

Abstract
Purpose.
How herpes simplex virus (HSV) is transported from the infected neuron cell body to the axon terminal is poorly understood. Several viral proteins are candidates for regulating the process, but the evidence is controversial. We compared the results of Us9 deletions in two HSV strains (F and NS) using a novel quantitative assay to test the hypothesis that the viral protein Us9 regulates the delivery of viral DNA to the distal axon of retinal ganglion cells in vivo. We also deleted a nine–amino acid motif in the Us9 protein of F strain (Us9-30) to define the role of this domain in DNA delivery.
Methods.
The vitreous chambers of murine eyes were infected with equivalent amounts of F or NS strains of HSV. At 3, 4, or 5 days post infection (dpi), both optic tracts (OT) were dissected and viral genome was quantified by qPCR.
Results.
At 3 dpi, the F strain Us9- and Us9-30 mutants delivered less than 10% and 1%, respectively, of the viral DNA delivered after infection with the Us9R (control) strain. By 4 and 5 dpi, delivery of viral DNA had only partially recovered. Deletion of Us9 in NS-infected mice has a less obvious effect on delivery of new viral DNA to the distal OT. By 3 dpi the NS Us9-strain delivered 22% of the DNA that was delivered by the NS wt, and by 4 and 5 dpi the amount of Us9-viral DNA was 96% and 81%, respectively.
Conclusions.
A highly conserved acidic cluster within the Us9 protein plays a critical role for genome transport to the distal axon. The transport is less dependent on Us9 expression in the NS than in the F strain virus. This assay can be used to compare transport efficiency in other neurotropic viral strains.
HSV DNA is delivered to infected neuron axons by two or more redundant mechanisms and the efficiency of delivery to the axon varies quantitatively with the HSV strain background. A nine–amino acid acidic cluster in the F-strain Us9 protein plays a critical role in this avenue of viral spread.
Introduction
The anterograde axonal transport of new herpes simplex virus (HSV) underlies the pathophysiology of both corneal keratitis and herpetic encephalitis. After initial infection of the corneal cells, virus spreads to innervating sensory nerve axon terminals and then travels by retrograde axonal transport to sensory or autonomic ganglion neuron cell bodies. In the neuron cell body the virus replicates, and new virus is re-delivered via anterograde axonal transport to axon terminals in the peripheral nervous system (PNS). Spread to mucous membranes leads to corneal blisters and epithelial scarring (see Al-Dujaili et al.1 for review). The same HSV-1 infection of trigeminal ganglion cells can also lead to the spread of new virus into the central nervous system (CNS), resulting in herpetic hemorrhagic encephalitis.2
Study of anterograde axonal transport of HSV in the trigeminal system is complicated for several reasons. The primary infection of the corneal epithelial cells is followed by infection and retrograde axonal transport in neurons of the trigeminal ganglion. Two to 3 days after corneal infection, virus continues to be transported in the retrograde direction, but now newly synthesized virus returns in the anterograde direction in the peripheral branches of trigeminal neurons. In contrast, the retinal ganglion cell model lets us study a unidirectional transport in a relatively elongated axon. New virus is delivered synchronously to retinal ganglion cell bodies and then transported unidirectionally from cell body toward terminal.
The precise viral mechanisms that direct selective anterograde axonal transport of virus are unclear.3,4 Even the identity of the viral protein(s) that contribute(s) to the functional delivery of new virus in anterograde transport is controversial.5–10 The controversy is due in part to the variety of assays, animal models, and viral strains used. To focus the question on the role of a single viral protein in the efficient delivery of new HSV viral DNA, we have compared transport of two different strains of HSV and two different mutants of a single protein in a single animal model with a single assay.
We have tested three main hypotheses: that Us9 expression plays different roles in different HSV strains, NS and F; that efficient anterograde axonal transport of viral DNA depends on the Us9 protein of F strain; and that an acidic domain within Us9 plays a critical role in DNA delivery.
Materials and Methods
Preparation of Viral Stocks
Five stocks of virus, including mutants on two different strain backgrounds (F and NS), were used (Fig. 1A). On the F background the wild type (wt) Us9 gene was modified in two ways. First, most of the gene was deleted (F Us9-), as previously characterized.8 Briefly, nucleotides (nts) 18 to 204 of the Us9 gene were deleted, leaving nts 1 to 17 intact and nts 205 to 270 frame shifted.8 A rescued version of the mutated Us9 (F Us9R) contained the deleted sequence reintroduced to the F Us9- genome (Fig. 1A).8 In the case of both Us9- and Us9R, PCR products for Us7 (gI), Us8A, and Us8 (gE) were normal.8 Both F Us9R and Us9- expressed glycoproteins gB, gE, and gI.5
Figure 1. .
