


Abstract
Besides the primary histone acetyltransferase (HAT)-mediated chromatin remodeling function, co-transcriptional factor, p300, is also known to play a distinct role in DNA repair. However, the exact mechanism of p300 function in DNA repair has remained unclear and difficult to discern due to the phosphorylation and degradation of p300 in response to DNA damage. Here, we have demonstrated that p300 is only degraded in the presence of specific DNA lesions, which are the substrates of nucleotide excision repair (NER) pathway. In contrast, DNA double-strand breaks fail to degrade p300. Degradation is initiated by phosphorylation of p300 at serine 1834, which is catalyzed by the cooperative action of p38 mitogen-activated protein kinases and Akt kinases. In depth, functional analysis revealed that (i) p300 and CBP act redundantly in repairing ultraviolet (UV) lesions, (ii) the phosphorylation of p300 at S1834 is critical for efficient removal of UV-induced cyclobutane pyrimidine dimers and (iii) p300 is recruited to DNA damage sites located within heterochromatin. Taken together, we conclude that phosphorylated p300 initially acetylates histones to relax heterochromatin to allow damage recognition factors access to damage DNA. Thereupon, p300 is promptly degraded to allow the sequential recruitment of downstream repair proteins for successful execution of NER.
INTRODUCTION
Of all the DNA repair systems, nucleotide excision repair (NER) is the most versatile in terms of lesion recognition. NER deals with a wide variety of helix-distorting lesions that interfere with base pairing and genetically impede transcription and normal replication. The most familiar substrates of NER include ultraviolet (UV)-induced cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PP) as well as cisplatin- and Benzo[a]pyrene Diol Epoxide-induced bulky adducts (1). It is believed that in mammalian cells, NER is mediated by the sequential assembly of repair proteins at the site of DNA lesions (2–4). Given the fact that the genomic DNA is tightly packaged through histone and non-histone proteins into chromatin, the cellular repair machinery has to circumvent this structural barrier to gain access to the deeply embedded damaged site. Generally, diverse histone modifications, especially lysine acetylation, help orchestrate the accessibility of DNA damage within chromatin (5).
p300 and CBP are general transcriptional co-activators and known to control many biological activities, e.g. cellular growth and differentiation, tumorigenesis and apoptosis. As a result, p300 and/or CBP dysfunction have been implicated in numerous disease processes, including several forms of cancers and cardiac hypertrophy (6,7). Although p300 and CBP have their unique functions, the high degree of homology between p300 and CBP suggests that these proteins could be functionally redundant in orchestrating the cognate cellular activities [Reviewed in (8)]. The inherent histone acetyltransferase (HAT) activity allows p300 and CBP to influence chromatin structure through histone modifications and impacting transcription (9). In addition, the HAT activity of p300 and CBP is also involved in DNA repair (10,11). It has been reported that p300 is recruited to double-strand break (DSB) sites. Ablation of p300 suppressed acetylation of histones H3 and H4 and co-suppressed the recruitment of key proteins of the non-homologous end joining (NHEJ) process (10). p300 was also suggested to play a role in NER through the p53-dependent recruitment to NER sites for chromatin relaxation (11). However, the exact role of p300 in DNA repair is still unclear and is further complicated by the phosphorylation and prompt degradation of p300 in response to exposures to various DNA-damaging agents.
Although p300 is phosphorylated in a cell-cycle-dependent manner during differentiation and in response to cytokines and extracellular cues, phosphorylated p300 can be detected in both quiescent and proliferating cells (12,13). p300 harbors several different phosphorylation sites available to extracellular signal-activated kinases including protein kinase A (PKA), PKC, Akt and mitogen-activated protein kinases (MAPK) (13,14). Site-specific p300 phosphorylations, besides resulting in distinct structural and functional alterations, also regulate p300 stability. For example, PKC- and salt-inducible kinase 2-mediated p300 phosphorylation at serine 89 represses intrinsic HAT activity (15,16), while phosphorylation at serine 1834 by Akt enhances HAT activity (17,18). In addition, p44/p42 MAPK-mediated p300 phosphorylation on the C-terminus (Ser-2279, Ser-2315 and Ser-2366) was also reported to stimulate its HAT activity (19). The interaction and phosphorylation by Akt help maintain cellular p300 steady-state levels (20,21). In contrast, the interaction with p38 MAPK resulted in p300 degradation (22). p300 phosphorylation and degradation are also functionally linked to DNA damage response. However, these linkages vary in response to treatments with different DNA-damaging agent. For instance, the ATM-mediated phosphorylation of serine 106 of p300 was found to be important for stabilization of p300 proteins in response to DSB-inducing ionizing radiation (IR) and Etoposide (23). However, Doxorubicin, a topoisomerase II poison also causing DSB, induced phosphorylation-dependent degradation of p300 in cardiomyocytes (22,24). Similarly, the DNA-crosslink inducer cisplatin also stimulated p300 degradation (25,26). Therefore, the distinguishing functional links between specific phosphorylation events and p300 in DNA damage repair remain largely unknown.
In this study, we have demonstrated that p300 is degraded only in response to DNA lesions that serve as substrates for the NER pathway. In contrast, DSB did not cause p300 degradation but increased the level of p300. In addition, DNA damage-induced p300 degradation paralleled p300 phosphorylation at serine 1834, which is mediated by both p38 MAPK and Akt. Furthermore, inhibition of either p38 or Akt blocked both p300 phosphorylation at serine 1834 as well as p300 degradation. The subsequent study indicated that p300 and CBP function redundantly in NER, with p300 phosphorylation at serine 1834, but not p300 itself, being critical to the efficient removal of UV-induced CPD and the recruitment of DDB2 to damage sites, especially in the heterochromatic regions of the genome.
MATERIALS AND METHODS
Cell culture and treatment
Normal human fibroblasts (NHFs) OSU-2 cells were established and maintained in culture as described earlier (27). Li-Fraumeni Syndrome MDAH 041 cells were kindly provided by Dr Michael Tainsky (MD Anderson Cancer Center). Both cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in a humidified atmosphere of 5% CO2. The primary NER-deficient fibroblasts XP-A (GM05509), XP-D (GM03248), XP-F (GM04313) and XP-E (GM01389) were purchased from NIGMS Human Genetic Cell Repository (Coriell Institute for Medical Research, Camden, NJ, USA) and were grown in minimum essential medium (MEM) with 2× essential amino acid, 2× non-essential amino acid and 2× vitamin supplemented with 15% FBS and antibiotics at 37°C in a humidified atmosphere of 5% CO2. For UV exposure, the cultures were washed with phosphate-buffered saline (PBS) and irradiated with UV followed by incubation for varying times. UV-C light (254 nm) was delivered from a germicidal lamp at a dose rate of 0.5 J/m2/s, as measured by a UVX digital radiometer connected to a UVX-31 sensor (UVP, Inc., Upland, CA, USA). For cisplatin treatment, cells were maintained in medium with the desired doses of cisplatin (Sigma, St Louis, MO, USA) for 1 h, and then washed with PBS and followed by incubation in fresh cisplatin-free medium for varying times post-treatment. For IR treatment, X-ray was delivered to cultured cells in a RS-2000 X-ray Biological Irradiator (Rad Source Technologies, Inc., Suwanee, GA, USA). For Etoposide treatment, cells were cultured in medium containing 5 µM of Etoposide (Sigma) for various time periods. To inhibit the activity of 26S proteasome, p38 MAPK or Akt, cells were first cultured in medium containing MG132 (Calbiochem, Billerica, MA, USA), SB203580 or LY294002 (Cell signaling Technology, Danvers, MA, USA) for 1 h, then treated with UV and further cultured in the medium containing inhibitors for various time periods.
