contributes to senescence in this setting, we evaluated the replicative capacity of IMR90 cells forced to express ME2. Compared with control cells, ME2-overexpressing cells could be cultured for extended passages with a greatly delayed onset of senescence (Fig. 3e and Supplementary Fig. 12a). As ME1 expression was maintained during replicative senescence, we were surprised to observe a delay in senescence when ME1 expression was forced (Fig. 3e and Supplementary Fig. 12a). In contrast, forced expression of any of the ME mutants did not delay senescence and instead moderately promoted senescence (Supplementary Fig. 12b–d), possibly through a dominant negative effect on the endogenous MEs. These results indicate that both MEs, and especially ME2, are capable of suppressing senescence and suggest that the decline in ME2 may contribute to replicative senescence.
To examine the effect of MEs on other scenarios of p53-regulated senescence, we found that culturing IMR90 cells in medium containing no glutamine resulted in p53-dependent senescence (Supplementary Fig. 13a). This senescence could be delayed by over-expression of either ME1 or ME2 (Supplementary Fig. 13b), or the addition of the ME substrate malate (Supplementary Fig. 13c). In contrast, exogenous ME expression did not influence premature senescence of IMR90 cells induced by the oncogene RasV12 (Supplementary Fig. 13d), which is not dependent on p53 (Ref. 14,15). These results suggest that ME1 and ME2 expression suppress the specific way in which p53 induces senescence.
We investigated the mechanism for ME down-regulation-induced senescence. In IMR90 and U2OS cells where the expression of either ME1 or ME2 was silenced by siRNA, even moderately, p53 levels were elevated, accompanied by enhanced phosphorylation of p53 and induction of its target gene p21 (Fig. 2a, 4a and Supplementary Fig. 5a, 5b, 10a). In contrast, over-expression of ME1 or ME2 in IMR90 cells substantially reduced p53 levels and activity in late passages (Fig. 3f). Over-expression of ME1 or ME2 also diminished DNA damage-induced p53 activation (Supplementary Fig. 14). These observations suggest a strong role of MEs in the suppression of p53. They also indicate the existence of a positive feedback loop for the p53-ME pathway: a higher p53 level leads to less ME expression, which alleviates the inhibition of MEs on p53, leading to even higher p53 activation.
Figure 4. Mechanisms of p53 activation induced by ME down-regulation and a role of MEs in tumor growth.
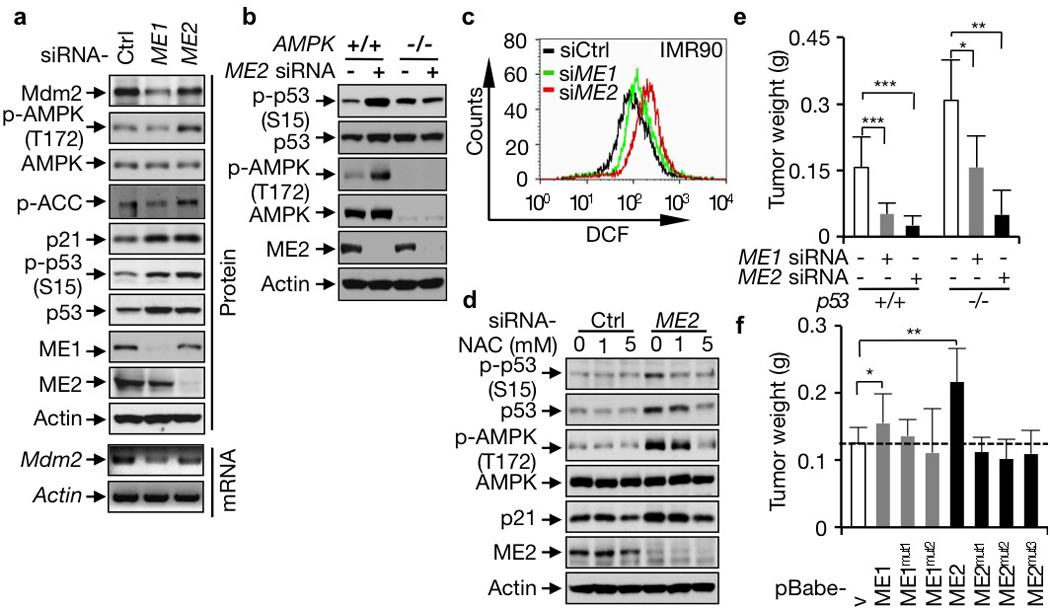
a, Effect of ME1 and ME2 knockdown on p53 and AMPK activation and Mdm2 expression. ACC: Acetyl-CoA carboxylase. b, p53 and AMPK activation in AMPK+/+ and AMPK−/− MEF cells transfected with control or ME2 siRNA. c, ROS levels in IMR90 cells transfected with ME1, ME2, or control siRNA. d, Effect of NAC on AMPK and p53 activation in IMR90 cells transfected with control siRNA or ME2 siRNA. e, f, Average weights of xenograft tumors (mean ± SD, n=6) generated by p53+/+ and p53−/− HCT116 cells transfected with ME1, ME2, or control siRNA (e), or p53+/+ HCT116 cells stably over-expressing wild-type or mutant MEs (f).
