
Fig. 3.
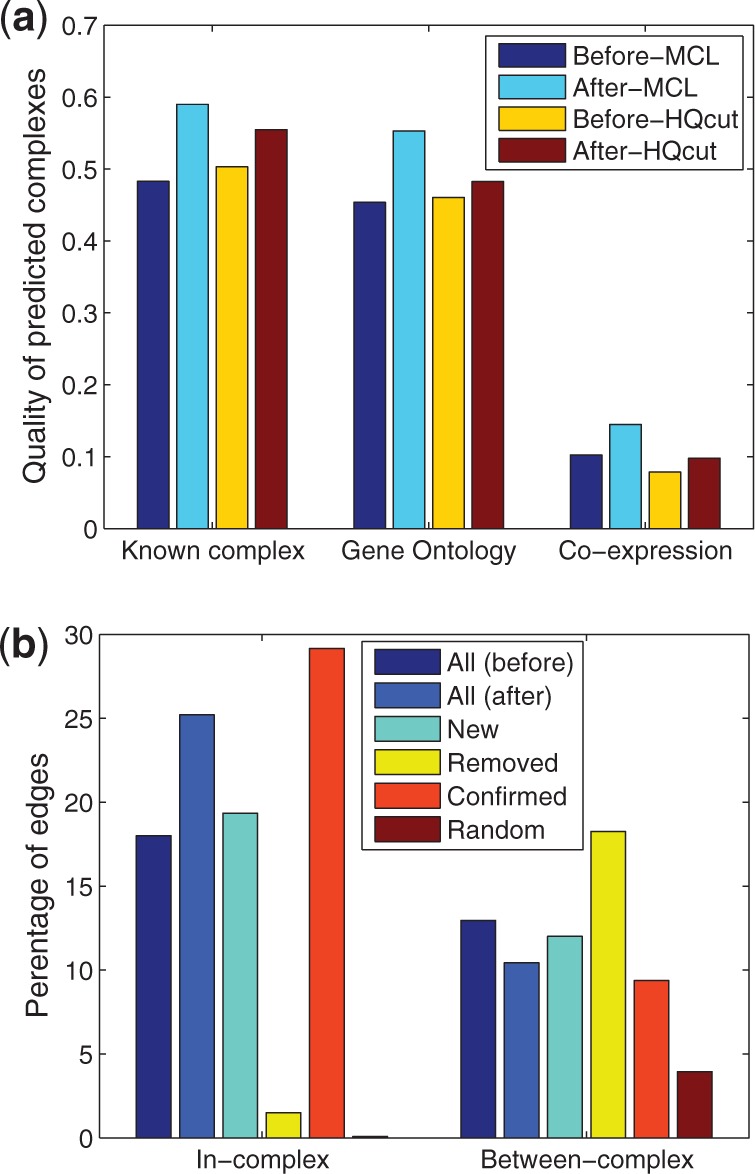
Evaluation of our method based on protein complexes. In (a), the original PPI network (before) or the reconstructed counterpart (after) is partitioned using two algorithms (MCL and HQcut) for protein complex predictions. Each prediction is then evaluated by its similarity (F–M index, see main text) to MIPS known complexes, by the average gene ontology similarity between pairs of nodes in the same complex, or by average gene co-expression correlation between pairs of nodes in the same complex. In (b), different edge groups are evaluated for the fraction of edges connecting edges in the same or different MIPS known protein complexes