Plasmids, siRNA and cell transfection
The plasmid encoding full-length p300-WT was kindly provided by Dr David Livingston (Harvard Medical School, Boston, MA, USA). The plasmid encoding p300S1834A mutant was kindly provided by Dr David Jones (University of Virginia) and Dr Terry Unterman TG (University of Illinois at Chicago). The plasmids were transfected into cells using FuGENE 6 (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s instruction. siRNA SMARTpools designed to target human p300 (M-003486) or human CBP (M-003477) and scramble non-targeting siRNA (5′-UUC UCC GAA CGU GUC ACG UdTdT-3′) were purchased from Dharmacon Inc. (Denver, CO, USA). 50 or 100 nM siRNA was transfected into cells using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacture’s instruction.
Immunoblotting and immunoprecipitation
Whole cell lysates were prepared by boiling cell pellets for 10 min in lysis buffer [2% sodium dodecyl sulfate (SDS), 10% glycerol, 62 mM2–4Tris–HCl, pH 6.8 and a complete mini-protease inhibitor cocktail (Roche Applied Science)]. After protein quantification with Bio-Rad Dc Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA), equal amounts of proteins were loaded, separated on a polyacrylamide gel and transferred to a nitrocellulose membrane. Protein bands were immuno-detected with appropriate antibodies, e.g. rabbit anti-p300 (N-15), rabbit anti-phospho-p300(S89), mouse anti-CBP, rabbit anti-HP1α, rabbit anti-HMGB1, rabbit anti-SNF5, mouse anti-Actin, mouse anti-Tubulin and goat anti-Lamin B from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA); rabbit anti-phospho-p300(S1834) from Thermo Scientific (Rockford, IL, USA); rabbit anti-phospho-MK2 and rabbit anti-phospho-Akt(S473) from Cell Signaling Technology and goat anti-DDB2 from R & D Systems (Minneapolis, MN, USA). For co-immunoprecipitation (co-IP), cells were lysed in RIPA buffer [50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% NP-40 and protease inhibitor cocktail] for 30 min at 4°C. After centrifugation at 14 000 rpm for 10 min, supernatant was collected for IP. One milligram of total protein was incubated with 2 µg of mouse anti-p300 antibody (clone RW128, Millipore, Billerica, MA, USA) or normal mouse IgG overnight at 4°C with continuous rotation. The immunoprecipitates were recovered after incubation for 2 h with 30 µl of Protein G plus/Protein A agarose beads (Calbiochem) and centrifugation. Western blot analysis was performed to visualize the presence of p300 and phospho-Akt.
Immuno-slot blot analysis
The extent of initial dimer formation and the damage remaining in DNA after cellular repair were quantified using non-competitive Immuno-slot blot (ISB) assay, essentially as described earlier (28,29). Briefly, OSU-2 cells were grown to confluence and further cultured in serum-free medium for 48 h. Cells were UV irradiated and further cultured in serum-free medium for the desired time periods. The nuclei were isolated and treated with RNase for 1 h. The genomic DNA was then isolated with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with ethanol and quantified using PicoGreen kit assay (Invitrogen). The same amounts of denatured DNA were applied to nitrocellulose membranes by mild vacuum blotting. UV-induced CPD and 6-4PP were detected with their corresponding antibodies (MBL International Corporation, Woburn, MA, USA). The intensity of each band was quantified, and the lesion concentrations were determined from a reference standard run in parallel to calculate the relative amounts of DNA lesions remaining at each time point.
Immunofluorescent staining
OSU-2 cells growing on glass coverslips were either transfected with siRNA or p300-expressing plasmids for 48 h. The cells were then washed with PBS and UV irradiated at 40 J/m2 through an isopore polycarbonate filter (Millipore, Bedford, MA, USA), containing pores of 5 µm diameter, as described previously (30). The cells were then double stained with mouse anti-DDB2 (ab51017, 1:100; Abcam, Cambridge, MA, USA) and rabbit anti-CPD antibodies (31). Fluorescence images were obtained with a Nikon Fluorescence Microscope E80i (Nikon, Tokyo, Japan) fitted with appropriate filters for FITC and Texas Red. The digital images were then captured with a cooled CCD camera and processed with the help of its SPOT software (Diagnostic Instruments, Sterling Heights, MI, USA). The intensity of DDB2 was analyzed in 100 separate foci by using ImageJ (NIH) and normalized to the intensity of CPD.
Chromatin segregation assay
Chromatin fractionation was based on the sensitivity to micrococcal nuclease (MNase) treatment as described by Goodarzi et al. (32) with modifications. Briefly, cells were lysed in low-salt buffer (10 mM HEPES, pH 7.4, 25 mM KCl, 10 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA, 0.5% NP-40 and protease inhibitor) for 10 min at 4°C. After centrifugation at 10 000 rpm for 5 min, the supernatant was saved as S10. The pellets were resuspended in high-salt (HS) buffer (50 mM Tris–HCl, pH 8.0, 1 mM EDTA, 10 mM MgCl2, 300 mM KCl and protease inhibitor) and immediately centrifuged at 10 000 rpm for 5 min. The supernatant is P10. The pellets were then resuspended in MNase buffer [10 mM HEPES, pH 7.9, 10 mM KCl, 1 mM CaCl2, 1.5 mM MgCl2, 0.35 M sucrose, 10% glycerol, 0.1% Triton X-100 and 1 mM dithiothreitol (DTT)], containing MNase (New England BioLabs, Ipswich, MA, USA) and incubated at 37°C for 30 min before an equal volume of solubilization buffer (MNase buffer + 2% NP40, 2% Triton X-100 and 600 mM NaCl) was added. Samples were mixed and centrifuged at 10 000 rpm for 5 min. The supernatant is C1. The remaining pellet was resuspended in MNase buffer and sonicated (C2). Ten percent of each fraction was precipitated with 10% trichloroacetic acid and redissolved in SDS loading buffer for western blot analysis.
RESULTS
p300 is degraded upon UV or cisplatin exposures, but not following treatment with IR or Etoposide
We have previously demonstrated that the cellular levels of p300 protein decreased appreciably upon cisplatin treatment in mouse embryonic fibroblasts (25). To extend this further and assess if other DNA-damaging agents also induce a similar decrease in p300 levels, cell lysates from NHF OSU-2 cells exposed to UV, cisplatin, IR or Etoposide were analyzed by western blotting for p300 protein. As reported earlier, the level of p300 protein showed the expected decrease immediately after 1 h treatment of cisplatin, and near full recovery to the initial levels at around 48 h post-exposure (Figure 1A). Analogous to this, UV irradiation also caused the reduction of p300 level, albeit this decrease peaked at ∼8 h of exposure and the restoration of p300 to its initial level occurred comparatively earlier at 24 h post-exposure (Figure 1A). This exact temporal pattern and degree of UV-induced p300 decrease were also observed in NIH 3T3 cells (Supplementary Figure S1). Surprisingly, however, we failed to find any p300 reduction following IR or Etoposide treatments of OSU-2 cells. Moreover, in contrast to the results with UV and cisplatin exposures, the level of p300 protein actually increased in response to IR or Etoposide treatments (Figure 1B). These data indicate that universally all DNA-damaging agents cannot result in an attenuation of cellular p300. Given that both cisplatin-induced DNA crosslinks and UV-induced DNA dimers are primarily repaired by NER pathway, while IR- and Etoposide-induced DNA DSBs are primarily repaired by homologous recombination (HR) and NHEJ, it is very likely that p300 down-regulation correlates strictly with lesion processing by NER. Moreover, to test if the decreased levels of p300 protein observed in UV-exposed cells were due to active degradation via ubiquitin-proteasomal system, OSU-2 cells were pretreated with proteasome inhibitor MG132 and analyzed for altered levels of p300 protein. As shown in Figure 1C, MG132 treatment blocked the UV-induced p300 reduction found at the 8 h time point, indicating that decreased p300 levels result from protein degradation through the 26S proteasome. Taken together, these results suggest that proteasome-mediated p300 degradation could be involved in the processing of DNA lesions by the NER pathway.