We examined the mechanism for the regulation of p53 by ME. In unstressed cells, Mdm2-mediated ubiquitination maintains a low basal level of p53 (Ref. 2). When ME1 was knocked down, the abundance of the Mdm2 protein and mRNA declined markedly (Fig. 4a and Supplementary Fig. 15a, b), suggesting that ME1 down-regulation activates p53 through a reduction in Mdm2 expression. ME2 knockdown did not significantly affect Mdm2 levels. Instead, it turned on AMP-activated kinase (AMPK) (Fig. 4a and Supplementary Fig. 15a, b), an intracellular energy gauge that activates p53 via phosphorylation16. We tested whether AMPK is required for induction of p53 by ME2 by knocking it down and by comparing AMPK-null and -wild-type MEFs. In both situations, loss of AMPK prevented ME2 knockdown from activating p53 (Fig. 4b and Supplementary Fig. 15c). Because silencing of ME2 did not influence cellular ATP levels (Supplementary Fig. 8c), we examined other pathways that could activate AMPK in ME2-depleted cells and observed a strong increase in reactive oxygen species (ROS) (Fig. 4c and Supplementary Fig. 16), correlating with a strong reduction in NADPH levels (Fig. 2a and Supplementary Fig. 5a–c). ROS are a known activator of AMPK17. Treatment with the ROS scavenger N-acetyl cysteine (NAC) blocked AMPK and p53 activation (Fig. 4d and Supplementary Fig. 17a, b), abrogated senescence, and restored growth of ME2-knockdown cells (Supplementary Fig. 17c, d). These results suggest that down-regulation of ME2 increases ROS levels leading to sequential activation of AMPK and p53 and the induction of senescence. In comparison, depletion of ME1 increased ROS levels moderately in IMR90 cells and minimally in U2OS cells (Fig. 4c and Supplementary Fig. 16), and NAC only slightly affected p53 activation, senescence, and growth arrest in ME1-depeleted cells (Supplementary Fig. 17b–d).
Previous studies on limited tumor samples suggest that the activity of ME2 is highly elevated in these tumors and correlates with tumor progression18–20. A survey of public gene expression databases (http://www.oncomine.org) showed that both ME1 and ME2 expression was significantly up-regulated in a variety of human cancers (Supplementary Fig. 18). We investigated whether MEs could influence tumor cell growth. Depletion of ME1 or ME2 in tumor cells, regardless of p53 status, strongly impaired their growth (Supplementary Fig. 19), and reduced the number of cells at the S phase of the cell cycle (Supplementary Fig. 20). In contrast, over-expression of ME1 or ME2, but none of the ME mutants, enhanced tumor cell growth (Supplementary Fig. 21). In a soft agar assay, tumor cells deprived of ME expression, unlike their control counterparts, failed to form anchorage-independent colonies (Supplementary Fig. 22a, b), while cells transduced with wild-type MEs, but not any of the ME mutants, showed enhanced anchorage-independent growth (Supplementary Fig. 22c, d).
