

Abstract
Trauma with hemorrhagic shock (T/HS), has been shown to result in liver injury marked by hepatocyte apoptosis and heart failure marked by cardiomyocyte apoptosis, both of which we have shown to be prevented by IL-6 administration at resuscitation, and Stat3 largely mediated this. As specific mediators have not been delineated, we investigated the unfolded protein response (UPR), which, with marked activation, can lead to apoptosis. Prior studies of hepatic and cardiac injury examined limited repertoires of UPR elements, making it difficult to assess the role of the UPR in T/HS. This study describes the first global examination of the UPR transcriptome in the liver and heart following T/HS, demonstrating organ-specific UPR transcriptome changes. The non-canonical UPR chaperone, Hsp70, was most dysregulated following T/HS and may contribute to hepatocyte protection via an IL-6-mediated pathway, identifying a potential new therapeutic strategy to prevent hepatocyte death and organ dysfunction in T/HS.
Trauma is a leading cause of morbidity and mortality in the United States for those under the age of 45 years, especially when complicated by hemorrhagic shock1. When trauma with hemorrhagic shock (T/HS) is accompanied with resuscitation, the end effect is essentially a systemic ischemia and reperfusion injury. Multiple organ failure is an important maladaptive sequelae contributing to late mortality in those who survive beyond 24 hrs following severe T/HS and resuscitation2.
Work done by our group, and others, in rodent models of T/HS, has shown that parenchymal cells within organs such as the liver, a key metabolic and homeostatic organ, and heart, an organ whose dysfunction often heralds post-traumatic mortality, undergo apoptosis3,4,5,6,7. The pathways leading to parenchymal cell apoptosis in these organs in T/HS are not fully understood. The classical mechanisms of apoptosis, such as the extrinsic and intrinsic apoptotic pathways, have been investigated in the liver and heart3,6. However, specific delineation of the pathways leading from T/HS to cell death and organ dysfunction is incomplete.
Prolonged or severe endoplasmic reticulum (ER) stress has recently been demonstrated to lead to apoptosis through the unfolded protein response (UPR). The canonical genes involved in ER stress and the UPR were first delineated in yeast including identification of the ER membrane bound sensors of ER stress8,9,10,11. Homologues for these sensors and their targets have been identified in mammals and their activation can reliably be assessed transcriptionally. While much of the focus of investigation on the UPR has centered around the three main signaling molecules inositol-requiring enzyme 1α (IRE1α), Activating Transcription Factor 4 (ATF4), and protein kinase RNA-like endoplasmic reticulum kinase (PERK), many non-canonical modulators of the UPR have been identified linking the UPR to pathways ranging from innate immunity to apoptosis. Emerging evidence has shown that prolonged ER stress and UPR activation leads to apoptosis that is an important mechanism of disease pathogenesis in a number of genetic disorders, such as lysosomal storage diseases, particularly within the liver12,13. Examination of the UPR as a potential cause of parenchymal cell apoptosis in metabolic and other derangements leading to ER stress initially focused on exocrine organs such as the liver14. The UPR and its contribution to liver disease has been investigated in liver diseases such as steatosis15,16, ischemia/reperfusion injury17,18 and T/HS19,20. The impact of the ER stress and the UPR on non-exocrine organs such as the heart, has only recently become a focus21,22. Studies of both the liver and heart are limited, however, since they have focused on isolated components of the UPR and did not provide direct evidence that would allow one to conclude that apoptosis or organ injury resulted from an insufficient adaptive UPR or that the UPR or components therein were, in fact, maladaptive.
We previously demonstrated that parenchymal cell apoptosis following T/HS in both the liver and heart is prevented by administration of IL-6, which mediates its effect through the actions of Stat33,6. In the current studies, we performed UPR transcriptome analysis of the liver and heart at a global level to identify candidate genes within the canonical and non-canonical UPR that contribute to apoptosis following T/HS. By tracking the direction and magnitude of changes in levels of these candidate genes that occurred following T/HS with IL-6 resuscitation, with or without Stat3 inhibition, we were able to clearly identify those genes most implicated in T/HS-induced apoptosis and its prevention by IL-6-activated Stat3. In particular, we demonstrated that Hsp70 and 40 were upregulated in the liver by T/HS, and that this response was adaptive and insufficient since IL-6 augmented it, thereby preventing apoptosis.
Results
T/HS-induced hepatocyte apoptosis is prevented by IL-6 resuscitation; the IL-6 effect is mediated, in part, by Stat3
To confirm our previous findings that T/HS induces liver apoptosis, we measured histone-associated DNA fragments (nucleosomes) in the livers of rats subjected to our T/HS protocol. Nucleosome levels were 13.5 times higher than sham (p < 0.001, ANOVA; Table 1). The nucleosome results were confirmed by TUNEL staining (Table 1), which also demonstrated that hepatocytes represented the overwhelming majority of cells undergoing apoptosis (data not shown).