Figure 1.
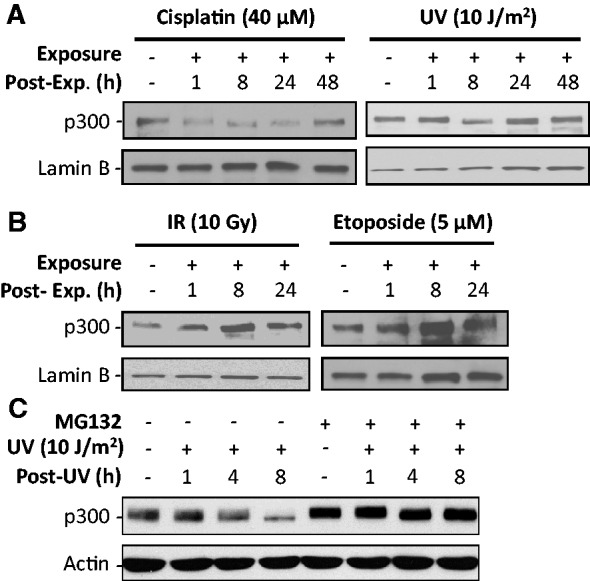
p300 is degraded upon UV or cisplatin, but not IR or Etoposide, treatments. (A and B) OSU-2 cells were exposed to UV, cisplatin (A), IR or Etoposide (B). The p300 level was detected by using western blotting. (C) OSU-2 cells were pre-treated with MG132 for 1 h, UV irradiated and the p300 protein was detected at the desired time point.
UV-induced p300 degradation is an early NER event, upstream of DDB2 recruitment to damage
p300 is already known to play an important role in the NER pathway (11). In order to understand whether p300 degradation is a consequence of NER-mediated damage processing, here we have undertaken a rigorous analysis of p300 protein following UV or IR treatments of various cell lines having a known NER-related defect (Figure 2A–C). UV-induced damage-dependent degradation of p300 was dramatically pronounced in all the NER-deficient XP-A, XP-D and XP-F cells. Thus, the trigger for p300 degradation must emanate from a processing event upstream of DNA lesion recognition and dual incision step of the NER pathway. Interestingly, in contrast to the NER-proficient cells, original p300 protein level in NER-deficient cells was not restored even after 48 h of UV irradiation. Most likely, this is attributable to the transcriptional repression accompanying unrepaired DNA lesions in the genomes of repair-deficient cells (33,34). Thus, failure of protein replenishment through normal transcription made the UV-induced p300 degradation more prominent as indicted by the complete disappearance of p300 at 24 and 48 h following UV irradiation of NER-deficient XP-A, XP-D and XP-F cells. Once again, as shown in Figure 1B, IR treatment failed to induce any p300 degradation in these NER-deficient cell lines, further reaffirming the inability of DSB processing to influence p300 degradation. Like other XP cell lines, the absence of DDB2 protein is responsible for the repair deficiency of the XP-E phenotype. However, DDB2 functions as the most upstream factor involved in the initial damage recognition. Besides, DDB2 is considered as a chromatin accessibility factor owing to its capacity to bind p300 (35). To investigate the impact of DDB2 on p300 degradation, we analyzed the p300 protein levels in DDB2-deficient XP-E cells upon UV irradiation. In this case, however, the results were similar to the repair-proficient NHF as DDB2 deficiency did not affect the ability of these cells vis-à-vis p300 degradation observed after 8 h UV post-exposure (Figure 2D). Due to the mild NER deficiency of XP-E cells and no impairment in transcriptional coupled repair (36), the transcriptional repression by unrepaired DNA lesions in XP-E cells is not as prominent as other XP cells. Thus, p300 protein level can readily recover after 24 h of UV irradiation. Finally, it has been reported that p53 mediates the p300 recruitment to NER sites (11). We also determined the effect of UV irradiation on p300 protein levels in p53-deficient 041 cells. Again, the results were similar to NER-proficient cells as UV irradiation instigated the typical p300 degradation in the absence of p53 (Figure 2E). Therefore, we conclude that UV-induced p300 degradation occurs earlier in the NER process and is independent of the initial damage recognition factor DDB2 or p53 protein.
Figure 2.
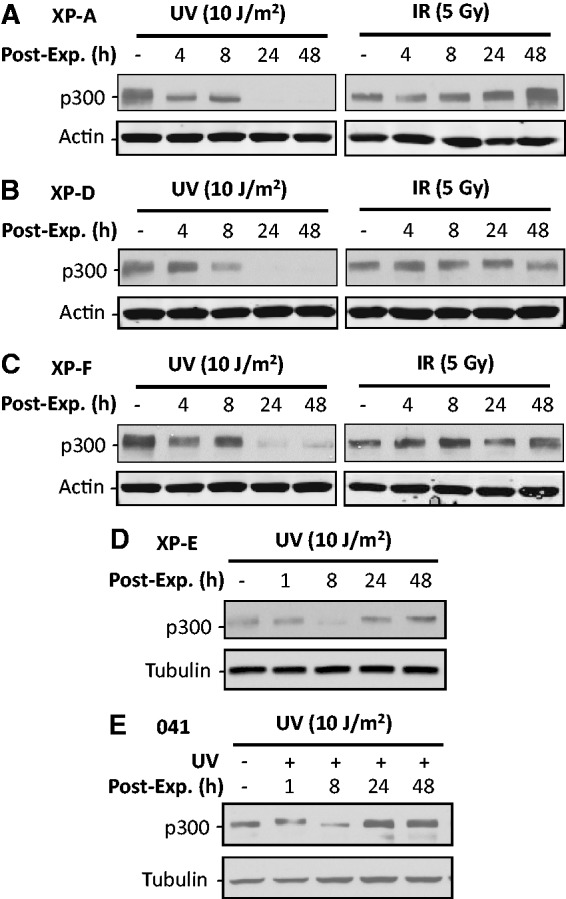
NER or DDB2 are not required for UV-induced p300 degradation. (A–C) NER-deficient XP-A (A), XP-D (B) and XP-F (C) cells were irradiated with either UV or IR. The p300 protein level was detected at various post-exposure times. (D and E) DDB2-deficient XP-E cells (D) and p53-deficient MDAH041 cells (E) were UV irradiated and further cultured for various time. The p300 protein level was detected using western blotting.