To analyze MEs’ function in the tumor xenograft model, we injected immunocompromised mice with p53+/+ and p53−/− HCT116 cells treated with ME or control siRNA. p53−/− HCT116 cells gave rise to tumors that were twice the weight of tumors generated by p53+/+ HCT116 cells. When ME1 or especially ME2 was silenced in these cells, the tumor sizes were markedly reduced (Fig. 4e and Supplementary Fig. 23a, b). p53+/+ HCT116 tumors devoid of MEs showed extensive senescence and were substantially smaller compared to the corresponding p53−/− HCT116 tumors (Supplementary Fig. 23c). Conversely, over-expression of wild-type ME1 or ME2, not mutant MEs, accelerated the growth of p53+/+ HCT116 tumors (Fig. 4f and Supplementary Fig. 23d). These observations indicate that MEs are essential for tumor growth through both p53-dependent and -independent mechanisms.
Although p53 is able to induce a range of anti-proliferative responses, emerging evidence indicates that senescence induction and metabolic regulation are central to its function as a tumor suppressor13,21–25. Our results demonstrate a positive feedback loop comprising of p53 and MEs that influences p53 activation and links metabolism with the onset of senescence (Supplementary Fig. 24). p53 suppresses all ME (1, 2, and 3) expression by directly binding to REs within these genes. Together with our recent finding that p53 targets the NADPH producer G6PD through a distinct direct catalytic mechanism4, the current study reveals p53 as a master immediate regulator of cellular NADPH levels. p53 is reciprocally regulated by ME1 and ME2. The marked stabilization of p53 upon ME1 and ME2 down-regulation is achieved through different mechanisms, via the decline in the Mdm2 levels and ROS-induced AMPK activation, respectively. These findings support the notion that p53 is a central sentinel for metabolic stresses and coordinates metabolic pathways with cell fate decision.
Mutual regulation of p53 and MEs is likely a major mechanism that modulates cellular senescence in both normal and tumor cells. Even moderate down-regulation of either ME1 or ME2 strongly induces p53 activation and senescence, while over-expression of either enzyme delays these processes. Thus, MEs modulate not only the amplitude but also the outcome of p53 activation. p53 is subjected to negative feedback regulation (e.g. the p53-Mdm2 feedback loop) that restrains its activity2. The p53-MEs positive feedback loop is likely important to alleviate the negative feedback regulation so that p53 can accumulate to high levels. This may be particularly relevant in situations where robust and persistent p53 activation is desirable, such as the induction and maintenance of senescence. The involvement of the p53-ME pathway in senescence demonstrates a close link between metabolism and this irreversible fate of the cell.
Methods
Antibodies and reagents
The antibodies against the following proteins/epitopes were purchased from the indicated sources: ME2, actin, and β-tubulin (Sigma, St Louis, MO); AMPK, phospho-AMPK (Thr172), phospho-p53 (Ser15), and phospho-acetyl-CoA carboxylase (Ser79) (Cell Signaling Technology, Danvers, MA); ME1, p21, and PML (Santa Cruz Biotechnology, Santa Cruz, CA); prohibitin (Thermo Scientific, Hudson, NH); GAPDH (Novus Biologicals, Littleton, CO); p53 (DO-1, Oncogene, Manhasset, NY and Santa Cruz Biotechnology); and Mdm2 (Calbiochem, San Diego, CA and Santa Cruz Biotechnology). The following reagents were purchased from Sigma: dimethyl L-malate, NADP+, NAD+, doxorubicin, etoposide, trichostatin A (TSA), [U-13C5] glutamine N-acetylcysteine (NAC), insulin, troglitazone, dexamethasone, and isobutylmethylxanthine.
