

Abstract
Studies of protein dynamics, structure and interactions using hydrogen/deuterium exchange mass spectrometry (HDX-MS) have sharply increased over the past 5–10 years. The predominant technology requires fast digestion at pH 2–3 to retain deuterium label. Pepsin is used almost exclusively, but it provides relatively low efficiency under the constraints of the experiment, and a selectivity profile that renders poor coverage of intrinsically disordered regions. In this study we present nepenthesin-containing secretions of the pitcher plant Nepenthes, commonly called monkey cups, for use in HDX-MS. We show that nepenthesin is at least 1400-fold more efficient than pepsin under HDX-competent conditions, with a selectivity profile that mimics pepsin in part, but also includes efficient cleavage C-terminal to “forbidden” residues K, R, H, and P. High efficiency permits a solution-based analysis with no detectable autolysis, avoiding the complication of immobilized enzyme reactors. Relaxed selectivity promotes high coverage of disordered regions and the ability to “tune” the mass map for regions of interest. Nepenthesin-enriched secretions were applied to an analysis of protein complexes in the nonhomologous end-joining DNA repair pathway. The analysis of XRCC4 binding to the BRCT domains of Ligase IV points to secondary interactions between the disordered C-terminal tail of XRCC4 and remote regions of the BRCT domains, which could only be identified with a nepenthesin-based workflow. HDX data suggest that stalk-binding to XRCC4 primes a BRCT conformation in these remote regions to support tail interaction, an event which may be phosphoregulated. We conclude that nepenthesin is an effective alternative to pepsin for all HDX-MS applications, and especially for the analysis of structural transitions among intrinsically disordered proteins and their binding partners.
Mass spectrometry has served the biochemical and biological communities by providing the capacity for protein identification and characterization, but in the last several years it has also become a powerful tool for interrogating protein structure and dynamics (1, 2). Solution-phase hydrogen/deuterium exchange (HDX)1, when coupled with mass spectrometry (MS), provides rich sets of data that can be mined to extract structural and dynamic parameters from proteins (3–7). In many cases, the technique is used in experimental situations in which x-ray diffraction analysis or other biophysical techniques (e.g. NMR, cryoEM) are difficult to apply, which is particularly true in the structure-function analysis of protein interactions (8). Most applications of the method involve deuteration of a protein in two or more states, and differential labeling data is extracted at the highest structural resolution possible. Although there have been some impressive developments in top-down protein analysis using newer ion fragmentation methods for label localization (9, 10), most studies continue to employ a bottom-up strategy, in which the protein is digested with an enzyme, and the label is quantified by mass analysis of the resulting peptides.
The reasons for the prominence of the bottom-up approach to HDX-MS are shared with the corresponding proteomics method. Peptides may be detected with more sensitivity than proteins, and samples of considerably higher complexity can be interrogated (11). Analytically, this shifts the focus toward optimizing protein digestion, to cover 100% of the sequence and generate a high degree of overlapping fragments to increase opportunities for localizing the deuterium label at high resolution (12–14).
The unique requirements of the HDX-MS workflow unfortunately place restrictions on the digestion enzymes that may be used. To avoid label loss through back-exchange to nondeuterated solvent, the kinetics of exchange must be dramatically slowed by pH and temperature reduction, and even under these conditions the digestion must be done rapidly. Conventional methods employ a pH of ∼2.5 and temperatures of 4–10 °C. The aspartic protease pepsin can function under such conditions, which has led to its prominence for HDX-MS applications. This does not mean the enzyme is ideal. Several laboratories have sought to identify other enzymes or develop analytical solutions that address its shortcomings, which include extensive autolysis, modest efficiency, and nonideal substrate specificity. Currently, most methods involve either enzyme microreactors presenting high concentrations of pepsin in a flow-through system (15), or solution-phase digestions using pepsin and protease XIII in separate experiments (16, 17). The latter enzyme is also an aspartic protease with a sequence specificity that partially overlaps pepsin, thus the two maps together tend to somewhat improve sequence coverage (18). A method that fuses the two strategies has recently been described, involving tandem pepsin and protease XIII microreactors (13).