p300 is phosphorylated at serine 1834 by p38 MAPK and Akt following cellular UV irradiation
The stability of p300 protein is believed to be regulated by its phosphorylation [Reviewed in (37)]. Multiple site-specific serine/threonine phosphorylations have been identified in p300 and are linked to its pleiotropic potential (15,19,38–41). To discern the underlying basis of p300 phosphorylation in response to UV damage, we irradiated NHF cells and analyzed p300 phosphorylations at serine 89 and serine 1834 by western blots with antibodies against site-specifically phosphorylated p300. As shown in Figure 3A, phospho-p300(S1834) increased while phospho-p300(S89) decreased as a function of time after UV irradiation. We then pretreated the cells with MG132 to inhibit proteasome activity and determined the changes of phospho-p300 protein in the absence and presence of irradiation insult. Inhibition of proteasomal protein degradation resulted in further enhancement of the UV-induced elevated levels of phospho-p300(S1834) (Figure 3B). Conversely, MG132 treatment did not influence the UV-induced reduction of phospho-p300(S89). Thus, phosphorylation of p300 at S1834 appears to be specifically related to the characteristic proteasome-dependent, UV-induced p300 degradation. More importantly, the UV-induced reduction of phospho-p300(S89) is not a consequence of proteasome-mediated degradation, but most likely an unrelated event of dephosphorylation. To identify the kinases that are responsible for UV-induced phosphorylation of p300 at S1834, we tested the function of p38 MAPK and Akt in mediating p300 phosphorylation and degradation by using their specific metabolic inhibitors, SB203580 (42) and LY294002 (43). The phosphorylation status of MK2 (pMK2) and Akt (pAkt) was utilized to determine the effects on p38 and Akt inhibition, respectively. Both p38 and Akt inhibitors caused a discernible reduction of UV-induced phospho-p300(S1834) levels, especially at 8 h time point (Figure 3C and D). More importantly, both inhibitors concomitantly blocked the characteristic p300 degradation observed at 8 h post-irradiation. Given that LY294002 is not a specific inhibitor for Akt, we examined the effect of siRNA-mediated down-regulation of Akt on UV-induced p300 phosphorylation and degradation. Similar to the Akt inhibitor, knockdown of Akt also decreased UV-induced p300 phosphorylation at S1834 as well as blocked the p300 degradation (Supplementary Figure S2). These results indicate that UV-induced phosphorylation of p300 at S1834 is catalyzed by both p38 MAPK and Akt, which appears to be instrumental in the degradation of p300 following cellular UV irradiation.
Figure 3.
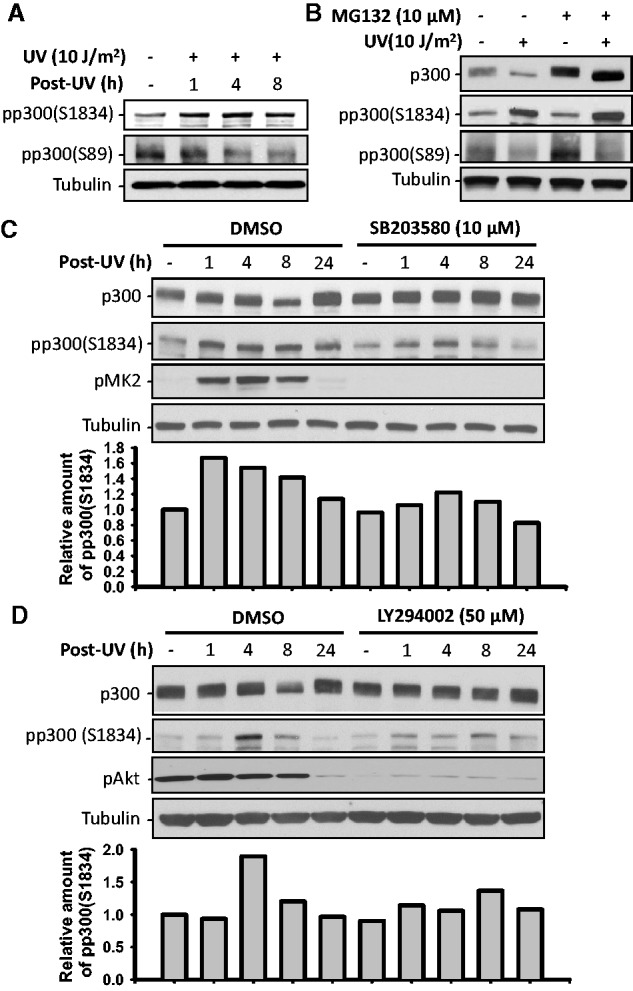
p300 is phosphorylated at S1834 upon UV irradiation by p38 MAPK and Akt. (A) OSU-2 cells were UV irradiated and further cultured for various time periods. p300 phosphorylation was detected using western blotting with anti-phospho-p300(S1834) or anti-phospho-p300(S89) antibodies. (B) OSU-2 cells were pretreated with MG132 for 1 h, UV irradiated and further cultured for 8 h, the p300 and phospho-p300 proteins were detected as (A). (C and D) OSU-2 cells were pretreated with either p38 MAPK inhibitor SB203580 (C) or Akt inhibitor LY294002 (D) for 1 h, UV irradiated at 10 J/m2 and further cultured in media containing inhibitors for various time periods. Whole cell lysates were prepared and subjected to western blotting to detect p300 and phospho-p300. pMK2 and pAkt were detected to show the inhibition of p38 MAPK and Akt activity, respectively. The intensity of phospho-p300(S1834) in each sample was quantified and normalized to Tubulin, the relative amount of phospho-p300(S1834) was calculated compared with control sample and plotted below each immunoblotting image.
Since both p38 MAPK and Akt are required for UV-induced p300 phosphorylation at S1834, it may be reasoned that simultaneous inhibition of both kinases would cause greater reduction of UV-induced phospho-p300(S1834). However, dual treatment of cells with p38 MAPK and Akt inhibitors exhibited inhibitory effect on UV-induced phospho-p300(S1834) comparable to that of single inhibitor (Figure 4A). This indicates that p38 MAPK and Akt are acting in tandem to phosphorylate p300. Earlier it has been reported that Akt interacts directly and phosphorylates p300 at S1834 (17,18). To understand whether p38 MAPK promotes p300 phosphorylation by facilitating the interaction between activated pAkt and p300, we pretreated OSU-2 cells with p38 MAPK inhibitor SB203580 or Dimethyl sulfoxide (DMSO) and followed it with UV irradiation at 10 J/m2. The IP analysis of cellular lysates obtained at 4 h post-irradiation showed the expected interaction between pAkt and p300 was dramatically decreased under conditions of p38 MAPK inhibition. The combined data suggest that activated p38 MAPK facilitates the interaction between p300 and activated Akt, which then catalyzes the causal phosphorylation of p300 at S1834.
Figure 4.
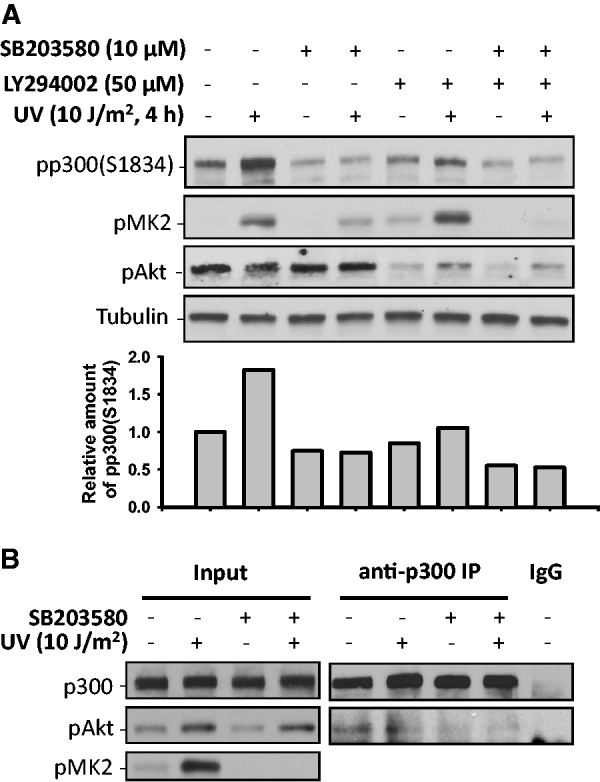
p38 MAPK and Akt cooperate to phosphorylate p300 at S1834 in response to UV irradiation. (A) OSU-2 cells were pretreated with either p38 inhibitor, Akt inhibitor or both for 1 h. Cells were subsequently UV irradiated at 10 J/m2, and further cultured for 4 h. The phospho-p300 at S1834, pMK2 and pAkt were detected using western blotting. The intensity of phospho-p300(S1834) was quantified as in Figure 3. (B) OSU-2 cells were pretreated with 10 µM of p38 inhibitor SB203580 for 1 h, UV irradiated at 10 J/m2 and further cultured for another 1 h. Whole cell extracts were prepared and subjected to IP with anti-p300 antibody or normal IgG. pAkt in the immunoprecipitates was detected using western blotting. pMK2 was detected in Input to show the inhibition of p38 MAPK activity.