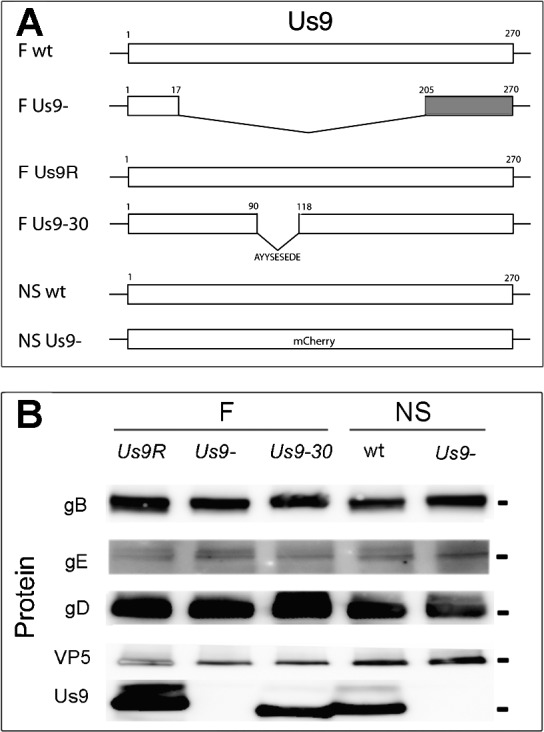
Construction and characterization of the HSV Us9 mutants. (A) Schematic of the HSV unique short (Us9) component. Most of the Us9 gene was deleted in F Us9-. The first 17 nts were intact and the final 205 to 170 nts were frame shifted (gray). The Us9R strain contained the reintroduced deleted sequences. In the Us9-30 strain, nine amino acids were deleted, and remaining portions of the gene were intact. The NS strains included the wt strain and a mutant strain, Us9-, in which the entire Us9 gene was replaced by a mCherry marker. (B) Protein expression. Vero cells were infected with either F strain Us9R, Us9-, or Us9-30 or NS strain wt or Us9- using 1 pfu/cell for 24 hours. Antibodies directed against gB, gE, gD, VP5, or Us9 were used to probe for protein expression. Us9 was not detected in F Us9- or in NS Us9- infected cells. However, Us9 was detected in Us9-30 infected cells at a slightly lower molecular weight location. Note that one of the higher molecular weight bands of Us9 in the Us9-30 appears to be reduced. Molecular markers are 100 (gB), 70 (gE), 58 (gD), 155 (VP5), and 17 (Us9) kDa.
A second mutant virus designed to eliminate nine amino acids in Us9 on the F background was created using bacterial artificial chromosome (BAC) mutagenesis (Us9-30) (Fig. 1A). The reagents used to create a recombinant Us9-30 virus were provided by Joel Baines with permission from Yashushi Kawaguch for the BAC containing HSV-1 and from Michael Kotlikoff for the plasmid pCAGGS-nlsCre. Baines also supplied the pBAD-I-sceI. The virus was constructed using a recombinant system described previously.11,12 The primers for production of a PCR amplicon for deletion of nts 91 to 117 from the Us9 gene in the HSV-1(F) containing BAC were as follows: forward, ATGTCCGTGCCGCTTTATCCCACGGCCTCGCCAGTTTCGGCCGCGGCCAACGACTTC- CTCGTATAGGGATAACAGGGTAATCGATTT; reverse, CGACTGTTGGCGGCCCATGCGTACGAGGAAGTCGTTGGCCGCGGCCGAAACTGGCGA- GGCCGTGCCAGTGTTACAACCAATTAACC. PCR products were gel purified and electroporated into competent EL250 cells, designated 30-EL250 BAC. DNA was extracted and analyzed using restriction fragment length polymorphisms (RFLP) to confirm the deletion by BamHI, ClaI restriction fragments. Using previously described methods the electrocompetent 30-EL250 cells were electroporated with pBAD-I-sceI, which contains arabinose-inducible expression of I-sceI. This removed the Kan marker from the 30-EL250 BAC. Ampicillin- and chloramphenicol-resistant colonies sensitive to kanamycin were selected. BAC DNA was co-transfected with pCAGGS-nls Cre using Superfect (Qiagen, Valencia, CA) and produced Us9-30 recombinant virus. Virus was plaque purified and titered. The deletion of the specific 27 bps was confirmed by PCR. Protein expression was confirmed by Western blots with antibodies to gB, gE, gD, VP5, and Us9 proteins (Fig. 1B).