Neither strategy is likely to be ideal when applied to protein complexes of far greater complexity, to support of structure-building activities or dynamics analysis of multiprotein “machines.” The microreactor approach complicates the front-end fluidic system, and can lead to sample loss and carryover (19). The solution-phase method requires large amounts of enzyme resulting in contamination because of enzyme autolysis (18). More importantly, neither fully overcomes the low efficiency of these enzymes. In many cases, changing the presentation of substrate can change the sequence map considerably. For example, the sequence map of a protein can be distorted when bound to a second protein (20). A map dependent on the protein load complicates the comparison of deuteration levels for the protein in different states. This alteration suggests a level of substrate inhibition (21), and probably reflects a wide range of specificity constants (kcat/Km) across the many individual cleavage sites presented in a protein substrate (22). Similar issues have been noted with trypsin (23). Overall, the sequence maps generated using these methods are serviceable for samples of lower complexity, but low enzymatic efficiency of the available proteases remains an important limitation and a key driver in the search for novel proteases (24).
In this study, we characterize the proteolytic activity of secretions from the Nepenthes genus (25), arising from the aspartic protease nepenthesin (26), and evaluate the enzyme for use within HDX applications. Nepenthesin displays remarkably high cleavage efficiency for a broad range of substrates at low pH and temperature, which promotes high sequence coverage for a collection of proteins selected from ongoing HDX projects in our laboratories. Globally, we demonstrate that it outperforms pepsin in sequence coverage and can be used in a simple workflow for broad sequence coverage, or targeted toward a desired area of protein sequence. This new tool was applied to an HDX-MS characterization of a protein complex involved in the nonhomologous end-joining (NHEJ) pathway of DNA damage repair, and the results support a model of the complex proposed from SAXS data (27).
EXPERIMENTAL PROCEDURES
Plants and Fluid Processing
Transplants of several Nepenthes varieties were acclimated in a small terrarium. On pitcher maturity, the plants were fed with one or two Drosophila per pitcher and the pitcher fluid harvested 1 week later. Pitchers and their secretions were left to recover for 1 week before a second round of feeding and extraction. All pitcher fluid was combined and clarified through a 0.22 μm filter, and then concentrated 80- to 100-fold using an Amicon Ultra 10 kDa molecular weight cutoff centrifugal filter (both from Millipore). Before use in digestions, the concentrate was acid-activated with 100 mm glycine HCl (pH 2.5) for 3 h, then washed 3 × with 100 mm glycine-HCl (pH 2.5) in the filtration device, using 10 × fluid volume for each wash. The final isolate was then rediluted to an 11 × concentration based on the original sampling of pitcher fluid. Additional details on plant horticulture, feeding and fluid extraction can be found in Supplementary Methods.
Digest Mapping by Mass Spectrometry
Digestions were carried out in solution using a HTX-PAL autosampler and dispensing system designed for HDX applications (Leap Technologies, Carrboro, NC), and data were collected using an AB Sciex Triple-TOF 5600 QqTOF mass spectrometer. Peptides were identified with Mascot (v2.3) from MS/MS data, from .mgf files created in Analyst TF v1.51. Data were mapped to sequence using the following search terms: a mass tolerance of 10 ppm on precursor ions and 0.6 Da on fragment ions, no modifications, and no enzyme specificity. A standard probability cutoff of p = 0.05 was implemented and matches near the cutoff manually verified. For nepenthesin-based digestion, 8 μm protein solutions (XRCC4, XLF, Ligase IV-tandem BRCT domains, PNK, myoglobin, or cytochrome C) were mixed with 11 × concentrated fluid for 2 min at 10 °C, in various ratios (see text). Information on the production of XRCC4, XLF, BRCT domains, and PNK can be found in Supplementary Methods. Myoglobin and cytochrome C were purchased from Sigma. After dilution to 1 μm substrate concentration, 15 μl were injected into the chilled reversed-phase LC system (4 °C) connected to the mass spectrometer. The peptides were trapped on a 5 cm, 200 μm i.d. Onyx C18 monolithic column (Phenomenex Inc., Torrance, CA) and eluted with an acetonitrile gradient from 3% to 40% in 10 min. Peptides detected in these analyses were selected for CID fragmentation in multiple information-dependent acquisitions of MS/MS spectra, akin to the gas-phase fractionation strategy (28). Spectra were searched against a miniature database containing the sequences for all six proteins. Sequencing results were manually verified.