p300 and CBP function redundantly in the removal of UV-induced DNA lesions
It is amply established that p300 is the major HAT which somehow influences the NER process (11). To clearly understand the underlying mechanism for the putative role of p300 in NER, especially in the context of our aforementioned results, we specifically knocked down the expression of p300 in repair-proficient OSU-2 cells and analyzed the removal rates of UV-induced CPD and 6-4PP by ISB analysis. Interestingly, we failed to find any measureable effect on the removal rates of CPD and 6-4PP under reduced cellular p300 level (Supplementary Figure S3A–C). Nevertheless, down-regulation of p300 did negatively affect the repair of IR-induced DSB (Supplementary Figure S4A and B). The absence of an effect on NER could be attributed to another homologous HAT, i.e. CBP, being functionally substituted in p300-compromised cells (8). To test whether p300 and CBP function redundantly in NER, we knocked down the expression of p300 and CBP, either singly or simultaneously, and determined the removal rate of UV-induced CPD and 6-4PP. The p300 siRNA specifically targeted the p300 protein level without an influence on CBP. The CBP siRNA not only knocked down the primary CBP protein but also targeted p300 to some extent (Figure 5A). As expected, double knockdowns of the two HATs, but not the single knockdowns of p300 or CBP, negatively affected the removal of CPD (Figure 5B and C). However, under the same conditions 6-4PP removal, which is known for its rapid repair rate, was unaffected (Supplementary Figure S5A and B). The reduction of acetylated histones H3 and H4 in cells with simultaneous double, but not the single, knockdown of p300 and CBP (Supplementary Figure S6), further confirmed the redundant function of p300 and CBP in NER. To investigate whether p300/CBP is required for the recruitment of DDB2 to UV-damaged sites, we performed the dual immunofluorescent staining to detect DDB2 and CPD within locally micropore UV-irradiated nuclear sites. As shown in Figure 5D, knockdown of either p300 or CBP did not affect the recruitment of DDB2 to CPD sites, as reflected by bright DDB2 foci (red) at almost all CPD sites (green). However, when p300 and CBP were simultaneously down-regulated, the intensity of DDB2 foci at CPD sites was seen to appreciably decrease both qualitatively and quantitatively. Since p300 and CBP are transcriptional co-activators, we wanted to know whether the effect of p300 and CBP double knockdown on NER is due to a reduced expression of NER genes. Western blotting analysis showed that none of the tested NER factors, e.g. XPA, XPC, XPF, XPG and DDB2, exhibited a reduction in their levels upon either single or double knockdown of p300 and CBP (Supplementary Figure S7). These results indicate that p300 and CBP redundantly function as the HATs influencing the recruitment of initial DNA-damage recognition factor DDB2 to damage sites in processing the lesions by NER pathway. Given that p300 is degraded upon UV irradiation, we wanted to know whether CBP protein also undergoes degradation to facilitate NER. Surprisingly, in contrast to p300, CBP protein did not exhibit a decrease upon UV irradiation (Supplementary Figure S8), indicating that CBP protein, after participating in NER, leaves the damage sites by mechanisms other than degradation.
Figure 5.

p300 and CBP function redundantly in NER. (A) OSU-2 cells were transfected with 50 nM of either control siRNA, p300 siRNA, CBP siRNA or both p300 and CBP siRNA for 48 h. The expression of p300 and CBP was detected using western blotting. (B) The siRNA transfected OSU-2 cells were UV irradiated at 10 J/m2 and further cultured for various time periods. The genomic DNA was isolated and subjected to ISB analysis to detect the amount of CPD with anti-CPD antibody (B). (C) The intensity of each band was quantified by scanning images and processing with Alphaimager-2000 software. The relative percentage of remaining CPD at different time points is an average of three independent repeats. (n = 3, bar: SD). (D) The siRNA transfected OSU-2 cells were UV irradiated at 40 J/m2 through a 5 -µm micropore filter and further cultured for 30 min. Cells were fixed, permeabilized and double stained with mouse anti-DDB2 and rabbit anti-CPD antibodies as described in ‘Materials and Methods’ section. The relative intensity of DDB2 focus was calculated by analyzing 100 foci and normalized to that of CPD (n = 100, bar: SD).
p300 phosphorylation at S1834 is critical for the removal of UV-induced DNA lesions
UV radiation-induced p300 phosphorylation at S1834 appears to be a pivotal modification with an exclusive role in NER. To further reinforce the distinct contribution of phospho-p300(S1834) to the NER process, we overexpressed wild-type p300 or its S1834A mutant (unable to phosphorylate at S1834) in OSU-2 cells (Figure 6A) and determined the removal rates of UV-induced CPD. As shown in Figure 6B and C, cells overexpressing p300-S1834A exhibited severely reduced efficiency in removing UV-induced CPD when compared with wild-type p300-overexpressing cells. Thus, p300 phosphorylation at S1834 is obviously important in NER. Notably, the p300-S1834A mutant protein exercised its NER attenuating influence in the presence of native wild-type p300 in OSU-2 cells. Thus, the mutant protein acts in a dominant negative manner to block the activity of the native protein. To further understand this phenomenon, we determined the effect of p300-S1834A overexpression on the recruitment of DDB2 to UV-damaged sites via local UV irradiation and immunofluorescence visualization. Figure 6D showed that overexpression of wild-type p300 did not affect the recruitment of DDB2 to CPD sites. However, similar to the effects of simultaneous down-regulation of p300 and CBP on DDB2 recruitment shown in Figure 5D, overexpression of p300-S1834A also reduced the recruitment of DDB2 to CPD sites, as demonstrated by a decreased intensity of DDB foci (red) at CPD (green) sites. Taken together, this set of data suggests that p300 phosphorylation at S1834 facilitates the removal of UV-induced CPD by promoting the recruitment of initial NER factors to damage sites.
Figure 6.

Phosphorylation of p300 at S1834 is critical to the repair of UV-induced DNA lesions. (A) OSU-2 cells were transfected with empty vector, wild-type p300 or mutant p300-S1834A for 48 h and western blotting was performed to detect the expression of p300. (B) OSU-2 cells transfected with either p300-WT or p300-S1834A expressing plasmids were UV irradiated at 10 J/m2 and further cultured for 8 or 24 h. The genomic DNA was isolated and an identical amount of DNA was loaded for ISB with anti-CPD antibody. Single-strand DNA was used as a loading control. (C) The relative remaining CPD was calculated as described in Figure 5. (D) OSU-2 cells transfected with either p300-WT or p300-S1834A were UV irradiated at 40 J/m2 through a 5 -µm micropore filter and further cultured for 30 min. Cells were processed for double staining of DDB2 and CPD as described in Figure 5.