Cell culture and gene knockdown with shRNA and siRNA
Cells were maintained in standard culture conditions without any antibiotic. Expression plasmids for p53, ME1, and ME2 shRNA were made in pLKO.1-puro vector. The targeted sequences for human and mouse p53 are 5’-GACTCCAGTGGTAATCTAC-3’ (ref. 26) and 5’-GTACTCTCCTCCCCTCAAT-3’ (Ref. 27), respectively. The targeted sequences for human ME1 and ME2 are 5’-GGGCATATTGCTTCAGTTC-3’ and 5’-GCACGGCTGAAGAAGCATATA-3’, respectively. Stable shRNA cell lines were established as previously described28. siRNAs for ME1, ME2, and AMPK were purchased form Invitrogen (Carlsbad, CA). siRNA sequences were AUAACAAUCAGGUAGAAUCUGGUCA (human ME1); UAUAGUUGAAGGCUUCAGUAUAUUC (human ME2); CCCUGUGGGUAAAUUGGCUCUAUAU (human ME1#2); CCUGACAAGCCAAUUGACAGAUGAA (human ME2#2); CGUUGAAAAUUGCAGUAAA (mouse ME1); GGGCACUGAUAACAUGGCACUAUUA (mouse ME2); and ACCAUGAUUGAUGAUGAAGCCUUAA (human AMPK). siRNAs were transfected into cells using Lipofectamine® RNAiMAX Transfection Agent (Invitrogen).
Semi-quantitative RT–PCR and real time RT-PCR
Total RNA was isolated from cells by TRIzol® Reagent (Invitrogen). Two micrograms of RNA for each sample were reversed to cDNA by First-strand cDNA Synthesis System (Marligen Biosciences), and 0.2 µg cDNA were used as a template to perform PCR. The primer pairs for human genes were: ME1, 5′-ACAGATAATATTTTCCTCACT-3’ and 5′-CTACTGGTCAACTTTGGT-3′; ME2, 5′-ATTAGTGACAGTGTTTTCCTA-3’ and 5′-CTATTCTGTTATCACAGG-3′; p21, 5′-CCGGCGAGGCCGGGATGAG-3’ and 5′-CTTCCTCTTGGAGAAGATC-3′; β-actin, 5′-GACCTGACTGACTACCTCATGAAGAT-3′ and 5′-GTCACACTTCATGATGGAGTTGAAGG-3′; p53, 5′-CACGAGCTGCCCCCAGG-3’ and 5′-TCAGTCGACGTCTGAGT-3′. Primer pairs for mouse genes were: ME1, 5’-GATGATAAGGTCTTCCTCACC-3’ and 5’-TTACTGGTTGACTTTGGTCTGT-3’; ME2, 5’-TTCTTAGAAG CTGCAAAGGC-3’ and 5’-TCAGTGGGGAAGCTTCTCTT-3’; p21, 5’-AACTTCGTCTGGGAGCGC-3’ and 5’-TCAGGGTTTTCTCTTGCAGA-3’; β-actin, 5’-ACTACATTCAATTCCATC-3’ and 5’-CTAGAAGCACTTGCGGTG-3’; p53, 5’-GAAGTCCTTTGCCCTGAAC-3’ and 5’-CTAGCAGTTTGGGCTTTCC-3’.
All real-time PCR reactions were performed using the 7900HT Fast Real-Time PCR System (Applied Biosystem) and the amplifications were done using the SYBR Green PCR Master Mix (Applied Biosystems). The thermal cycling conditions were: 50 °C for 2 min followed by an initial denaturation step at 95 °C for 10 min, 45 cycles at 95 °C for 15s, 60 °C for 1 min, and a dissociation curve at 95 °C for 15s and 60 °C for 15s. The experiments were carried out in triplicate for each data point. Using this method, we obtained the fold changes in gene expression normalized to an internal control gene.
Cell lysate fractionation and ME activity
Cell fractionation was carried out as described29. Cells were homogenized in 20 mM HEPES-KOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, and 1 mM dithiothreitol in the presence of 250 mM sucrose and protease inhibitor cocktail (Roche Diagnostics, Meylan, France). Homogenates were centrifuged at 500 g for 5 min at 4 °C, and the supernatant was collected and centrifuged again at 10,000 g for 20 min to obtain cytosolic and mitochondrial fractions.