HD Exchange of a Complex Involved in DNA Damage Repair
Stock solutions of XRCC4(1–200) with BRCT, and XRCC4(full length) with BRCT were diluted in buffer (10 mm Tris-HCl, pH 7.5) to equimolar concentrations (10 μm), and incubated at 4 °C for a minimum of 30 min to promote complexation. The samples were held at 4 °C until HDX analysis. Aliquots were deuterated for 2 min at 20 °C with the addition of D2O (25% v/v). Aliquots were then digested in two ways. In the first digestion strategy, protein deuteration was quenched by adding the sample to chilled 100 mm glycine-HCl (pH 2.5), and the quenched protein solution was injected into a pepsin microreactor. This microreactor was installed in the HTX-PAL system between the injector valve and the C18 column. Protein digest was captured on the monolithic C18 capillary column and eluted into the mass spectrometer. All fluidic elements, including the microreactor, were chilled at 4 °C to minimize deuterium back-exchange during the analysis time (<15 min). In the second digestion strategy, an equivalent amount of deuterated protein was simultaneously quenched and digested with 3 or 5 μl of 11 × nepenthes fluid for 3 or 5 min, respectively, at 10 °C. The samples were then injected into the chilled LC-system connected to the mass spectrometer.
Replicate mass shift measurements were made (four or more) and referenced to control protein states—free XRCC4(1–200), free XRCC4(full length), and free LigIV-BRCT. The average deuterium level for each peptide was determined using Mass Spec Studio (manuscript in preparation), which is a rebuild of Hydra v1.5 (29). Perturbed mass shifts were considered significant if they (a) passed a two-tailed t test (p < 0.05) using pooled standard deviations from the analyses of each state, (b) passed a distribution analysis to guard against spectral overlap, and (c) exceeded a threshold shift value (±2 s.d.) based on a measurement of the shift noise and assuming its normal distribution (30).
Chemicals
Water and acetonitrile, HPLC grade from Burdick and Jackson, were purchased from VWR. Formic acid, Tris, and glycine were purchased from Sigma Aldrich.
RESULTS
Pitcher Fluid Extract
The fluidic secretions of the pitcher plant were filtered, concentrated and the nepenthesin activated by pH reduction (pH 2.5), approximately the same pH as found in the wild. A low complexity and protein content of the activated fluid was observed by SDS-PAGE, with the presence of nepenthesin confirmed by LC-MS/MS (see Supplementary Methods). The total protein concentration of the activated and 80 × enriched fluid was measured by a BCA assay to be 22 ng/μl. The simplicity of the fluid proteome is expected based on earlier studies (31, 32), in which nepenthesin was found to be a major component. For the purposes of our study, we assume 22 ng/μl represents the nepenthesin concentration in the 80 × enriched fluid, recognizing that it is likely an overestimate. Yields of this magnitude are consistent with earlier reports (33).
We then digested a series of proteins with the enriched fluid under conditions suitable for HDX-MS experiments, and characterized the digestion specificity of the concentrate at the P1 and P1′ positions (Fig. 1), to support a comparison with similar studies applied to pepsin (34). In our experiments, the enzyme-to-substrate ratio was 1:85 based on the above assumption that all the measured protein in the enriched fluid is nepenthesin. The nepenthesin data represents an assessment of 1612 residues and although not as extensive as the corresponding pepsin data (13,766 residues), the sequence diversity is sufficiently high in the protein set to warrant a comparison at the level of P1 and P1′ positions at least. The greatest specificity for pepsin is clearly in the P1 position. It presents high-efficiency cleavage for the hydrophobic residues F, L, and M but cleavage after P, H, K, and R is essentially forbidden. Nepenthesin cleaves after most residues with the exception of G, S, T, V, I, and W. It supports a high rate of cleavage after the expected pepsin P1 residues but also at the residues forbidden in pepsin digestion, notably K, R, and P. In the P1′ position, pepsin shows a preference for hydrophobic residues in general, including any residue with aromaticity. Conversely, nepenthesin demonstrates little in the way of selectivity at the P1′ position, except perhaps against G, P, and H. The significantly relaxed specificity relative to pepsin is remarkable for an aspartic protease, and the selectivity data provides an early indication of very high enzymatic efficiency.