p300 is recruited to nuclease-resistant chromatin fraction to facilitate the relocation of DDB2
Participation of p300 in NER entails that it is somehow directly associated with DNA damage sites. Indeed, two research teams have attempted to demonstrate the discernible co-localization of p300 at actual NER sites (11,44). Although the analysis of pixel intensity profiles seems to indicate the co-localization of p300 and CPD or NER sites, the immunofluorescence images were ambiguous and inconclusive for co-localization. Similarly, our attempts to simultaneously visualize both CPD and p300 did not show a clear recruitment of the p300 protein to UV-induced DNA damage sites (Supplementary Figure S9). Since p300 is distributed throughout the entire nucleus, its level of recruitment to damage sites by delocalization/relocalization appears to be outside the sensitivity of detection by the immunofluorescent staining schemes. Hence, we utilized an alternate approach to demonstrate the possible association of p300 with damaged DNA. It is believed that p300 contributes to DNA repair through histone acetylation. This process relaxes the condensed chromatin and increases accessibility to DNA lesions (11). Thus, p300 could very well function in the repair of DNA lesions located in heterochromatic regions. To address this, we separated the cellular protein extracts into fractions enriched for highly soluble, open chromatin or compacted chromatin segments based on the sensitivity to MNase digestion and salt extraction, as outlined by Goodarzi et al. (32) (Figure 7A and B and Supplementary Figure S10A and B). Successful fractionation is revealed by the presence of HP1 in both relaxed and condensed chromatin as well as the absence of its release by 0.3 M KCl without MNase treatment. In contrast, due to its loose association with chromatin, HMGB1 was completely released by the HS and was absent from the chromatin fraction. Neither of the two proteins, HP1 and HMGB1, showed any UV-induced translocation between different fractions. In the absence of stress, p300 and CBP exhibited a discrete localization and was abundant in the fraction constituting nucleoplasm. UV irradiation caused a portion of p300 and CBP to specifically relocate to the compacted chromatin, but not to the open chromatin fraction, indicating p300 and CBP are required for processing DNA lesions within the compacted chromatin compartment. Our results also showed that upon UV irradiation two NER factors, DDB2 and XPA, as well as a chromatin remodeling factor, SNF5, translocated to the compacted chromatin fraction. These combined results indicate that the repair of UV-induced DNA lesions and chromatin remodeling occur in the structurally distinct nuclease-resistant chromatin compartment.
Figure 7.
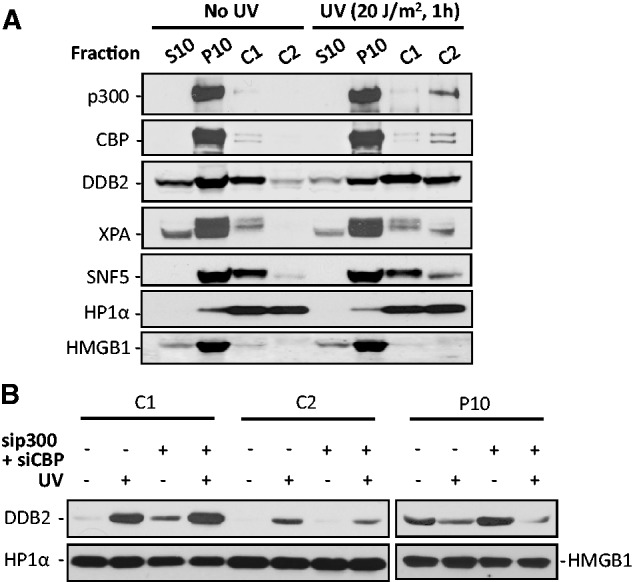
p300 is recruited to nuclease-resistant chromatin upon UV irradiation to facilitate the relocation of DDB2. (A) OSU-2 cells were UV irradiated at 0 or 20 J/m2 and further cultured for 1 h. Cells were processed for chromatin segregation as described in ‘Materials and Methods’ section. Each fraction, corresponding to equivalent cell numbers, was subjected to western blot analysis. S10: cytoplasmic proteins; P10: nucleoplasmic proteins and proteins bound to chromatin loosely; C1: proteins bound to chromatin that are accessible to MNase and C2: proteins bound to chromatin that areresistant to MNase. (B) OSU-2 cells were transfected with 50 nM of p300 and 50 nM of CBP siRNA simultaneously for 48 h, UV irradiated at 10 J/m2 and further cultured for 1 h. Cells were then processed for chromatin segregation, and the individual fractions were subjected to western blot analysis with anti-DDB2, anti-HP1α and anti-HMGB1 antibodies.
To determine whether p300/CBP is required for the translocation of DDB2 to compacted chromatin, we simultaneously down-regulated the expression of p300 and CBP in OSU-2 cells and assessed the specific protein partitioning in segregated chromatin following cellular UV irradiation. As shown in Figure 7B, dual knockdown of p300/CBP did not affect the translocation of DDB2 to nuclease-sensitive open chromatin (C1), but negatively affected the translocation of DDB2 to nuclease-resistant compacted chromatin (C2), indicating that p300/CBP is critical to NER factor assembly in the heterochromatic regions.
DISCUSSION
UV and cisplatin, but not IR or Etoposide, transiently induce p300 degradation
The level of p300 protein in the cellular nucleus is tightly regulated to maintain normal development and cell proliferation (45). Effective termination of its activity after transcriptional activation is essential for cells to ensure precisely controlled gene expression. Similarly, if p300 is recruited to participate in DNA repair, the effective removal of p300 from a damage site is also essential for the recruitment of subsequent DNA repair machinery. According to the theory of sequential assembly of repair factors in the process of NER (3,46,47), initially assembled proteins must vacate from the repair site either through degradation, such as DDB2 and XPC (48,49), or via dissociation, such as TFIIH, to make space for the recruitment of other subsequent repair factors. Thus, the degradation of p300 following UV irradiation could serve as the mechanism for evicting p300 from repair sites. Alternatively, p300 degradation, following UV and cisplatin treatment, could serve as the function of transcriptional repression. It is well known that transcription by RNA polymerase II is globally repressed following DNA damage (33,34). As a transcription co-activator, p300 degradation may contribute to DNA damage-induced transcription inhibition. However, we failed to find p300 degradation in response to IR or Etoposide treatment. It was reported that IR or Etoposide treatment induced p300 phosphorylation at serine 106 in an ATM-dependent manner (23). This kind of phosphorylation was found to stabilize p300 protein in response to DNA damage, and thus explain why no p300 degradation was found after IR and Etoposide treatments in our study. IR and Etoposide induce DSB, which are repaired by HR and NHEJ. It has been reported that p300 is recruited to DSB sites to acetylate histones H3 and H4 and facilitate SWI/SNF chromatin remodeling (10). Since we did not observe p300 degradation after IR or Etoposide treatments, we speculate that soon after its action, p300 must vacate the DSB sites through dissociation from repair machinery. In essence, it is clear that while p300 participates in both NER and DSB repair, the fate of p300 is dramatically different.
Akt-mediated p300 phosphorylation at S1834 correlates with p300 degradation
Multiple signaling pathways have been identified in the regulation of p300 turnover. Phosphorylation of p300 has been shown to either enhance the degradation of p300 protein through the 26S proteasome pathway or maintain p300 protein stability. It has been reported that p300 stability is modulated by Akt activation, and the phosphorylation at S1834 is required for this p300 stabilization (20,21). However, our data clearly demonstrated that inhibition of Akt activity totally blocked the UV-induced p300 degradation, as well as p300 phosphorylation at S1834. Apparently, Akt-mediated p300 phosphorylation at S1834 is critical to the stability of p300 in the absence of an exogenous assault, while Akt-mediated p300 phosphorylation promotes its degradation in the presence of NER-specific DNA damage. Clearly, therefore, the phosphorylation-mediated p300 degradation would play an important role in NER.