Table 1. Impact of T/HS without and with IL-6 on markers of apoptosis in the heart and liver.
| Liver | Heart | |||
|---|---|---|---|---|
| Intervention | Nucleosomea | TUNELb | Nucleosomea | TUNELb |
| Sham | 139 ± 67* | 0.9 ± 0.4** | 0‡ | 1.3 ± 0.2‡‡ |
| T/HS | 1874 ± 127*† | 27 ± 3.6**,†† | 63 ± 8Δ,‡ | 16.2 ± 2Δ,‡‡ |
| T/HS-IL6 | 264 ± 36†¥ | 1.9 ± 0.5††,¥¥ | 4 ± 1◊,Δ | 8.5 ± 0.2◊◊,ΔΔ |
| T/HS-IL6-GQ | 1556 ± 241¥ | 12.3 ± 1.1¥¥ | 24 ± 5◊ | 16.5 ± 1◊◊ |
§ *,†,¥,**,††,¥¥,‡,Δ,◊,‡‡,ΔΔ,◊◊ indicate group comparisons with statistical significance of p < 0.05, one-way ANOVA.
aNucleosome data presented as mU/mg total protein.
bTUNEL data presented as number of TUNEL-positive nuclei per high power field.
Nucleosome levels in the IL-6-resuscitated rats were decreased 7.1 times compared to those of the T/HS group (p < 0.001) and were similar to sham levels (Table 1). TUNEL staining confirmed these results (Table 1). The number of TUNEL-positive nuclei/hpf in the IL-6 group was decreased 14.2 times compared to the placebo group (p < 0.001), to levels statistically similar to those of the sham group (Table 1).
Pretreatment of rats with the G-rich, quartet-forming oligonucleotide Stat3 inhibitor (T40214) was accompanied by a return of nucleosomes to levels similar to those of the placebo treated group and 5.9 fold higher that those of the IL-6 treated group (p < 0.001; Table 1). Similarly, the number of TUNEL-positive nuclei/hpf in livers of rats from the T/HS-IL6-GQ group was 6 fold higher than that of the T/HS-IL6-treated group (p < 0.0001); Table 1). Nucleosome levels and number of TUNEL-positive nuclei/hpf in livers of rats pre-treated with a NS-ODN before T/HS and IL-6 resuscitation were indistinguishable from those of the IL-6 group (data not shown). Thus, pharmacological inhibition of Stat3 using T40214 in rats subjected to severe HS resuscitated with IL-6 completely blocked IL-6-mediated prevention of liver apoptosis.
Liver UPR transcriptome is significantly altered in T/HS
We investigated the impact of T/HS on the ER stress response at the transcriptome level, and then defined the role of this ER stress response on the observed reversible hepatic apoptosis. Unbiased hierarchical clustering of our experimental animals based on intervention group and entity clustering with the UPR transcriptome demonstrated the reproducible nature of the impact of T/HS on the UPR transcriptome (Figure 1). Of the broad 185-gene UPR-associated entity list generated via literature review and Ingenuity Pathway Analysis (IPA®), 113 distinct gene entities were annotated and expressed across our chips after spot duplicates were removed. Using this list of 113 genes, 63 (56%) were significantly altered in one-way ANOVA (p < 0.05) among all three-group comparisons, T/HS vs. Sham, T/HS-IL6 vs. T/HS, and T/HS-IL6-GQ vs. T/HS-IL6. When the impact of T/HS was looked at specifically, 31 (27%) of those gene entities were significantly dysregulated in the T/HS group when compared to sham, with 55% (17 of 31) significantly upregulated and 45% of gene transcripts downregulated. When asking the question of potential mediators of the protective effect of IL-6, 17 entities were significantly altered in both group comparisons. Taking known apoptotic function of these genes into context, we demonstrated that all UPR-associated genes with known potential pro-apoptotic function were upregulated following T/HS and subsequently normalized with IL-6 (Table 2). The most dysregulated genes within this intergroup comparison were the chaperones, Heat Shock Protein 70 (25.6-fold), and Heat Shock Protein 40 (5.9-fold), the UPR transcription factor ATF4 (3.1-fold), and endoplasmic oxidoreductin-1-like protein (Ero1l) (9.8-fold) suggesting a strong impact on the protein folding mechanics both in the cytoplasm and the endoplasmic reticulum. Indeed, when assessed by Real-Time PCR (RTPCR), Hsp70 and Hsp40 demonstrated significantly increased transcript levels in T/HS animals when compared to Sham, with 5.1 fold (p = 0.004) and 3.5 fold (p = 0.001) increase, respectively (Figure 2). Likewise confirming the findings of the microarray analysis, Hsp70 and Hsp40 were found to be significantly further increased in animals resuscitated with IL-6 when compared to T/HS animals that did not receive IL-6 at resuscitation with 11.2 fold (p = 0.04) and 4.5 fold (p = 0.026) increases, respectively (Figure 2.)
Figure 1. Unbiased hierarchical heatmap clustering based on both UPR entity and experimental intervention of animals confined to 113 UPR-associated gene entities on whole liver preparations.

Clustering performed using Hierarchical analysis using Euclidean similarity measure, expression data normalized to chip standards for clustering.