Fig. 1.
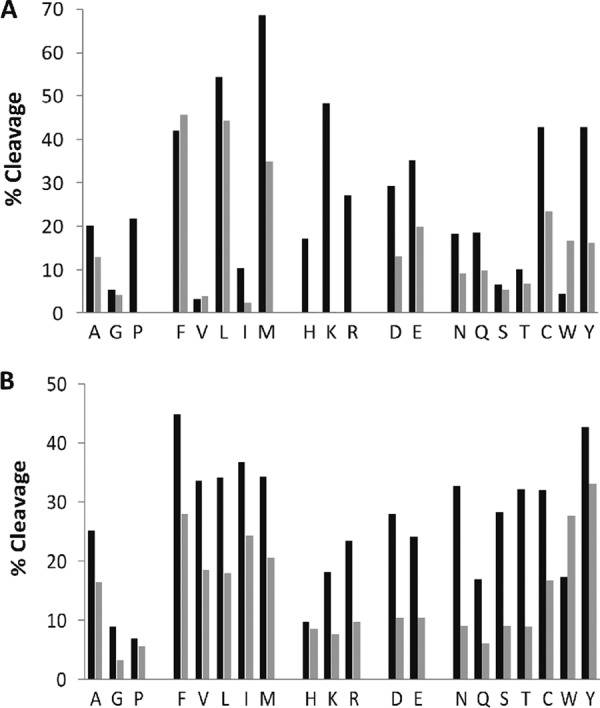
Nepenthesin cleavage preferences at (A) the P1 or N-terminal side of the cleavage site and at (B) the P1′ or C-terminal side of the cleavage site. Data is grouped according to amino acid type and compared with a similar rendering of pepsin data from Hamuro et al. (34) Black bars indicate nepenthesin digestion and the gray bars pepsin digestion. The % cleavage represents the number of observed cleavages at the given residue, relative to the total number of the given residues in the set. Nepenthesin data were obtained from 2 min digests of six denatured proteins, as described in the text.
To determine if this relaxed specificity translates into improved sequence mapping for HDX-MS applications, we profiled full-length XRCC4, a protein that contains a globular domain, an extended helical stalk, and a long disordered C-terminal (27, 35). Such multidomain proteins are challenging to encompass in a single digestion protocol, and in particular, intrinsically disordered regions tend to digest poorly with pepsin as they are relatively depleted in hydrophobic residues and enriched in proline, and charged residues (36). The pepsin and nepenthesin maps for this protein are displayed in Fig. 2. In this comparison, an exhaustive mapping was pursued for both enzymes, using a range of different protease/substrate ratios, and recursive MS/MS experiments. Nepenthesin provides superior coverage of the full length protein: 357 peptides for nepenthesin compared with 187 for pepsin, but with the same average peptide length (11 residues). Both enzymes represent the globular and stalk regions with a large number of overlapping peptides but the complementarity provided by nepenthesin is evident. For example, nepenthesin offers considerably deeper coverage of a β-sheet region in the globular domain (residues 1–30). The disordered C-terminal region is covered to a much greater extent as well, and to a considerably higher level of redundancy. On average, each residue in this disordered tail region receives 16X coverage using nepenthesin and only 4.7 × coverage with pepsin.
Fig. 2.