NS strain stocks were obtained from Harvey M. Friedman (University of Pennsylvania). NS (NS wt) is a low-passage clinical isolate (Fig. 1A).5 The NS US9null (NS Us9-) was a gift to his laboratory from Lynn W. Enquist (Princeton University) (Fig. 1A). The Us9 gene in the NS Us9- mutant has been completely replaced by a mCherry marker (Fig. 1A).5
All viruses were propagated in Vero cells as described previously.13 Viral titers were determined in triplicate by standard plaque assay.14 No significant difference in single-cycle kinetics of growth between mutants and parent strain was found at 6 or 24 hours post infection.5,8 Thus, neither F Us9 nor NS Us9 protein was necessary for normal production of infectious virus in Vero cells. Furthermore, the nine–amino acid segment that was deleted in the F Us9-30 was also unnecessary for normal growth kinetics (Draper JM, unpublished data, 2011).
Intraocular Injections
Male BALB/c mice (5- to 6-weeks old) from Simonsen's Laboratories (Gilroy, CA) were used for all experiments. Avertin was injected intraperitoneally for anesthesia and proparacaine and atropine (1:1) eye drops were delivered for analgesia.15 The vitreous chamber of each eye of each animal was infected with 4 μL of virus diluted in sterile minimal essential medium (MEM) using viral concentrations of approximately 107 plaque-forming units (pfu)/mL, according to a standard procedure.16 To reduce the complications of viral secondary infection of glial and supporting cells in the optic tract (OT), we introduced an antiviral drug, valacyclovir hydrochloride (1 mg/mL) (Valtrex; Glaxo Wellcome, Inc., Greenville, NC) in the drinking water after 24 hours of replication.13,17 The reduction in replication at 3 dpi was similar for both F and NS retinal infections (Draper JM, unpublished data, 2012). All procedures involving animals adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the guidelines of the UCSF Committee on Animal Research.
Tissue Isolation
Surgical instruments were autoclaved before collection of tissues. The animals were anesthetized with avertin and perfused intracardially with saline. Both OTs were collected at 3, 4, or 5 dpi.18 Each OT was dissected from the hypothalamus to avoid contamination from bloodborne virus at the median eminence.19–21 The OT was approximately 6 mm in length and was dissected to the point where the fibers enter the dorsal thalamus. Each experiment was repeated at least twice. As a negative control, optic pathways of uninfected mice were similarly prepared. Aliquots of viral stocks were used as positive controls.
DNA Extraction and Total DNA Quantification
The OTs of each mouse were collected in 0.15 mL of PicoPure DNA extraction buffer with 1 mg/mL proteinase K (Arcturus, Mountain View, CA). The samples were sonicated for 15 minutes in a water bath (Branson 2510; Branson, Danbury, CT), incubated for 3 hours at 65°C, and then for 10 minutes at 95°C.
Total DNA quantification was performed using the Quant-iT PicoGreen DNA kit (Molecular Probes, Inc., Eugene, OR) in conjunction with the Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA). Dilute PicoGreen solution (Quant-iT PicoGreen reagent, 10 mM Tris-HCl, 1 mM EDTA, pH 7.5; Molecular Probes, Inc.) was freshly prepared and protected from light. A standard curve of serial dilutions from 10 ng/μL to 10 pg/μL of lambda DNA in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) was prepared. Samples were plated in duplicate, and extrapolated fluorescence data was normalized against the standard curve with an R2 greater than 0.98. TE with diluted PicoGreen solution was used to define the baseline. Quantitative assays for total DNA with PicoGreen and fluorescent detection have been shown to provide accurate and repeatable results.22–24
Determination of Viral Copy Number by Quantitative PCR
Copy numbers of viral genomes in the murine OTs were determined by quantitative PCR. Primers and probe were specific for sequences within the HSV-1 thymidine kinase gene (TK1): forward 5′-AAA ACC ACC ACC ACG CAA CT-3′, reverse 5′-TCA TCG GCT CGG GTA CGT A-3′, probe 5′-FAM-TG GGT TCG CGC GAC GAT ATC G-TAMRA-3′ (Integrated DNA Technology, Coralville, IA). Each reaction contained Taqman Universal PCR Master Mix (Applied Biosystems), forward and reverse primers, fluorescent probe, and 5 μL of OT sample prepared as above. Samples were run in duplicate using the Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems) under standard cycle conditions. A standard curve based on 10-fold dilutions of purified HSV-1 Us9R DNA from 101 to 107 copies of genome and with R2 greater than 0.98 was used to determine copy numbers.