Table 2. Liver UPR Transcripts Significantly Altered in Both T/HS vs. Sham and T/HS/IL6 vs. T/HS Comparisons.
| Regulation | Fold Change | |||||
|---|---|---|---|---|---|---|
| Gene Symbol | UPR Function | Apoptosis Function | T/HSvs Sham | T/HS-IL6 vs T/HS | T/HS vs Sham | T/HS-IL6 vs T/HS |
| Hspa1a/Hspa1b (Hsp70) | Chaperone | Anti | up | up | 25.6 | 5.5 |
| Hspa1b (Hsp70-1b) | Chaperone | Anti | up | up | 18.4 | 5.9 |
| Ero1I | Disulfide Bond Formation | Anti | up | down | 9.8 | −3.8 |
| Dnajb1 (Hsp40 Subunit b1) | Chaperone | Anti | up | up | 5.9 | 2.7 |
| Atf4 | Transcription Factor | Anti/Pro | up | down | 3.1 | −2.0 |
| Casp3 (caspase 3) | Apoptosis Signalling | Pro | up | down | 1.8 | −1.7 |
| Eif2s1 (Eif2α) | Protein Transtation | Anti | up | down | 1.7 | −1.6 |
| Sels | Modulation of ATF6 | Uknown | up | down | 1.5 | −1.3 |
| Eif2ak3 (PERK) | UPR Sensory Molecule | Anti/Pro | up | down | 1.4 | −1.3 |
| Psmb3 | Proteasome Degradation | Anti | down | up | −1.2 | 1.3 |
| Calr (calreticulin) | Chaperone | Anti | down | up | −1.2 | 1.2 |
| Uba1 | Ubiquitination | Anti | down | up | −1.3 | 1.2 |
| Psme2 | Proteasome Degradation | Anti | down | up | −1.3 | 1.4 |
| Psme1 | Proteasome Degradation | Anti | down | up | −1.3 | 1.2 |
| Dyt1 | ATPase | Anti | down | up | −1.4 | 1.4 |
| Tmbim6 (Baxinhibitor 1) | Apoptosis Signalling | Anti | down | up | −1.5 | 1.3 |
| Ccnd1 | Cell Cycle Signalling | Anti | down | up | −3.4 | 2.3 |
Figure 2. Q-RT-PCR using TaqMan® (Life Technologies) for (A) heat shock protein 70 (Hsp70; Hspa1a) and (B) heat shock protein 40 (Hsp40; Dnajb1) performed on whole liver samples from Sham (n = 6), trauma with hemorrhagic shock (T/HS, n = 4), and T/HS animals resuscitated with IL-6 (T/HS-IL6, n = 4).
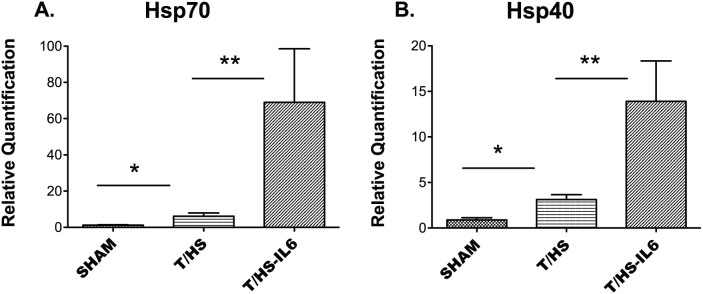
Transcript values reported as relative quantification (RQ) in comparison to a normal rat liver. Values expressed as mean RQ ± SEM. “*”, “**” indicate group comparisons which are statistically different (p < 0.05) by T-test.
To assess which of these dysregulated genes may be impacted via IL-6 through Stat3, we incorporated animals pre-treated with a pharmacologic Stat3 inhibitor (GQ T40214) then resuscitated with IL-6 to animal resuscitated with IL-6 alone. Using this combined intergroup approach, we found 12 gene entities with significant dysregulation across all three-group comparisons (Table 3). Interestingly, we find that of the most dysregulated transcripts, the chaperones Hsp70 and Hsp40 demonstrate upregulation in T/HS. In animals in which hepatocyte apoptosis was prevented by receiving IL-6 at resuscitation, we find that Hsp70 and Hsp40 were further upregulated, suggesting a contribution to prevention of hepatocyte apoptosis. When Stat3 is pharmacologically inhibited, however, we find downregulation of these chaperones, suggesting IL-6 acts to upregulate Hsp70/40 via a Stat3-dependent mechanism not previously described.
Table 3. Liver UPR Transcripts Significantly Altered T/HS vs. Sham, T/HS/IL6 vs. T/HS and T/HS/IL6/GQ Comparisons.
| Regulation | Fold Change | |||||
|---|---|---|---|---|---|---|
| Gene Symbol | T/HS vs Sham | T/HS-IL6 vs T/HS | T/HS-IL6-GQ vs T/HS-IL6 | T/HS vs Sham | T/HS-IL6 vs T/HS | T/HS-IL6-GQ vs T/HS-IL6 |
| Hspa1a/Hspa1b (Hsp70) | up | up | down | 25.6 | 5.5 | −9.0 |
| Hspa1b (Hsp701b) | up | up | down | 18.4 | 5.9 | −10.8 |
| Ero1l | up | down | down | 9.8 | −3.8 | −1.3 |
| Dnajb1 (Hsp40 subunit) | up | up | down | 5.9 | 2.7 | −3.4 |
| Casp3 (Caspase 3) | up | down | up | 1.8 | −1.7 | 1.4 |
| Sels | up | down | up | 1.5 | −1.3 | 1.4 |
| Eif2ak3 (PERK) | up | down | down | 1.4 | −1.3 | −1.3 |
| Psmb3 | down | up | up | −1.2 | 1.3 | 1.9 |
| Uba1 | down | up | up | −1.3 | 1.2 | 1.8 |
| Psme2 | down | up | up | −1.3 | 1.4 | 2.9 |
| Psme1 | down | up | up | −1.3 | 1.2 | 2.2 |
| Tmbim6 (Bax inhibitor 1) | down | up | down | −1.5 | 1.3 | −1.2 |
T/HS-induced cardiomyocyte apoptosis is prevented by IL-6 resuscitation; the IL-6 effect is mediated, in part, by Stat3
To confirm our previous findings that T/HS induces cardiomyocyte apoptosis, we measured histone-associated DNA fragments (nucleosomes) in the hearts of rats subjected to our T/HS protocol. Nucleosome levels were significantly increased in comparison to sham in T/HS rats (p < 0.01, ANOVA; Table 1). The nucleosome results were confirmed by TUNEL staining with a 12 fold increase in T/HS rats (p < 0.01, ANOVA; Table 1), which also demonstrated that cardiomyocytes represented the overwhelming majority of cells undergoing apoptosis (data not shown).