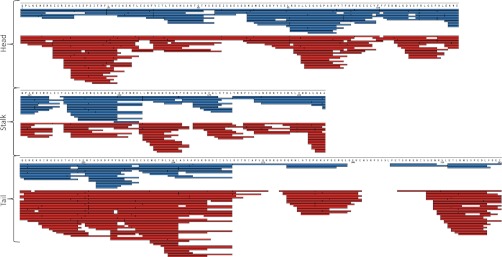
XRCC4 composite peptide sequence map, arranged according to domain type. Peptides obtained using pepsin digestion at four different enzyme to substrate ratios (65:1 to 520:1, blue bars), and using nepenthesin digestion at four different enzyme to substrate ratios (0.0075:1 to 0.38:1, red bar).
The high cleavage efficiency C-terminal to basic residues prompted us to explore if there exists any bias in peptide detection. This could be tested in several ways, but we chose to select average search score as the metric (Fig. 3). The approach emphasizes confidence in sequence identification as the principal means by which sequence maps are defined. There is only one outlier, R. The higher scores for peptides terminating in R likely reflect a combination of higher average peptide intensity and better fragmentation, which is consistent with what we know from trypsin-based bottom-up proteomics (37).
Fig. 3.

Average MASCOT score of peptides obtained after nepenthesin digestion, grouped by C-terminal amino acid. The number of peptides used for each calculation is associated with the terminal amino acid, above the bar. Peptides were obtained from the digests of six denatured proteins, as described in the text.
We then examined enzyme efficiency in greater detail and the degree to which the peptide mass map could be varied, or tuned, simply by altering the enzyme-to-substrate ratio (Fig. 4). Nepenthesin load was varied over a 50-fold range for in-solution digestions. For the pepsin experiment, immobilized pepsin in a slurry format was used rather than free pepsin so that we avoided extensive pepsin autolysis. The enzyme load was varied over an eightfold range; lower amounts led to poor peptide intensities and higher amounts had no effect on the map. We note that nepenthesin generated a very low autolysis profile even at the higher loads (see supplemental Fig. S1). We used an aggregate peptide ion chromatogram (PIC) as a measure of effective digestion, reasoning that a distribution peaking toward low elution times presents a digest with a bias toward smaller peptides, which are more useful for HDX-MS applications. Comparing the relatively similar distributions found at 0.38:1 (nepenthesin/substrate) with 520:1 (pepsin/substrate) represents a remarkable 1400-fold improvement in efficiency for nepenthesin over pepsin in HDX-like applications. Similarly high efficiencies have been realized in the digestions of other proteins (not shown), thus the effect is not limited to XRCC4.
Fig. 4.

Peptide ion chromatograms (PICs) of XRCC4 digested with (A) nepenthesin and (B) pepsin over a range of enzyme to substrate ratios (shown in the legends). PICs for enzymatic digestion were generated from the same mass-load of substrate on column.
The nepenthesin digest could be more readily tuned from large fragments to small by varying the enzyme load, which generated variable representation of XRCC4. This is demonstrated in Fig. 4A by the transition in the PIC from long retention times at low load, to short retention times at high load. This transition correlated with a decrease in the average peptide length for the most abundant peptides, from >12 at low enzyme load to 10 at high enzyme load. Conversely, varying pepsin load did not significantly alter the PIC or average peptide length (Fig. 4B). A forced-flow pepsin microreactor may improve tuning but would likely not generate smaller fragments.
To explore the utility of nepenthesin in HDX-MS, we applied it to an analysis of Ligase IV interactions with XRCC4, a scaffolding protein in a DNA damage repair complex of the nonhomologous end-joining (NHEJ) pathway. The tandem BRCT domains of Ligase IV were complexed with both full length XRCC4 and a truncated form of XRCC4(1–200), in which the intrinsically disordered region was removed (supplemental Fig. S2). The analysis of full-length XRCC4 used a conventional pepsin-based approach, and as expected the regional changes in deuteration for each protein correlate with the stabilization of XRCC4, and the binding interface in general (Fig. 5 middle, and supplemental Fig. S3). The coiled coil “stalk” domain of the XRCC4 dimer is strongly stabilized on complexation to the XRCC4 dimer, as is the clamp-shaped helix-loop-helix in the linker between the tandem BRCT domains. The stalk domain is slightly stabilized beyond the binding site proper, which is consistent with earlier studies (38). There is no structure of the interaction involving the full-length XRCC4, so we compared deuteration results with a structure using a truncated form (39). We expected to observe reduced deuteration in other regions of BRCT beyond the clamp region, as the binding interface extends beyond the clamp and is not contiguous. We see reductions in the 3:5 β-hairpin of the BRCT1 domain, as well as in loop 1 and α1′ of the BRCT2 domain, which indeed is consistent with the map of the interface (39). However, it was unexpected to see significantly reduced deuteration in regions of the tandem BRCT domains not involved in the interface (Fig. 5 middle, blue boxes). To determine if these reflect an allosteric effect on binding to the stalk, or perhaps a novel binding interface, we compared these results to an HDX-MS analysis of tandem BRCT complexed to the truncated form of XRCC4(1–200). The change profile is almost identical, except we now see an increased deuteration in remote regions, relative to the free protein (Fig. 5 left, blue boxes). That is, BRCT binding to the truncated XRCC4 destabilizes these regions, but they become stabilized when the XRCC4 tails are present (see also supplemental Fig. S3).