Statistical Analysis
In live animal experiments, the viral load in the pathway can be measured at only one time point. Multiple animals are needed to obtain sufficient sample sizes. The variability within data sets was similar within the entire F or NS groups. Because the number of mutant or control values in F or NS groups was too small and not normally distributed for analysis with parametric tests, the nonparametric Mann-Whitney U rank-sum test was chosen.25 The level of statistical significance was defined as a confidence level of P less than 0.05 or P less than 0.01.
Antibodies and Western Blots
Equivalent amounts of infected Vero cells for each viral strain were loaded onto precast 4% to 15% Tris-HCl SDS-PAGE gels, transferred to Immuno-Blot PVDF membrane (BioRad, Hercules, CA), and immunoblotted according to previously published procedures.13 The monoclonal antibodies specific for HSV-1 gD (HA025), for gE (H1A054), and for gB (HA056) were from Virusys Corporation (Gaithersburg, MD). The antibody for HSV-1 VP5 (C05014M) was from Biodesign International (Saco, ME). Donkey antirabbit IgG/HRP (NA934V) and sheep antimouse IgG/HRP (NA931V) were from GE Healthcare UK limited (Chalfont St. Giles, UK). A polyclonal rabbit antisera that was raised against a peptide containing the first 17 amino acids of Us9 was previously described.8
Results
Five stocks of virus including mutants on two different strain backgrounds (F and NS) were used (Fig. 1A). On the F background, the Us9 gene was modified in two ways. The entire Us9 protein expression was blocked by genetically eliminating most of the Us9 gene, as previously described, and by removing nine amino acids from the Us9 sequence. On the NS background, the Us9 gene was entirely replaced with a fluorescent marker, mCherry.5 We injected the same amount of each viral stock into the vitreous chambers of both eyes of mice, and thus infected the cell bodies of retinal ganglion cells. The anterograde transport of newly synthesized virus was assayed by dissecting distal OTs and measuring the amount of viral and cellular DNA in the tissues.
We compared the total amount of cellular and viral DNA in the infected OTs to confirm that our dissections of OTs were consistent. The totals did not differ significantly between animals infected with F Us9R, F Us9-, or Us9-30 (P > 0.05, n = 90), nor did they differ between NS wt or NS Us9- (P > 0.05, n = 56) (Table 1). To facilitate the comparison between individual experiments, we measured the copies of viral DNA in each sample and divided that by the total viral and cellular DNA for that sample. We expressed this ratio as copies of viral DNA/ng total DNA in each sample.
Table 1. .
The Dissections of OT after Infection with Viral Strains and Mutants Are Consistent (Total DNA ng/OT)
|
Strain |
Mutation |
3 dpi |
4 dpi |
5 dpi |
| F | Us9R | 126.83 ± 11.35 (10) | 134.65 ± 11.93 (10) | 214.21 ± 23.34 (10) |
| F | Us9- | 118.52 ± 11.95 (10) | 141.59 ± 14.81 (10) | 153.60 ± 21.73 (10) |
| F | Us9-30 | 167.88 ± 18.43 (10) | 132.79 ± 9.30 (10) | 153.63 ± 15.37 (10) |
| NS | wt | 199.91 ± 27.29 (10) | 312.02 ± 26.57 (10) | 288.17 ± 35.33 (9) |
| NS | Us9-null | 172.45 ± 20.74 (9) | 235.07 ± 26.97 (9) | 247.44 ± 17.93 (9) |
Values are mean ± SEM (no. mice/experiment).