Nucleosome levels in hearts from IL-6 resuscitated rats were reduced by more than 15 fold compared to placebo treated rats undergoing T/HS (p < 0.05, ANOVA). TUNEL assays of sections of rat hearts confirmed these findings with a similar 1.9-fold reduction (p < 0.05, ANOVA; Table 1).
Pretreatment of rats with a Stat3 inhibitor was accompanied by a return of nucleosomes to levels similar to those of the placebo treated group. Nucleosome levels in the hearts of T/HS-IL6-GQ rats (Table 1) were increased 6-fold compared to hearts from IL-6 resuscitated rats (p < 0.05, ANOVA; Table 1). Thus, pharmacological inhibition of Stat3 using T40214 in rats subjected to severe T/HS resuscitated with IL-6 completely blocked IL-6-mediated prevention of cardiomyocyte apoptosis.
Heart UPR transcriptome is significantly altered in T/HS
The results above demonstrate that cardiomyocyte apoptosis caused by T/HS is largely prevented with administration of IL-6 at time of resuscitation (Table 1). We investigated the impact of T/HS on the ER stress response at the transcriptome level, and then defined the role of this ER stress response on the observed reversible cardiomyocyte apoptosis. Using the previously described UPR gene entity list, we found that of the 113 genes present on the chip, 86 (76%) were significantly altered in one-way ANOVA (p < 0.05) among all three-group comparisons, T/HS vs. Sham, T/HS-IL6 vs. T/HS, and T/HS-IL6-GQ vs. T/HS-IL6. When the impact of T/HS was looked at specifically, 29 (26%) of those gene entities were significantly dysregulated when compared to sham, with the majority, 79% (23 of 29) significantly upregulated and 6 gene transcripts downregulated. When asking the question of potential mediators of the protective effect of IL-6, 16 entities were significantly altered in both group comparisons (Table 4). The direction of dysregulation induced by T/HS was reversed by IL-6 in all transcripts identified. When taking known apoptotic functions of these genes and the impact of our experimental model into context, we demonstrated that 4 of the 5 genes with known pro-apoptotic function are upregulated following T/HS and subsequently normalized with IL-6 (Table 4). The most dysregulated genes, those genes with > 2 fold change, within this intergroup comparison were the chaperones, Hsp70 (10 fold), Hsp40 (3 fold), and Hsp105 (2.5 fold), and the negative regulator of PERK, phosphoinositide-3-kinase interacting protein 1 (Pik3ip1) (−3.0 fold), suggesting, as in the liver, a strong impact on the protein folding mechanics both in the cytoplasm and the endoplasmic reticulum. In contrast to the liver however, the heat shock protein chaperones, Hsp70 and Hsp40, were downregulated in the hearts of IL-6-treated animals, indicating they likely are not contributing to the apoptotic protection conferred by IL-6. When adding the comparison of GQ T40214 to IL-6 group, we found 11 gene entities with significant dysregulation across all three group comparisons, and, of those, 8 suggest potential IL-6 mediated effect through Stat3 (Table 5).
Table 4. Heart UPR Transcripts Significantly Altered in Both T/HS vs. Sham and T/HS/IL6 vs. T/HS Comparisons.
| Regulation | Fold Change | |||||
|---|---|---|---|---|---|---|
| Gene Symbol | UPR Function | Apoptosis Function | T/HS vs Sham | IL6 vs T/HS | T/HSvs Sham | IL6 vs T/HS |
| Hspa1a/Hspa1b (Hsp 70) | Chaperone | Anti | up | down | 10 | −6.4 |
| Hspa1b (Hsp701b) | Chaperone | Anti | up | down | 7.5 | −4.9 |
| Cebpb | Transcription factor | Pro | up | down | 5.1 | −1.7 |
| Dnaja1 (Hsp40 subunit) | Co-chaperone | Anti | up | down | 3.1 | −2.3 |
| Hsph1 (Hsp 105) | Chaperone | Anti | up | down | 2.5 | −2.1 |
| Dnajb1 (Hsp40 subunit) | Co-chaperone | Anti | up | down | 2.2 | −2 |
| Nfe2l2 | Transcription factor | Anti | up | down | 2 | −1.4 |
| Ppp1r15a (GADD34) | Transcription factor | Pro | up | down | 2 | −1.4 |
| Xbp1 (X-box-protein 1) | Transcription factor | Pro | up | down | 1.5 | −1.2 |
| Tra1 (Hsp90b1) | Chaperone (ERAD) | Anti | up | down | 1.4 | −1.3 |
| Calr (calreticulin) | Chaperone | Anti | up | down | 1.4 | −1.3 |
| Ddit3 (CHOP) | Transcription factor | Pro | up | down | 1.4 | −1.3 |
| Serp1 | protects unfolded proteins from ERAD | Anti | up | down | 1.3 | −1.2 |
| Sp1 | Transcription factor | Pro | down | up | −1.2 | 1.3 |
| Sels | modulates ATF6 | Unknown | down | up | −1.5 | 1.3 |
| Pik3ip1 | negative regulator of PERK | Anti | down | up | −3 | 1.4 |
Abbreviation: ERAD, endoplasmic reticulum-associated degradation.