Fig. 5.

Differential deuteration levels mapped to the available structure of XRCC4(1–200) complexed with the tandem BRCT domains (PDB 3II6). Middle structure: comparing full-length XRCC4 and free BRCT(631–911) domains to the complex. Left structure: comparing truncated XRCC4(1–200) and free BRCT(631–911) domains to the complex. Right structure: differential deuteration as in the middle structure, with a model of the disordered tails folding back onto the BRCT domains. For clarity, the corresponding altered deuteration in the tails is omitted. Red represents decreased labeling on complexation in all figures, green represents increased labeling, and the blue boxes mark the distal regions in the tandem BRCT domains described in the text. The approximate location of XRCC4 phosphorylation sites are indicated with a blue P.
The pepsin map did not permit full analysis of the disordered tail (as shown in Fig. 2), so we used nepenthesin and repeated the HDX-MS analysis. The controllable nature of nepenthesin digestion permitted a region specific optimization of the map, which we tuned to elements of the tail region (Fig. 6). On confirming that the nepenthesin digestion strategy generated comparable deuteration results for the binding interface (supplemental Fig. S4), we compared the full-length XRCC4-BRCT to the corresponding free proteins. A segment of the tail shows significant reductions in deuteration on complexation (residues 213–251), but the remaining portions of the tail remained highly deuterated (supplemental Fig. S4). These findings suggest that the tails fold back on the tandem BRCT domains, in the regions we observed to undergo tail-induced reductions in deuteration (modeled in Fig. 5, right). An interaction of this nature is consistent with structural models built on SAXS data (27). The asymmetrical orientations of the tandem BRCT domains on the stalk suggest that the tails would interact differently. Reduced deuteration in residues 213–223 is consistent with one tail binding to one BRCT domain, and reduced deuteration at residues 242–251 with the other tail binding to the other BRCT domain.
Fig. 6.

Peptide map for the N-terminal portion of the XRCC4 tail region. Increasing the enzyme concentration 2-fold and the digestion time from 3 to 5 min improves the map in this region of the tail. Red map corresponds to the weaker digestion condition and the blue map to the stronger digestion conditions.
DISCUSSION
The only endopeptidases that are currently used for bottom-up HDX-MS experiments are drawn from the acid protease class. These enzymes are chosen primarily for their digestion efficiency in the 2–3 pH range, where HDX-MS must be performed, to suppress loss of peptide deuteration into nondeuterated solvent during the sample workup stage. Unfortunately, acid proteases have not offered up many alternatives to pepsin, and these are needed. Using a single enzyme leads to biased sequence maps. Protein regions may simply not process well, because the selectivity of pepsin may not match with the amino acid composition of the region. This is especially true for intrinsically disordered proteins, an area of great interest in protein biochemistry. Such regions are depleted in pepsin cleavage sites and enriched in forbidden sites (36). Equally important is the general inefficiency of pepsin relative to the demands of the HDX-MS workflow. Most methods employ very large amounts of enzyme to compensate, and this will likely limit HDX-MS methods to protein complexes and systems of low to moderate complexity.