Deletion of Us9 in the F strain significantly reduced the amount of viral DNA delivered to the OT as measured by real time PCR (qPCR) at all time points (Table 2). Specifically by 3 dpi with F Us9-, there is roughly a 14-fold reduction in the amount of viral DNA delivered to the OT, as compared with that delivered after infection with F Us9R (P < 0.01) (Fig. 2A). By 4 and 5 dpi the amounts of F Us9- DNA were 1.5- to 2.0-fold less than that detected after infection with the control, Us9R (4 day, P < 0.05; 5 day, P < 0.01) (Fig. 2A).
Table 2. .
The Delivery of Viral DNA to the OT Depends on Viral Strain and Mutation of Us9 (Viral Copy/ng of Total DNA)
|
Strain |
Mutation |
3 dpi |
4 dpi |
5 dpi |
| F | Us9R | 112,450 ± 25,250 (10) | 142,007 ± 22,718 (10) | 113,838 ± 16,788 (10) |
| F | Us9- | 7,820 ± 3,077 (10) | 92,174 ± 21,833 (10) | 53,656 ± 12,546 (10) |
| F | Us9-30 | 1,057 ± 417 (10) | 10,102 ± 6,412 (10) | 57,547 ± 20,054 (10) |
| NS | wt | 207,635 ± 51,274 (9) | 214,209 ± 29,621 (10) | 110,605 ± 32,081 (10) |
| NS | Us9- | 46,579 ± 23,088 (9) | 204,861 ± 8,393 (9) | 89,892 ± 13,371 (9) |
Values are mean ± SEM (no. mice/experiment).
Figure 2. .

Comparison of the concentrations of viral DNA in mutated Us9. (A) Quantitative PCR values for F-strain infected mice at 3, 4, and 5 dpi. The amount of DNA transported after infection with Us9- and Us9-30 mutants (light blue) is expressed as a percentage of the DNA transported after infection with Us9R (dark blue). (B) Quantitative PCR values for NS-infected mice at 3, 4, and 5 dpi. The amount of DNA transported after Us9- infection (orange) is expressed as a percentage of DNA transported with the wt (brown). *P < 0.05; **P < 0.01.
At 3 and 4 dpi the Us9-30 mutant delivered less HSV DNA to the OT than the amount found after infection with either Us9R or Us9- (Table 2). By 3 dpi there was less than 1% of DNA than that delivered by the Us9R or 1057 ± 418 copies/ng of Us9-30 DNA (P < 0.01) (Fig. 2A, Table 2). At 4 dpi we detected approximately 7% of the HSV DNA delivered by the Us9R or only 10,102 ± 6412 copies/ng (P < 0.01); however, by 5 dpi the Us9-30 and Us9- mutants had transported equivalent amounts of DNA into the OT (Fig. 2A). This delivery was still significantly less than that measured after Us9R infection, that is, 57,547 ± 20,054 copies/ng or 51% of the viral genome transported with a fully expressed Us9.
Next, we consider the Us9 deletion in the NS background (Table 2). By 3 dpi the DNA from NS Us9- strain had spread to the OT, but the amount was less than that recovered after infection with the wt parent strain. Specifically, the number of copies of viral DNA delivered to the OT was approximately 22% of that delivered after NS wt infection, or a 4- to 5-fold reduction (Fig. 2B). At 4 and 5 dpi, the concentration of NS Us9- viral DNA was approximately 96% and 81%, respectively, of that delivered by NS wt. This was not a significant decrease. Only at 3 dpi was there a significant difference between NS Us9- and NS wt (P < 0.01). This suggests that in the NS background deletion of Us9 affects the delivery of viral DNA, but not to the same extent as that seen in the case of Us9- in the F strain.
In sum, our results are consistent with the model that viral capsids are transported in the axon by two or more redundant mechanisms and that the efficiency of delivery to the axon varies quantitatively with the HSV strain background. We have also identified that a highly conserved acidic cluster of nine amino acids is necessary for this function of Us9 in HSV.