Table 5. Heart UPR Transcripts Significantly Altered Across T/HS vs. Sham, T/HS/IL6 vs. T/HS, and T/HS/IL6/GQ vs. T/HS/IL6 Comparisons.
| Regulation | Fold Change | |||||
|---|---|---|---|---|---|---|
| Gene Symbol | T/HS vs Sham | T/HS-IL6 vs T/HS | T/HS-IL6-GQ vs T/HS-IL6 | T/HS vs Sham | T/HS-IL6 vs T/HS | T/HS-IL6-GQ vs T/HS-IL6 |
| Dnaja1 (Hsp40 subunit) | up | down | up | 3.1 | −2.3 | 2.0 |
| Hsph1 (Hsp105) | up | down | up | 2.5 | −2.1 | 1.5 |
| Nfe2I2 | up | down | up | 2.0 | −1.4 | 2.0 |
| Ppp1r15a (GADD34) | up | down | up | 2.0 | −1.4 | 1.4 |
| Xbp1 (X-box-protein 1) | up | down | up | 1.5 | −1.2 | 1.8 |
| Tra1 | up | down | up | 1.4 | −1.3 | 1.2 |
| Calr (calreticulin) | up | down | up | 1.4 | −1.3 | 1.4 |
| Ddit3 (CHOP) | up | down | up | 1.4 | −1.3 | 5.5 |
| Sp1 | down | up | up | −1.2 | 1.3 | 1.6 |
| Sels | down | up | up | −1.5 | 1.3 | 3.1 |
| Pik3ip1 | down | up | up | −3.0 | 1.4 | 1.8 |
Discussion
Our findings provide the first-ever global description of the UPR transcriptome of the heart and liver following T/HS. We demonstrated that T/HS leads to significant cardiomyocyte and hepatocyte apoptosis, which is prevented through the Stat3-dependent actions of IL-6. We examined the UPR transcriptome to identify candidate gene transcripts responsible for T/HS-induced apoptosis. By utilizing an expanded repertoire of UPR members, both canonical and non-canonical, and the reproducible and measurable outcome of IL-6-preventable apoptosis in our model of T/HS, we were able to identify potential UPR modulators that significantly impact T/HS-induced hepatocyte and cardiomyocyte apoptosis.
In the liver, members of the heat shock family of protein folding chaperones, Hsp70 and Hsp40, emerged as significant potential non-canonical UPR modulators of hepatocyte apoptosis in our model of T/HS. This compares with findings in other models of organ injury, such as work done by Wang et al., which demonstrated that Hsp70 and its induction with geranylgeranylacetone (GGA) can protect against primary proximal tubule apoptosis and acute kidney damage in an ischemic injury model27, and work done by Kuboki et al., which demonstrated in a partial liver I/R model that induction of Hsp70 with sodium arsenite reduced liver injury, as determined by transaminase levels and histology28. Besides their role in protein folding in the cytoplasmic space, heat shock proteins have been linked to the canonical UPR pathways of the endoplasmic reticulum. One example is Hsp72, a Hsp 70 family member, which has been shown to interact with the cytosolic domain of IRE1α, enhancing XBP1 splicing, and attenuating apoptosis in vitro29. Heat shock protein chaperones have also been shown to prevent CHOP-induced apoptosis through the Hsp70-DnaJ chaperone pair inhibiting translocation of Bax to mitochondria in vitro30.
Our findings provide in vivo data linking the heat shock protein family to hepatocyte apoptosis possibly via a Stat3-dependent mechanism in T/HS. These findings are supported by previous work linking IL-6/Stat3 transcriptional regulation of heat shock protein family members31. Hsp70 and Hsp40 appear to contribute to an adaptive and protective process in the liver, demonstrating upregulation in T/HS and further upregulation in livers of IL-6-resuscitated animals, correlating with prevention of apoptosis. However, when animals were pretreated with a Stat3 inhibitor that blocked IL-6's prevention of apoptosis, these chaperone transcripts were downregulated. Thus, these findings suggest that IL-6, via a Stat3-dependent pathway, acts to superinduce Hsp70 and 40 transcripts in T/HS. These findings are supported by the work of Masumichi et al32, which demonstrate that IL-6 is necessary for upregulation of heat shock protein members, including Hsp70/40, in a model of acetaminophen-induced hepatic injury.