ME1 activity was determined using cytosolic extracts as described30. The reaction buffer contained 67 mM triethanolamine, 3.3 mM L-malic acid, 0.3 mM β-NADP+, and 5.0 mM manganese chloride. For measuring ME2 activity, mitochondria were purified as described29 and re-suspended in mitochondrial lysis buffer (20 mM MOPS-KOH pH7.4, 250 mM sucrose, 80 mM KCl, 5 mM EDTA, 1 mM PMSF, 1% Triton X-100, and protease inhibitor cocktail) on ice for 30 min by gentle vortexing for 5 s at 5 min intervals. Lysates were centrifuged for 10 min at 14,000 rpm at 4°C. The enzyme reaction mixtures contained 50 mM Tris-HCl, pH7.4, 10 mM MgCl2, 0.3 mM NAD+, and 3.3 mM L-malic acid. The reactions were started by adding either cytosolic and mitochondrial extracts, and were monitored by absorbance at 340 nm every 5 s for up to 10 min. Background control was run without L-malic acid as substrate. Enzyme activity was determined by subtracting the activity of the background control to each sample. The resulting changes of absorbance versus time were normalized to protein contents, which were determined using the Bio-Rad protein assay.
Analysis of ME gene sequence and chromatin immunoprecipitation (ChIP) assay
We used the Genomatix Promoter Inspector software (URL: http://www.genomatix.de) to search in ME genes for potential p53 REs with the consensus sequence 5’-RRRCWWGYYY-(0–13bp spacer)-RRRCWW GYYY-3’, where R is a purine, Y a pyrimidine, and W either A or T8. The sequences for the putative p53 REs in ME genes are: ME1-RE, 5’-ttacctggttaactaggacttgccc-3’; ME2-RE1, 5’-aggcatgcaccaccatgccc-3’; ME2-RE2, 5’-agaccagtcaaaaacatgtcc-3’; ME2-RE3, 5’-gggcatgatggcacatgcct-3’, and ME3-RE, 5’-tgacttggtttggctttcttgtcc-3’.
For ChIP assay, cells were washed with PBS and cross-linked with a 1% formaldehyde solution for 15 min at RT. The cross-linking reaction was stopped by the addition of glycine to 125 mM final concentration. Cell lysates were sonicated to generate DNA fragments with the average size below 1000 bp and followed by immunoprecipitation with indicated antibodies. Bounded DNA fragments were eluted and amplified by PCR. The primer pairs were: ME1-RE: 5’-GCCTTAGTATGTGGATTC-3’ and 5’-GGAAAGCGTAGGGAAGGA-3’; ME2-ME1: 5’-GTTGCCCAGGCTGGAGTG-3’ and 5’-CTGTAATCCCAGCACTTT-3’; ME2-RE2: 5’-TCAGCACTTTGGGAGG-3’ and 5’-GCGACAGAGTCTTGCC-3’; ME2-RE3: 5’-GGCTCAGTGGCTCACG-3’ and 5’-GTGCAGTGGCATG-3’; ME3-RE: 5’-GTTGCGATCCCGTGGCTG-3’ and 5’-ACCGCAGGTCAGACTGAC-3’; p21: CTGAAAACAGGCAGCCCAAG-3’ and 5’-GTGGCTCTGATTGGCTTTCTG-3’ 28.
Reporter assay
The DNA fragment containing the potential p53-binding region was amplified by PCR with primers used in the ChIP assay and was cloned into a pGL3-promoter vector (Promega). 293T cells were plated 18 h prior to transfection in 24-well plates and transiently transfected with 450 ng of the reporter plasmid and/or 100 ng of the p53 plasmid using Lipofectamine 2000 (Invitrogen). The Luciferase activity was determined according to the manufacturer's instructions (Promega). Transfection efficiency was normalized on the basis of the Renilla luciferase activity.
Measurements of metabolites and lipid accumulation
The levels of NADPH, NADH, NAD+, ATP, and ADP in cultured cells were determined using a NADP+/NADPH Quantification Kit, NAD+/NADH Quantification Kit, ATP assay kit, and ADP assay kit (all from BioVision, Milpitas, CA, USA) respectively, following the manufacturer's instructions. Glucose and glutamine consumption was determined using YSI 7100 Multiparameter Bioanalytical System (YSI Life Sciences, Yellow Springs, OH, USA). Triglyceride was measured using a Triglyceride Assay Kit (BioVision). Total lipids were measured using Oil Red O staining10. For this, confluent cells were grown in medium with 10% FBS supplemented with insulin (5 µg/ml), dexamethasone (1 μM), troglitazone (5 μM) and isobutylmethylxanthine (0.5 mM) for 2 days, and in medium supplemented with insulin and rosiglitazone for an additional 5 days. The medium was changed every other day. Cells were then fixed with 4% paraformaldehyde for 30 min at room temperature (RT), washed with dH2O and 60% isopropanol, and stained with a filtered Oil Red O work solution at RT. Stain was then removed and cells were washed 4 times in dH2O.