Pepsin is a member of the aspartic acid protease subclass, and several members of the class have been assessed for use within the HDX-MS workflow. Aside from protease XIII, no other enzymes have entered common use. In our assessment of various candidates, we considered nepenthesin (EC 3.4.23.12) based on its phylogenetic remoteness from pepsin (40). This enzyme is produced in carnivorous plants of the Nepenthes genus. Plants in this genus have long fascinated botanists interested in mechanisms of insect trapping and nutrient uptake (25, 41), and more recently, they have captured the imagination of materials researchers developing new lubrication technology (42). The proteome of the unfractionated fluid is remarkably simple, containing three to nine major proteins in the unstimulated secretions (31, 32). Two major components are nepenthesin I and II, where I is present at ∼10-fold higher levels than II (43). Initial characterizations of the enzyme suggested a selectivity profile similar to pepsin, but including cleavage on either side of D and at K, R (26, 33). Early evidence also indicated an optimum digestion pH of 2.6 (44), and that the enzyme may be more efficient than pepsin (33). For these reasons, we evaluated it for HDX-MS applications as an alternative to pepsin. Proteome analysis confirmed the presence of nepenthesin I/II and the overall simplicity of the fluid, at the protein level. Activating the fluid by pH reduction proved an effective and reproducible means of “scrubbing” the fluid. It seemed to promote digestion of contaminating protein before protein concentration (from bacterial and drosophila sources), as seen in the very low background in the fluid. The low background also serves to highlight very low levels of autolysis.
Nepenthesin enriched as described is significantly more efficient than pepsin in processing protein substrates for HDX-MS. Enzyme levels need to be 1400-fold higher for pepsin-based experiments to achieve comparable peptide maps, assuming that nepenthesin is the only protein in the enriched pitcher fluid proteome. Our experiments using pepsin were performed at 4 °C, and 10 °C for nepenthesin, but we have not found the digestion efficiency of pepsin to increase significantly at 10 °C. It would be of some benefit to decrease digestion temperatures further for nepenthesin digestions, but we note a modest drop in efficiency at 4 °C. We believe this is due either to extensive enzyme glycosylation, the presence of complex carbohydrate in the enriched fluid fraction increasing viscosity, or both. This will be explored in the future.
The current study improves our understanding of the substrate specificity for nepenthesin. It is significantly broader than pepsin, and much less specific than first reported, which leads to a sequence map that can be highly tuned as a result. This degree of relaxed specificity is unique among the acid proteases tested to date for HDX-MS. It raises the potential for “over-digestion” to individual amino acids but overdigestion is not very common for single endoproteases. Even with high levels of enzyme we still observe a large number of sequenceable peptides, which probably highlights that secondary interaction sites remote from the actual catalytic site are needed for processing, as has been noted with pepsin (22, 45). There is no structure available for nepenthesin I aside from a homology model derived from pepsin (43). If accurate, the model provides support for an extended binding site, and further suggests that the impressive pH and temperature stability of the enzyme derives in part from extensive disulfide bridges. We propose that the broader specificity results not from an altered catalytic site, but from the stabilization of a more active enzyme conformer. For pepsin, kcat values vary over a much wider range than their corresponding Km values, suggesting that an enzyme conformational change is needed to promote hydrolysis of certain substrates (the “induced fit” model) (22). It is possible that nepenthesin I/II is less dependent on such a change, where the mechanism of hydrolysis is closer to “lock and key.”
The high efficiency and resistance of nepenthesin to autolysis improves the HDX-MS workflow. Automated analyses do not require online enzyme microreactors, but simple solution dispensing and incubation. At least with the monolithic columns used in our LC-MS analyses, there was no evidence the enzyme preparation clogged columns, even after hundreds of runs. “Tuning” the sequence map to optimize the analysis can therefore be readily controlled by dispensing different volume ratios of the enzyme extract. In our hands, pepsin is not amenable to such tuning, in either free or immobilized form.