Besides Akt, we also demonstrated that p38 MAPK participated in the phosphorylation of p300 at S1834 and its degradation following UV irradiation. Most importantly, our data suggest that activated p38 MAPK facilitates the interaction between p300 and activated Akt, which catalyzes the phosphorylation of p300 at S1834. We have previously reported that p38 MAPK is involved in UV-induced chromatin relaxation and histone acetylation (50). Given that p300 phosphorylation at S1834 stimulates its HAT activity (18), in all likelihood the reported p38-mediated chromatin relaxation and histone acetylation can be attributed to the enhancement of p300 HAT activity caused by p300 phosphorylation.
It is also possible that Akt may affect the interaction between MAPK and p300. However, unlike Akt, although p38 MAPK was reported to phosphorylate p300, the phosphorylation site is in N-terminus (aa 1-596), but not the C-terminus (aa 1572-2414) (41). Thus, Akt is more likely than p38 MAPK to interact with and catalyze p300 phosphorylation at S1834.
p300 phosphorylation, but not p300 itself, is important for efficient NER
It is well known that the complete NER reaction involves several biochemical steps including damage recognition, dual incision and gap-filling DNA synthesis (51). Existing information seems to suggest that p300 influences NER by participating at all of these steps. First, p300 interacts with both DDB2 and DDB1 (35,52), and it is suggested that p300 could be recruited to the damaged chromatin by the DDB complex to help chromatin remodeling at the damage sites to allow recruitment of repair complexes to recognize DNA damage. Second, p300 is required for optimal activation of the dual incision reaction in chromatinized template in vitro (53). Third, p300 is associated with newly synthesized DNA and PCNA after UV irradiation, leading to suggestion that p300 facilitates PCNA function in DNA repair synthesis (44,54). In this study, we have assessed the role of p300 in efficiently removing UV-induced photolesions and, not surprisingly, found that p300 is not absolutely required for the removal of UV damages due to the well-established redundant role of p300 and CBP HATs. Instead, we were able to demonstrate that p300 phosphorylation at S1834 is essential to NER. As discussed above, phosphorylation-mediated p300 degradation seems to be the key for the important role of p300 in DNA repair. These data suggest that p300 is obviously recruited to UV-induced DNA damage sites (11,44). Once used, the eviction of p300 via the degradation of the phosphorylated form (p300-S1834) could facilitate the subsequent recruitment of other repair factors. On the other hand, failure to clear out, due to the sustained occupancy of DNA damage sites by the un-phosphorylated form (p300-S1834A mutant), impedes the recruitment of these factors and consequently results in a repair deficiency following UV exposure. Our previous finding, that inhibiting p38 activity compromised the recruitment of XPC and TFIIH to UV-induced DNA damage sites (50), could also be attributed to such a repair deficiency due to inhibited p300 phosphorylation and degradation. Protein sequence comparisons show that the amino acid residues containing S1834 in p300 protein (MAS1834M) can also be found in CBP protein (MAS2184M). Given that p300 phosphorylation at S1834 is critical to NER, it will be interesting to understand whether the CPB protein can also be phosphorylated at serine 2184 and if it plays a unique role in DNA repair.
p300 participates in the repair of UV-induced CPD in heterochromatic regions
Structurally, chromatin in mammalian cells is organized as the highly condensed heterochromatin and loosely packed euchromatin, representing two distinct entities from the standpoint of DNA damage sensitivity and repair processing. As such, chromatin creates a natural barrier for access to DNA during damage repair. Our chromatin segregation analysis demonstrated that p300 redistributed to the MNase-resistant heterochromatin compartment but not to the MNase-sensitive euchromatin, indicating that p300 may play a role in the repair of heterochromatic DNA lesions. According to our data, the majority of p300 resides in the nucleus, and only a small portion of p300 is redistributed to MNase-resistant chromatin, explaining why it has been difficult to demonstrate p300 recruitment to local UV-damage sites by immunofluorescence. Given the differences in the extent of chromatin condensation within the euchromatin and heterochromatin, we can reason that the processing of UV-induced photolesions in the structurally recalcitrant heterochromatin requires additional layers of regulation. In this vein, the redistribution of p300 and SNF5, and possibly other factors, to the MNase-resistant chromatin suggests that these factors are important in overcoming the constraints imposed by the heterochromatin on the repair machinery.
In conclusion, our data suggest that in response to UV irradiation, p300 is phosphorylated at serine 1834 by the cooperative effect of p38 and Akt and is specifically recruited to DNA lesions in heterochromatic regions to facilitate the relaxation of chromatin. The phospho-p300(S1834) is subsequently evicted through proteasomal protein degradation to allow the space needed for the recruitment of downstream repair factors and efficient removal of DNA lesions (Figure 8).
Figure 8.

Schematic representation of p300 phosphorylation and degradation in NER. After UV irradiation, p300 is recruited to the damage sites in the heterochromatin and phosphorylated at S1834 by the cooperation of p38 MAPK and Akt. The enhanced HAT activity of p300 catalyzes the acetylation of histones H3 and H4, resulting in relaxed local chromatin surrounding the damage sites. Phosphorylated p300 is then degraded to make space for the subsequent recruitment of NER factors, e.g. DDB2 and XPC.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–10.
FUNDING
National Institute of Health (NIH) [CA151248 to Q.E.W., CA93413, ES2388 and ES12991 to A.A.W.]. Funding for open access charge: NIH [CA93413].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Michael Tainsky for providing LFS 041 cells, Drs David Livingston, Yuan Liu, David Jones and Terry Unterman for kindly providing the plasmids encoding full-length p300-WT or p300-S1834A mutant.