Measurement of glutaminolytic flux
Malic enzyme-dependent glutaminolytic flux (ME flux) was determined by labeling the malate pool with 13C from [U-13C5] glutamine and monitoring the conversion of malate to pyruvate indirectly by detecting 13C-labeled lactate. 13C enrichments were determined with gas chromatography-mass spectrometry (GC-MS). We observed that this approach results in approximately 50% 13C enrichment and allows for the determination of glutaminolytic flux through malic enzyme with high sensitivity. Cells cultured in 10 cm plates with around 60% confluence were transfected with control, ME1, or ME2 siRNA. Cells were cultured in regular medium for 60 h and in DMEM containing 12 mM glucose, 3 mM [U-13C5] glutamine, and 10% dialyzed fetal bovine serum (Sigma, St. Louis, MO, USA) for an additional 9 h. After medium was removed, cells were immediately quenched with cold 80% methanol. The methanol/cell mass mixtures were centrifuged. The insoluble (protein/lipid) fraction was analyzed for protein content. The soluble fraction was dried under a stream of gaseous nitrogen at 40 °C and silylated with N-tertbutyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA; Regis, Morton Grove, IL, USA). The silylated cell extracts were analyzed with an Agilent 7890A gas chromatograph/5975C mass spectrometer (Agilent, Santa Clara, CA, USA). Mass spectra were quantified with the MSD ChemStation software (Agilent) and corrected for natural abundance contributions from 13C, 29Si and 30Si using Isocor (www.python.org)31. The total lactate level was determined with the YSI 7100 Multiparameter Bioanalytical System. The glutaminolytic flux through malic enzymes was calculated from the equation: FME= FL × (Lm+3)/(Mm+4), where FME = malic enzyme flux, FL = total lactate flux, Lm+3= fraction of lactate enriched in all 3 carbons, and Mm+4= fraction of malate enriched in all 4 carbons.
Measurements of ROS
ROS levels were determined as described32. Cells were incubated at 37 °C for 30 min in phosphate buffered saline (PBS) containing 10 µM 2’,7’-dichlorodihydrofluorescein diacetate (H2-DCFDA, Sigma). Afterwards, the cells were washed twice in PBS, treated with trypsin, and re-suspended in PBS. Fluorescence was immediately measured using a FACScan Flow Cytometer (Becton Dickinson, San Jose, CA).
Senescence-associated β-galactosidase (SA-β-gal) activity
The SA-β-gal activity in cultured cells was determined using a Senescence Detection Kit (BioVision) following the manufacturer's instructions. Percentages of cells that stained positive were calculated by counting 1,000 cells in random fields per cell line.
Immunofluorescence
Cells treated with siRNA for 48 h were washed with 1×PBS and fixed in 4% paraformaldehyde. After being treated with 0.1% Triton X-100, cells were stained using anti-PML antibody followed by Texas-red conjugated anti-mouse IgG antibody, and mounted with 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). The images were acquired with a confocal microscope. 200 nucleuses were selected randomly and PML-NBs with each nucleus were counted.
Cell proliferation assay
Cells were treated with siRNAs for 24 h and seeded in 6-well cell culture dishes in triplicates at a density of 20,000 cells/well in 2 ml of medium containing 10% FBS. The medium was changed everyday. Cells were counted and cell number at the indicated time points was determined.