Discussion
The molecular and cell biological basis for intracellular viral transport has remained elusive, despite intense interest (for review, see Enquist26 and Diefenbach and Cunningham27). Previously, we tested the role of Us9 in F strain of HSV anterograde axonal transport using genetic and biochemical tools and in vivo, unfixed neurons.8 We found that F strain Us9 plays an important role after retinal infection in the transfer of HSV to the OT and lateral geniculate nucleus of the brain, and that Us9 is likely involved specifically in viral DNA transport.8 However, different model systems, assays, and particular strains of virus have produced varied results.6,28–37 Our current method using in vivo tissue and DNA quantification has several advantages. First, the retinal infection model allows synchronous infection of only the cell body of the retinal ganglion cell. Second, dissection of the OT allows us to resolve the movement of virus over time and over a greater distance than is currently possible in cell culture models. Third, the DNA assay is sensitive, quantitative, and can be completed rapidly using very small amounts of tissue. We have chosen to focus on DNA delivery to the distal axon because the viral genome (contained in a capsid) is the essential component to affect encephalic spread. Whether Us9 is critical to delivery of other viral components to the axon (e.g., envelope membrane, viral light particles that lack capsid and DNA, or cellular membrane vesicles that may include viral proteins) remains to be determined.7
The spread of viral DNA differed between F and NS viral strains. Expression of F Us9 was necessary for delivery of F strain viral DNA to the distal OT at all times but most critically at 3 dpi; NS Us9, however, was necessary only at 3 dpi. This suggests that Us9 affects DNA loading or axonal delivery differently on different genetic backgrounds. One possible explanation is that the NS variant may be rescued by background compensatory mechanisms.38 Alternatively, the F strain may be attenuated with mutations that accumulate by repeated passage through cultured cells.
Brideau et al.39,40 found that an acidic cluster at amino acid residues 46 to 55 in Pseudorabies virus (PRV) Us9 was necessary for axonal spread in vivo. Based on this finding, we deleted a similar, although not identical, cluster of amino acids in the Us9 of F strain HSV (i.e., the Us9-30 HSV mutant). Using this highly sensitive viral DNA assay we found that this acidic region of Us9 protein of HSV plays a critical role for genome transport to the distal axon. The cluster includes two putative tyrosine kinase phosphorylation sites, two potential casein kinase II sites, and an N-linked glycosylation sequence.39 The absence of these potential activation sites may affect multiple steps, including assembly of capsid in the cell body or the association of nucleocapsids with cytoplasmic motor proteins.41
Several facts must be considered in evaluating the role of NS Us9 in viral spread to distal axons of retinal ganglion cells. We recognize that NS wt was an imperfect control virus for the NS Us9-. NS wt is not a genetically repaired mutant virus, although it expresses an intact Us9 protein on the same background. Secondary compensatory mutations in NS Us9- may account for the differences we found between NS wt and NS Us9-, although at 4 and 5 dpi, viral delivery is nearly identical. Because the NS wt and NS Us9- have been compared in earlier studies, we chose to use the same agents to study axonal transport in our murine model.5 Our findings provide important clarifications of previous results.
Other studies have raised the possibility that additional proteins may cooperate with Us9 in targeted transport.9,38 In our study, the Us9 gene was the targeted mutation, and expression of other genes, for example, gE, was not affected.8 Glycoproteins gE and gI as a complex may be able to direct delivery of viral DNA to the axons in the absence of Us9, although perhaps not as efficiently as in the presence of Us9.6,42 Further experiments using our current assay may clarify possible synergy and redundant mechanisms between gE/gI and Us9.
This is the first quantitative demonstration of how a gene mutation can have profoundly different phenotypic effects on the in vivo axonal delivery of virus. The complicated nature of egress and the synergy between proteins are neither fully understood nor within the scope of this article; however, the variance between the F and NS deletion mutant strains supports the complexity of the mechanism and the necessity to evaluate quantitatively the function of genes in HSV. At its simplest, the time of arrival of new viral DNA to axon endings in the CNS varies with HSV strain. This may be one factor underlying the virulence phenotypes of different strains.43
Last, the assay permits standardization of data and opens the field of experimental pathogenesis not only to a quantitative evaluation of the basic mechanisms of viral transport, but also to the study of the effects of additional variables, including viral genetic and host cell neuronal differences. In vitro evidence suggests that the Us9 protein may partner with cytoplasmic microtubule-dependent motors, such as kinesin, to transport HSV viral capsids and DNA.41,44 Once a particular binding domain is confirmed, the relevant Us9 mutant could be rapidly analyzed for its transport phenotype in this assay. The same approach would be valuable for tests of long distance, intracellular movement of cargo by other neurotropic DNA viruses, such as HSV-2, varicella virus, adenovirus, and possibly by bacteria.45–47
Acknowledgments
We thank the reviewers for helpful suggestions. Joel Baines and Kui Yang provided helpful discussions. Anatol Sucher, Chanyuan Dong, and Suling Wang supplied technical and artistic assistance.