Interestingly, the canonical members of the UPR, while altered, were not the most dysregulated transcripts in the liver. CCAAT/-enhancer-binding protein homologous protein (CHOP), PERK, alpha subunit of eukaryotic initiation factor 2 (Eif2α), activating transcription factor 4 (ATF4), and calreticulin were significantly dysregulated (6 to −1.2 fold change) in T/HS (Supplemental Table 1). When considering those entities altered > 2-fold and taking into account known UPR and apoptotic functions of the canonical UPR members, only the transcriptional profile of ATF4 suggested a maladaptive contribution to hepatocyte apoptosis. However, this maladaptive role does not appear to be mediated through Stat3.
The heart demonstrated a distinctly different UPR transcriptional profile in comparison to the liver. When one considers the nature and functions of these organs, this is not unexpected. The liver is the largest glandular mass of tissue in the body and is highly secretory with both exocrine and endocrine function, whereas the heart, with myocyte predominance, is largely non-secretory with maintenance of biophysical function more paramount. The impact of T/HS on the canonical UPR transcriptome was even less in magnitude in the heart than in the liver. Significantly dysregulated canonical UPR transcripts included CHOP, PERK, X-box binding protein 1 (XBP1), Eif2α, and calreticulin (2.5 to −1.2 fold change) with only ATF4 dysregulated by more than 2 fold in response to T/HS (Supplemental Table 2). When taking into account known UPR and apoptotic function, CHOP, XBP1, and GADD34 exhibit transcriptional profiles suggestive of an adaptive role in T/HS-induced cardiomyocyte apoptosis. Given the fold-change was nominal (1.4 to 2-fold) however, further investigation is required to determine their true contribution to T/HS-induced apoptosis.
The protein folding chaperones, Hsp70 and Hsp40, which proved important modulators of apoptosis in the liver, were upregulated in the heart following T/HS, but were downregulated in animals in which IL-6 prevented cardiomyocyte apoptosis, suggesting these chaperones may play a maladaptive role in T/HS-induced cardiomyocyte apoptosis. The dichotomous nature of these chaperones' roles in the liver and heart in T/HS is supported by work in other models of organ injury. Indeed, previous studies have suggested that Hsp70 family proteins may serve to augment cardiac inflammation and contractile dysfunction33,34, and its downregulation in IL-6 treated animals would support this hypothesis, as we have previously demonstrated that IL-6 acts to preserve contractile function following T/HS3. However, Yao et al., have recently shown that Hsp70 upregulation may contribute to the cardioprotection against ischemia/reperfusion injury observed with lipopolysaccharide (LPS) pretreatment35. Thus, the role of Hsp70 in myocardial ischemia/reperfusion injury may be specific to the insult and requires further study to clarify the adaptive versus maladaptive role it may play in ischemia/reperfusion events such as resuscitated hemorrhagic shock.
In addition to providing a global description of the UPR transcriptome of the heart and liver following T/HS, our findings demonstrate that IL-6, when utilized as a resuscitation adjuvant, may augment a physiologic protective role of Hsp70 and Hsp40 via a Stat3-dependent mechanism, thereby protecting against hepatocyte apoptosis. These findings support the concept that modulators of Hsp70 or 40 may offer a therapeutic strategy for prevention of apoptosis and ultimately hepatic dysfunction following T/HS.
Methods
Rat T/HS protocol
For the rat experiments in this study, 8-week old male Sprague-Dawley rats (200–250 gm) were used. Rats were subjected to the sham or T/HS protocols, as described3,6,23 with modifications. Blood was withdrawn into a heparinized syringe to achieve and then maintain the target MAP at 35 mmHg until blood pressure compensation failed. Blood was then returned as needed to maintain the target MAP. The amount of shed blood returned (SBR) defined shock severity as reflected in the duration of hypotension, and the animals used in this analysis received 50% SBR (SBR50; duration of hypotension, 273 ± 24.9 minutes). At the end of the hypotensive period, rats were resuscitated as described3,6,23 and humanely sacrificed 60 minutes after the start of resuscitation in order to capture the first wave of transcriptional changes. Where indicated, rats received 10 μg/kg of recombinant human IL-6 in 0.1 ml PBS at the initiation of the resuscitation or PBS alone. Sham rats were anesthetized and cannulated for 250 minutes but were not subjected to hemorrhage or resuscitation. Rat livers and hearts were harvested immediately after sacrifice and snap frozen in liquid nitrogen for nucleosome and RNA extraction steps.
In vivo pharmacological inhibition of Stat3
To achieve pharmacological inhibition of Stat3 activity within the rats, the G-rich, quartet-forming oligodeoxynucleotides (GQ-ODN), T4021424 (2.5 mg ODN/kg) was given by tail vein injection, 24 hours prior to subjecting them to the SBR50 protocol with IL-6 treatment. The half-life of T40214 in tissues is ≥ 48 hours25.
Nucleosome ELISA
Levels of histone-associated DNA fragments (nucleosomes) were determined in homogenates of snap-frozen liver using an ELISA method (Cell Death Detection ELISAplus; Roche Diagnostics, Manheim, Germany), as described6,23. The nucleosome concentration for each liver sample was normalized for total protein concentration determined by Bradford assay (Bio-Rad Protein Assay, Bio-Rad Laboratories, Inc., Hercules, CA). The final nucleosome concentration for each liver sample was the average of duplicate determinations.
Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) staining
TUNEL staining to enzymatically detect the free 3′-OH DNA termini was performed using the ApopTag Plus Peroxidase in situ Apoptosis Detection Kit from Chemicon International (now Millipore, Billerica, MA). Slides were rehydrated from xylene to PBS through a series of decreasing concentrations of ethanol and digested in proteinase K (20 μg/ml) for 3 minutes at 23°C. Endogenous peroxidases were quenched for 30 minutes in 3% hydrogen peroxide in PBS. TdT enzyme was diluted in TUNEL solution buffer then used as suggested by the manufacturer. Slides were counterstained with hematoxyllin. TUNEL positive cells were assessed microscopically by counting the total nuclei and the number of TUNEL-positive nuclei in twenty random 1000× fields by an experienced histologist, blinded to the treatment each rat received. Data is presented as the number of TUNEL positive cells per high power field (hpf).
RNA isolation and oligonucleotide microarray hybridization
Total RNA was isolated from 4–5 micron cryotome sections of liver using TRIzol® Reagent (Invitrogen, Carlsbad, California) single step RNA isolation protocol followed by purification with RNeasy® Mini Kit (QIAGEN, Hilden, Germany) as instructed by the manufacturer. Gene expression profiling was performed with the Affymetrix Rat Array RAE 230A chips following Affymetrix protocols used within the Baylor College of Medicine Microarray Core Facility.
Microarray analysis
We used GenespringGX (Agilent Technologies Inc., Santa Clara CA) software package for quality assessment, statistical analysis and annotation. Low-level analyses included background correction, quartile normalization and expression estimation using RMA-based analysis within Genespring. One-way analysis of variance (ANOVA) with contrasts was used for group comparisons on all genes and on the list of UPR entities. P-values were adjusted for multiple comparisons using the Benjamini-Hockberg method. The adjusted p-values represent false discovery rates (FDR) and are estimates of the proportion of “significant” genes that are false or spurious “discoveries”. We used a FDR = 5% as cut-off. The genechip used, RAE 230A, contained 15,923 probe sets representing 13,521 annotated genes or expressed sequence tags. A UPR gene entity list was created using both Ingenuity Pathway Analysis (IPA® Redwood City, CA) and the Gene Ontology Database©, with keywords “endoplasmic reticulum stress, unfolded protein response”. Three or more chips for each organ were hybridized using mRNA isolated from hearts and livers, respectively for each group comparison: Sham (4), T/HS-PBS (4) and T/HS-IL6 (4) and T/HS-IL6-GQ (3) groups.
Quantitative (Q) RT-PCR
Two-step Q-RT-PCR was performed using the ABI Prism 7700 sequence detection system (Perkin-Elmer/Applied Biosystems, Foster City, CA) as described previously3,26. Briefly, total RNA (1 μg) was reverse transcribed using reverse transcription reagents (BioRad catalog no. 170-8842; Hercules, CA); 20% of each RT reaction was used in duplicate PCR reactions using TaqMan® Universal Master Mix II, with uracil N-glycosylase (PN 4440038) and specific primer and probe sets designed by the manufacturer (TaqMan Gene Expression Assay, Applied Biosystems, Darmstadt, Germany)—Hsp70 (Hspa1a; catalog no. Rn04224718_u1), Hsp40 (Dnajb1; catalog no. Rn 01426952_g1), and 18S rRNA (catalog no.Rn03928990_g1). Each PCR amplification run consisted of incubation for 5 min at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The cycle threshold of each duplicate determination was normalized by subtraction of the cycle threshold for its corresponding 18S rRNA (ΔCT). Each ΔCT was then calibrated by subtracting the ΔCT value for control rat tissue (ΔΔCT). RNA amount was expressed as relative units calculated as 2− ΔΔCT, as described26.
Statistical analysis
Statistical differences between experimental groups were analyzed using one-way ANOVA and post-hoc analysis was performed using Student-Newman-Keuls test. T-test analysis performed using unpaired Student's T-test.
Author Contributions
S.T. and D.T. contributed to experimental design. S.T. and A.A. performed animal hemorrhagic shock protocols. S.T., P.R. and D.T. contributed to all tables. S.T. and D.T. contributed to figure 1. S.T., D.T. and P.R. contributed to figure 2, S.T. and D.T. wrote the main manuscript text. All authors reviewed the manuscript.
Acknowledgments
Supported, in part, by grant R01 HL07619 (DJT) from the National Heart, Lung and Blood Institute of the National Institutes of Health, grant DM090899 (DJT) from the Department of Defense, and grant T32 AI055413 (SAT) from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.