Soft agar assay and xenograft tumor models
For soft agar assay, cells were suspended in 1 ml of 10% FBS DMEM medium containing a 0.3% agarose and plated on a firm 0.6% agarose base in 6-well plates (5,000 cells/well) as described previously33. Cells were then cultured in a 37°C and 5% CO2 incubator for 2 weeks. Images were obtained and colonies were counted under a microscope. Each experiment was done in triplicate. For mouse xenograft experiment, cells (2×106) were injected subcutaneously into the flanks of 4- to 5-week-old athymic Balb-c nu/nu male mice (Taconic Farms, Germantown, NY, USA). Tumor growth was evaluated at 2 weeks post-injection. All animal experiments were performed in accordance with relevant guidelines and regulations and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
Supplementary Material
Acknowledgments
We thank M. J. Birnbaum, B. Vogelstein, W. El-Deiry, and M. Lazar for reagents; M. J. Bennett, S. Patel, A. Stonestrom, and M. Brewer for technical assistance; and A. Stonestrom for help with manuscript preparation. Supported by grants from the National Institutes of Health (CA088868) and the US Department of Defense (W81XWH-10-1-0468) to X.Y.
Footnotes
Author contributions P.J., W. D., and X. Y. designed the study, interpreted the data, and wrote the manuscript. P.J. and W. D. performed the experiments. K. E. W. helped with the metabolic studies and data interpretation. A. M. designed the glutaminolytic flux procedure and performed the experiment with the help from P.J.
Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Jiang P, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu RY. Pigeon liver malic enzyme. Mol Cell Biochem. 1982;43:3–26. doi: 10.1007/BF00229535. [DOI] [PubMed] [Google Scholar]
- 6.Chang GG, Tong L. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry. 2003;42:12721–12733. doi: 10.1021/bi035251+. [DOI] [PubMed] [Google Scholar]
- 7.Pongratz RL, Kibbey RG, Shulman GI, Cline GW. Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J Biol Chem. 2007;282:200–207. doi: 10.1074/jbc.M602954200. [DOI] [PubMed] [Google Scholar]
- 8.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 9.Murphy M, et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999;13:2490–2501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferbeyre G, et al. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015–2027. [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson M, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 13.Brady CA, et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 15.Wei W, Hemmer RM, Sedivy JM. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol Cell Biol. 2001;21:6748–6757. doi: 10.1128/MCB.21.20.6748-6757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 17.Blattler SM, Rencurel F, Kaufmann MR, Meyer UA. In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104:1045–1050. doi: 10.1073/pnas.0610216104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wasilenko WJ, Marchok AC. Malic enzyme and malate dehydrogenase activities in rat tracheal epithelial cells during the progression of neoplasia. Cancer Lett. 1985;28:35–42. doi: 10.1016/0304-3835(85)90089-8. [DOI] [PubMed] [Google Scholar]
- 19.Sauer LA, Dauchy RT, Nagel WO, Morris HP. Mitochondrial malic enzymes. Mitochondrial NAD(P)+-dependent malic enzyme activity and malate-dependent pyruvate formation are progression-linked in Morris hepatomas. J Biol Chem. 1980;255:3844–3848. [PubMed] [Google Scholar]
- 20.Nagel WO, Dauchy RT, Sauer LA. Mitochondrial malic enzymes. An association between NAD(P)+-dependent malic enzyme and cell renewal in Sprague-Dawley rat tissues. J Biol Chem. 1980;255:3849–3854. [PubMed] [Google Scholar]
- 21.Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 22.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 25.Li T, et al. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 27.Ventura A, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godar S, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang P, Du W, Heese K, Wu M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol. 2006;26:9071–9082. doi: 10.1128/MCB.01025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guay C, Madiraju SR, Aumais A, Joly E, Prentki M. A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J Biol Chem. 2007;282:35657–35665. doi: 10.1074/jbc.M707294200. [DOI] [PubMed] [Google Scholar]
- 31.Millard P, Letisse F, Sokol S, Portais JC. IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics. 2012;28:1294–1296. doi: 10.1093/bioinformatics/bts127. [DOI] [PubMed] [Google Scholar]
- 32.Cossarizza A, et al. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat Protoc. 2009;4:1790–1797. doi: 10.1038/nprot.2009.189. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, et al. AFAP-110 is overexpressed in prostate cancer and contributes to tumorigenic growth by regulating focal contacts. J Clin Invest. 2007;117:2962–2973. doi: 10.1172/JCI30710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.