Footnotes
Supported by Public Health Service Grant R01 EY019159 from the National Eye Institute, as well as by funds from Research to Prevent Blindness and That Man May See, Inc.
Disclosure: J.M. Draper, None; G. Huang, None; G.S. Stephenson, None; A.S. Bertke, None; D.A. Cortez, None; J.H. LaVail, None
References
- 1. Al-Dujaili LJ, Clerkin PP, Clement C, et al. Ocular herpes simplex virus: how are latency, reactivation, recurrent disease and therapy interrelated? Future Microbiol. 2011; 6: 877–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Margolis TP, LaVail JH, Setzer PY, Dawson CR. Selective spread of herpes simplex virus in the central nervous system after ocular inoculation. J Virol. 1989; 63: 4756–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol. 2011; 9: 382–394 [DOI] [PubMed] [Google Scholar]
- 4. Antinone SE, Zaichick SV, Smith GA. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J Virol. 2010; 84: 13019–13030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGraw HM, Awasthi S, Wojcechowskyj JA, Friedman HM. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J Virol. 2009; 83: 8315–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snyder A, Polcicova K, Johnson DC. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J Virol. 2008; 82: 10613–10624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor MP, Kramer T, Lyman MG, Kratchmarov R, Enquist LW. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. mBio. 2012; 3: 2e00063-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. LaVail JH, Tauscher AN, Sucher A, Harrabi O, Brandimarti R. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience. 2007; 146: 974–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howard PW, Howard TL, Johnson DC. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J Virol. 2013; 87: 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Snyder A, Wisner TW, Johnson DC. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J Virol. 2006; 80: 11165–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang K, Homa F, Baines JD. Putative terminase subunits of Herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J Virol. 2007; 81: 6419–6433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tischer B, von Einem J. Two-step RED-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques. 2006; 40: 191–197 [DOI] [PubMed] [Google Scholar]
- 13. LaVail JH, Tauscher AN, Aghaian E, Harrabi O, Sidhu SS. Axonal transport and sorting of herpes simplex virus components in mature mouse visual system. J Virol. 2003; 77: 6117–6127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burleson FG, Chambers T, Wiedbrauk DL. Virology: A Laboratory Manual. San Diego: Academic Press, Inc.; 1992: 250 [Google Scholar]
- 15. Lumb WV. Small Animal Anesthesia. Philadelphia, PA: Lea and Febiger; 1963. [Google Scholar]
- 16. Garner JA, LaVail JH. Differential anterograde transport of HSV type 1 viral strains in the murine optic pathway. J NeuroVirol. 1999; 5: 140–150 [DOI] [PubMed] [Google Scholar]
- 17. Field HJ, Tewari D, Sutton D, Thackray AM. Comparison of efficacies of famciclovir and valaciclovir against herpes simplex virus type 1 in a murine immunosuppression model. Antimicrob Agents Chemother. 1995; 39: 1114–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Winkler BS. The electroretinogram of the isolated rat retina. Vision Res. 1972; 12: 1183–1198 [DOI] [PubMed] [Google Scholar]
- 19. Broadwell RD, Brightman MW. Entry of peroxidase into neurons of the central and peripheral nervous systems from extracerebral and cerebral blood. J Comp Neurol. 1976; 166: 257–283 [DOI] [PubMed] [Google Scholar]
- 20. Canteras NS, Ribeiro-Barbosa ER, Goto M, Cipolla-Neto J, Swanson LW. The retinohypothalamic tract: comparison of axonal projection patterns from four major targets. Brain Res Rev. 2011; 65: 150–183 [DOI] [PubMed] [Google Scholar]
- 21. Smeraski CA, Sollars PJ, Ogilvie MD, Enquist LW, Pickard GE. Suprachiasmatic nucleus input to autonomic circuits identified by retrograde transsynaptic transport of pseudorabies virus from the eye. J Comp Neurol. 2004; 471: 298–313 [DOI] [PubMed] [Google Scholar]
- 22. Blotta I, Prestinaci F, Mirante S, Cantafora A. Quantitative assay of total dsDNA with PicoGreen reagent and real-time fluorescent detection. Ann Ist Super Sanita. 2005; 41: 119–123 [PubMed] [Google Scholar]
- 23. McMichael GL, Highet AR, Gibson CS, et al. Comparison of DNA extraction methods from small samples of newborn screening cards suitable for retrospective perinatal viral research. J Biomol Tech. 2011; 22: 5–9 [PMC free article] [PubMed] [Google Scholar]