References
- Minino A., Anderson R., Fingerhut L., Boudreault M. & Warner M. Deaths: injuries, 2002. Natl Vital Stat Rep 54, 1–124 (2006). [PubMed] [Google Scholar]
- Dewar D., Moore F. A., Moore E. E. & Balogh Z. Postinjury multiple organ failure. Injury 40, 912–918 (2009). [DOI] [PubMed] [Google Scholar]
- Alten J. et al. Prevention of hypovolemic circulatory collapse by IL-6 activated Stat3. PLoS One 3, e1605 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J., Jin D.-D., Jin L.-J. & Lu Q. Apoptosis in organs of rats in early stage after polytrauma combined with shock. J Trauma 52, 104–111 (2002). [DOI] [PubMed] [Google Scholar]
- Jaskille A. et al. Hepatic apoptosis after hemorrhagic shock in rats can be reduced through modifications of conventional Ringer&'s solution. Journal of the American College of Surgeons 202, 25–35 (2006). [DOI] [PubMed] [Google Scholar]
- Moran A. et al. Prevention of trauma and hemorrhagic shock-mediated liver apoptosis by activation of stat3alpha. Int J Clin Exp Med 1, 213–247 (2008). [PMC free article] [PubMed] [Google Scholar]
- Sundar S. V., Li Y.-Y., Rollwagen F. M. & Maheshwari R. K. Hemorrhagic shock induces differential gene expression and apoptosis in mouse liver. Biochemical and Biophysical Research Communications 332, 688–696 (2005). [DOI] [PubMed] [Google Scholar]
- Cox J. S., Shamu C. E. & Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 (1993). [DOI] [PubMed] [Google Scholar]
- Mori K., Ma W., Gething M. J. & Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756 (1993). [DOI] [PubMed] [Google Scholar]
- Normington K., Kohno K., Kozutsumi Y., Gething M. J. & Sambrook J. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57, 1223–1236 (1989). [DOI] [PubMed] [Google Scholar]
- Shamu C. E. & Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. The EMBO journal 15, 3028–3039 (1996). [PMC free article] [PubMed] [Google Scholar]
- Dara L., Ji C. & Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology (Baltimore, Md) 53, 1752–1763 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawless M. W. et al. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. Journal of immunology (Baltimore, Md: 1950) 172, 5722–5726 (2004). [DOI] [PubMed] [Google Scholar]
- Malhi H. & Kaufman R. J. Endoplasmic reticulum stress in liver disease. Journal of hepatology 54, 795–809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K., Herzig S., Kulkarni R. N. & Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300, 1574–1577 (2003). [DOI] [PubMed] [Google Scholar]
- Ota T., Gayet C. & Ginsberg H. N. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest 118, 316–332 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly-Maitre B. et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America 103, 2809–2814 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakon M., Ariyoshi H., Umeshita K. & Monden M. Ischemia-reperfusion injury of the liver with special reference to calcium-dependent mechanisms. Surgery today 32, 1–12 (2002). [DOI] [PubMed] [Google Scholar]
- Duvigneau J. C. et al. Reperfusion does not induce oxidative stress but sustained endoplasmic reticulum stress in livers of rats subjected to traumatic-hemorrhagic shock. Shock (Augusta, Ga) 33, 289–298 (2010). [DOI] [PubMed] [Google Scholar]
- Jian B. et al. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim Biophys Acta 1782, 621–626 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S., Thuerauf D. J., Marcinko M. C., Belmont P. J. & Glembotski C. C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem 284, 29735–29745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H. Y. et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 122, 361–369 (2010). [DOI] [PubMed] [Google Scholar]
- Moran A. et al. IL-6-Mediated Activation of Stat3α Prevents Trauma/Hemorrhagic Shock-Induced Liver Inflammation. PLoS One 6, e21449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing N. et al. G-quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res 64, 6603–6609 (2004). [DOI] [PubMed] [Google Scholar]
- Jing N., Sha W., Li Y., Xiong W. & Tweardy D. J. Rational drug design of G-quartet DNA as anti-cancer agents. Curr Pharm Des 11, 2841–2854 (2005). [DOI] [PubMed] [Google Scholar]
- Ono M., Yu B., Hardison E. G., Mastrangelo M.-A. A. & Tweardy D. J. Increased susceptibility to liver injury after hemorrhagic shock in rats chronically fed ethanol: role of nuclear factor-kappa B, interleukin-6, and granulocyte colony-stimulating factor. Shock (Augusta, Ga) 21, 519–525 (2004). [DOI] [PubMed] [Google Scholar]
- Wang Z. et al. Induction of heat shock protein 70 inhibits ischemic renal injury. Kidney Int 79, 861–870 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboki S. et al. Role of heat shock protein 70 in hepatic ischemia-reperfusion injury in mice. Am J Physiol Gastrointest Liver Physiol 292, G1141–9 (2007). [DOI] [PubMed] [Google Scholar]
- Gupta S. et al. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol 8, e1000410 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T., Terada K., Oyadomari S. & Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ 11, 390–402 (2004). [DOI] [PubMed] [Google Scholar]
- Stephanou A. & Latchman D. S. Transcriptional regulation of the heat shock protein genes by STAT family transcription factors. Gene expression 7, 311–319 (1999). [PMC free article] [PubMed] [Google Scholar]
- Masubuchi Y. et al. Role of interleukin-6 in hepatic heat shock protein expression and protection against acetaminophen-induced liver disease. Biochemical and Biophysical Research Communications 304, 207–212 (2003). [DOI] [PubMed] [Google Scholar]
- Mathur S., Walley K. R., Wang Y., Indrambarya T. & Boyd J. H. Extracellular Heat Shock Protein 70 Induces Cardiomyocyte Inflammation and Contractile Dysfunction via TLR2. Circulation journal : official journal of the Japanese Circulation Society (2011). [DOI] [PubMed] [Google Scholar]
- Zou N. et al. Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia-reperfusion. Am J Physiol Heart Circ Physiol 294, H2805–13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.-W. et al. Lipopolysaccharide pretreatment protects against ischemia/reperfusion injury via increase of HSP70 and inhibition of NF-κB. Cell Stress & Chaperones 16, 287–296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]