REFERENCES
- 1.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Araujo SJ, Nigg EA, Wood RD. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol. Cell. Biol. 2001;21:2281–2291. doi: 10.1128/MCB.21.7.2281-2291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volker M, Mone MJ, Karmakar P, Van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, Van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 4.Sugasawa K, Ng JMY, Masutani C, Iwai S, Van der Spek P, Eker A, Hanoaka F, Bootsma D, Hoeijmakers JHJ. Xeroderma pigmentosum group C complex is the initiator of global genome nucleotide excision repair. Mol. Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Q, Wani AA. Histone modifications: crucial elements for damage response and chromatin restoration. J. Cell Physiol. 2010;223:283–288. doi: 10.1002/jcp.22060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000;24:300–303. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- 7.Gusterson RJ, Jazrawi E, Adcock IM, Latchman DS. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J. Biol. Chem. 2003;278:6838–6847. doi: 10.1074/jbc.M211762200. [DOI] [PubMed] [Google Scholar]
- 8.Kalkhoven E. CBP and p300: HATs for different occasions. Biochem. Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 9.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 10.Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30:2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- 11.Rubbi CP, Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003;22:975–986. doi: 10.1093/emboj/cdg082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yaciuk P, Moran E. Analysis with specific polyclonal antiserum indicates that the E1A-associated 300-kDa product is a stable nuclear phosphoprotein that undergoes cell cycle phase-specific modification. Mol. Cell. Biol. 1991;11:5389–5397. doi: 10.1128/mcb.11.11.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 14.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 15.Yuan LW, Gambee JE. Phosphorylation of p300 at serine 89 by protein kinase C. J. Biol. Chem. 2000;275:40946–40951. doi: 10.1074/jbc.M007832200. [DOI] [PubMed] [Google Scholar]
- 16.Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 2010;120:4316–4331. doi: 10.1172/JCI41624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Denlinger CE, Rundall BK, Smith PW, Jones DR. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J. Biol. Chem. 2006;281:31359–31368. doi: 10.1074/jbc.M604478200. [DOI] [PubMed] [Google Scholar]
- 18.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell. Biol. 2005;25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen YJ, Wang YN, Chang WC. ERK2-mediated C-terminal serine phosphorylation of p300 is vital to the regulation of epidermal growth factor-induced keratin 16 gene expression. J. Biol. Chem. 2007;282:27215–27228. doi: 10.1074/jbc.M700264200. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Halappanavar SS, St-Germain JR, Tsang BK, Li Q. Role of Akt/protein kinase B in the activity of transcriptional coactivator p300. Cell Mol. Life Sci. 2004;61:1675–1683. doi: 10.1007/s00018-004-4103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howie HL, Koop JI, Weese J, Robinson K, Wipf G, Kim L, Galloway DA. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011;7:e1002211. doi: 10.1371/journal.ppat.1002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol. Cell. Biol. 2005;25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang ER, Choi JD, Jeong G, Lee JS. Phosphorylation of p300 by ATM controls the stability of NBS1. Biochem. Biophys. Res. Commun. 2010;397:637–643. doi: 10.1016/j.bbrc.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 24.Poizat C, Sartorelli V, Chung G, Kloner RA, Kedes L. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol. Cell. Biol. 2000;20:8643–8654. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang QE, Han C, Milum K, Wani AA. Stem cell protein Piwil2 modulates chromatin modifications upon cisplatin treatment. Mutat. Res. 2011;708:59–68. doi: 10.1016/j.mrfmmm.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duyndam MC, van Berkel MP, Dorsman JC, Rockx DA, Pinedo HM, Boven E. Cisplatin and doxorubicin repress Vascular Endothelial Growth Factor expression and differentially down-regulate Hypoxia-inducible Factor I activity in human ovarian cancer cells. Biochem. Pharmacol. 2007;74:191–201. doi: 10.1016/j.bcp.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Venkatachalam S, Denissenko MF, Wani AA. DNA repair in human cells: quantitative assessment of bulky anti-BPDE DNA adducts by non-competitive immunoassays. Carcinogenesis. 1995;16:2029–2036. doi: 10.1093/carcin/16.9.2029. [DOI] [PubMed] [Google Scholar]
- 28.Wani AA, D’Ambrosio SM, Alvi NK. Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem. Photobiol. 1987;46:477–482. doi: 10.1111/j.1751-1097.1987.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang QE, Milum K, Han C, Huang YW, Wani G, Thomale J, Wani AA. Differential contributory roles of nucleotide excision and homologous recombination repair for enhancing cisplatin sensitivity in human ovarian cancer cells. Mol. Cancer. 2011;10:24. doi: 10.1186/1476-4598-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang QE, Zhu Q, Wani MA, Wani G, Chen J, Wani AA. Tumor supressor p53 dependent recruitment of nucleotide excision repair ractors XPC and TFIIH to DNA damage. DNA Repair. 2003;2:483–499. doi: 10.1016/s1568-7864(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 31.Wani AA, Gibson-D’Ambrosio RE, D’Ambrosio SM. Antibodies to UV irradiated DNA: the monitoring of DNA damage by ELISA and indirect immunofluorescence. Photochem. Photobiol. 1984;40:465–471. doi: 10.1111/j.1751-1097.1984.tb04619.x. [DOI] [PubMed] [Google Scholar]
- 32.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 33.Proietti-De-Santis L, Drane P, Egly JM. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J. 2006;25:1915–1923. doi: 10.1038/sj.emboj.7601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rockx DA, Mason R, van HA, Barton MC, Citterio E, Bregman DB, Van Zeeland AA, Vrieling H, Mullenders LH. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc. Natl Acad. Sci. USA. 2000;97:10503–10508. doi: 10.1073/pnas.180169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T, Raychaudhuri P. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat. Res. 2001;486:89–97. doi: 10.1016/s0921-8777(01)00082-9. [DOI] [PubMed] [Google Scholar]
- 36.Wittschieben BB, Wood RD. DDB complexities. DNA Repair (Amst) 2003;2:1065–1069. doi: 10.1016/s1568-7864(03)00113-7. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Li Q. Life and death of transcriptional co-activator p300. Epigenetics. 2011;6:957–961. doi: 10.4161/epi.6.8.16065. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz C, Beck K, Mink S, Schmolke M, Budde B, Wenning D, Klempnauer KH. Recruitment of p300 by C/EBPbeta triggers phosphorylation of p300 and modulates coactivator activity. EMBO J. 2003;22:882–892. doi: 10.1093/emboj/cdg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gusterson R, Brar B, Faulkes D, Giordano A, Chrivia J, Latchman D. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J. Biol. Chem. 2002;277:2517–2524. doi: 10.1074/jbc.M104626200. [DOI] [PubMed] [Google Scholar]
- 40.See RH, Calvo D, Shi Y, Kawa H, Luke MP, Yuan Z, Shi Y. Stimulation of p300-mediated transcription by the kinase MEKK1. J. Biol. Chem. 2001;276:16310–16317. doi: 10.1074/jbc.M008113200. [DOI] [PubMed] [Google Scholar]
- 41.Bratton MR, Frigo DE, Vigh-Conrad KA, Fan D, Wadsworth S, McLachlan JA, Burow ME. Organochlorine-mediated potentiation of the general coactivator p300 through p38 mitogen-activated protein kinase. Carcinogenesis. 2009;30:106–113. doi: 10.1093/carcin/bgn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 43.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2 -(4-morpholinyl)-8-phenyl-4 H-1-benzopyran-4-one (LY294002) J. Biol. Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 44.Cazzalini O, Perucca P, Savio M, Necchi D, Bianchi L, Stivala LA, Ducommun B, Scovassi AI, Prosperi E. Interaction of p21(CDKN1A) with PCNA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res. 2008;36:1713–1722. doi: 10.1093/nar/gkn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brouillard F, Cremisi CE. Concomitant increase of histone acetyltransferase activity and degradation of p300 during retinoic acid-induced differentiation of F9 cells. J. Biol. Chem. 2003;278:39509–39516. doi: 10.1074/jbc.M307123200. [DOI] [PubMed] [Google Scholar]
- 46.Wakasugi M, Sancar A. Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc. Natl Acad. Sci. USA. 1998;95:6669–6674. doi: 10.1073/pnas.95.12.6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakasugi M, Sancar A. Order of assembly of human DNA repair excision nuclease. J. Biol. Chem. 1999;274:18759–18768. doi: 10.1074/jbc.274.26.18759. [DOI] [PubMed] [Google Scholar]
- 48.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 49.Wang QE, Praetorius-Ibba M, Zhu Q, El-Mahdy MA, Wani G, Zhao Q, Qin S, Patnaik S, Wani AA. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res. 2007;35:5338–5350. doi: 10.1093/nar/gkm550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Q, Barakat BM, Qin S, Ray A, El-Mahdy MA, Wani G, Arafa,e., Mir SN, Wang QE, Wani AA. The p38 mitogen-activated protein kinase augments nucleotide excision repair by mediating DDB2 degradation and chromatin relaxation. J. Biol. Chem. 2008;283:32553–32561. doi: 10.1074/jbc.M803963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petit C, Sancar A. Nucleotide excision repair: from E. coli to man. Biochimie. 1999;81:15–25. doi: 10.1016/s0300-9084(99)80034-0. [DOI] [PubMed] [Google Scholar]
- 52.Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 2002;30:2588–2598. doi: 10.1093/nar/30.11.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frit P, Kwon K, Coin F, Auriol J, Dubaele S, Salles B, Egly JM. Transcriptional activators stimulate DNA repair. Mol. Cell. 2002;10:1391–1401. doi: 10.1016/s1097-2765(02)00732-3. [DOI] [PubMed] [Google Scholar]
- 54.Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature. 2001;410:387–391. doi: 10.1038/35066610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.