- 24. Burns MJ, Nixon GJ, Foy CA, Harris N. Standardisation of data from real-time quantitative PCR methods—evaluation of outliers and comparison of calibration curves. BMC Biotechnol. 2005; 5: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pett MA. Nonparametric Statistics for Health Care Research: Statistics for Small Samples and Unusual Distributions. Thousand Oaks, CA: Sage Publications; 1997. [Google Scholar]
- 26. Enquist LW. Five questions about viral trafficking in neurons. PLoS Pathog. 2012; 8: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diefenbach RJ, Cunningham AL. eds Viral Transport, Assembly and Egress. 1st ed. Kerala, India: Research Signpost; 2011: 199 [Google Scholar]
- 28. Lee GE, Murray JW, Wolkoff AW, Wilson DW. Reconstitution of Herpes simplex virus microtubule-dependent trafficking in vitro. J Virol. 2006; 80: 4264–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ibiricu I, Huiskonen JT, Döhner K, Bradke F, Sodeik B, Grünewald K. Cryo electron tomography of Herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 2011; 7: e1002406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Antinone SE, Smith GA. Two modes of herpesvirus trafficking in neurons: membrane acquisition directs motion. J Virol. 2006; 80: 11235–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Curanovic D, Enquist L. Directional transneuronal spread of alpha-herpesvirus infection. Future Virol. 2009; 4: 591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. LaVail JH, Draper JM, Chang WC, Sretavan DW. The anterograde axonal transport of herpesviruses: a review of experimental approaches. In: Diefenbach RJ, Cunningham AL. eds Viral Transport, Assembly and Egress. Kerala, India: Research Signpost; 2011: 1–20 [Google Scholar]
- 33. Miranda-Saksena M, Boadle RA, Aggarwal A, et al. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J Virol. 2009; 83: 3187–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang J, Lazear HM, Friedman HM. Completely assembled virus particles detected by transmission electron microscopy in proximal and mid-axons of neurons infected with herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology. 2011; 409: 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Negatsch A, Granzow H, Maresch C, et al. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J Virol. 2010; 84: 13031–13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wisner TW, Sugimoto K, Howard PW, Kawaguchi Y, Johnson DC. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J Virol. 2011; 85: 5919–5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Oliveria AP, Glauser DL, Laimbacher AS, et al. Live visualization of Herpes simplex virus type 1 compartment dynamics. J Virol. 2008; 82: 4974–4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGraw HM, Friedman HM. Herpes simplex virus type 1 glycoprotein E mediates retrograde spread from epithelial cells to neurites. J Virol. 2009; 83: 4791–4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brideau AD, Eldridge MG, Enquist LW. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J Virol. 2000; 74: 4549–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brideau AD, Card JP, Enquist LW. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J Virol. 2000; 74: 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology. 2008; 378: 347–354 [DOI] [PubMed] [Google Scholar]
- 42. Wang F, Tang W, McGraw HM, Bennett J, Enquist LW, Friedman HM. Herpes simplex virus type 1 glycoprotein E is required for axonal localization of capsid, tegument, and membrane glycoproteins. J Virol. 2005; 79: 13362–13372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brandt CR. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Exp Eye Res. 2004; 80: 607–621 [DOI] [PubMed] [Google Scholar]
- 44. Radtke K, Kieneke D, Wolfstein A, et al. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010; 6: e1000991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salinas S, Bilsland LG, Henaff D, et al. CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog. 2009; 5: e1000442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Antal EA, Loberg EM, Bracht P, Melby KK, Maehlen J. Evidence for intraaxonal spread of Listeria monocytogenes from the periphery to the central nervous system. Brain Pathol. 2001; 11: 432–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dons L, Jin Y, Kristensson K, Rottenberg ME. Axonal transport of Listeria monocytogenes and nerve-cell induced bacterial killing. J Neurosci Res. 2007; 85: 2529–2537 [DOI] [PubMed] [Google Scholar]