We also show that nepenthesin-based HDX-MS analyses are broadly equivalent to the conventional pepsin-based method, and that nepenthesin provides unique opportunities to analyze disordered regions. HDX-MS is one of few methods with the potential to study structural transitions in “disordered” proteins, but the limitation of pepsin in these regions is problematic. The high coverage we observe in the of XRCC4 results from cleavage at proline and charged residues, which are sites with no to low cleavage probability using pepsin. Functionally, the tail has been proposed to sequester XRCC4 until association with the BRCT domains of DNA Ligase IV is needed (27), an enzyme that is responsible for DNA end ligation in the NHEJ repair mechanism. This sequestration is thought to involve interaction of the tails with or near the XRCC4 head domain, so we might expect the tails to increase in deuteration when the BRCT domains bind. We do not observe this, but rather reduced deuteration in segments of the tail. These reductions correlate with a similar reduction in two distal regions of the tandem BRCT domains. Intriguingly, when these tandem BRCT domains bind to the stalk region of XRCC4 without the tails, the distal regions appear to reorganize first, suggested by their higher deuteration profiles. We propose that stalk-binding creates a permissive binding groove in each BRCT domain, which then engages the C-terminal tails of XRCC4 for a coordinated, multisite interaction between the two proteins. This type of tail interaction is supported by a modeling exercise based on SAXS data (27). Interestingly, the proposed binding regions in the tail span one known CK2 phosphorylation site (T233) (46, 47) and one predicted site (S232), which suggests a mechanism for regulating an interaction that may have important functional significance to the DNA end-joining mechanism.
Overall, nepenthesin is an important addition to HDX technology and has replaced pepsin in our laboratory as the enzyme of choice. The high digestion efficiency coupled with a more broadly distributed amino acid specificity should render most protein substrates amenable to processing for HDX-MS. We note that membrane proteins in addition to disordered proteins may be better suited to nepenthesin as well. Our initial efforts with integral membrane proteins are suggestive of this, perhaps due in part to detectable lipase activity in the pitcher fluid (33). Future studies will explore this in greater depth. Sufficient enzyme for a large number of projects is easily accessible to any lab willing to grow and feed a modest number of plants.
Supplementary Material
Footnotes
* The work was supported by NSERC Discovery Grant 298351-2010 (DCS) and CIHR grant 13639 (SPLM). DCS acknowledges the additional support of the Canada Research Chair program, Alberta Ingenuity-Health Solutions and the Canada Foundation for Innovation.
 This article contains supplemental S1 S4.
This article contains supplemental S1 S4.
1 The abbreviations used are:
- HDX-MS
- hydrogen/deuterium exchange mass spectrometry
- NHEJ
- nonhomologous end-joining
- XRCC4
- X-ray repair cross-complementing protein 4
- BRCT
- BRCA1 (breast cancer suppressor protein) C-terminus
- XLF
- XRCC4-like factor
- PNK
- polynucleotide kinase
- NMR
- nuclear magnetic resonance
- cryoEM
- cryo electron-microscopy
- SAXS
- small-angle x-ray scattering
- CK2
- casein kinase 2.
REFERENCES
- 1. Konermann L., Pan J., Liu Y. H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 [DOI] [PubMed] [Google Scholar]
- 2. Xu G., Chance M. R. (2007) Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 107, 3514–3543 [DOI] [PubMed] [Google Scholar]
- 3. Soon F. F., Ng L. M., Zhou X. E., West G. M., Kovach A., Tan M. H., Suino-Powell K. M., He Y., Xu Y., Chalmers M. J., Brunzelle J. S., Zhang H., Yang H., Jiang H., Li J., Yong E. L., Cutler S., Zhu J. K., Griffin P. R., Melcher K., Xu H. E. (2012) Molecular Mimicry Regulates ABA Signaling by SnRK2 Kinases and PP2C Phosphatases. Science 335, 85–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Devarakonda S., Gupta K., Chalmers M. J., Hunt J. F., Griffin P. R., Van Duyne G. D., Spiegelman B. M. (2011) Disorder-to-order transition underlies the structural basis for the assembly of a transcriptionally active PGC-1 alpha/ERR gamma complex. Proc. Natl. Acad. Sci. U.S.A. 108, 18678–18683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang J., Chalmers M. J., Stayrook K. R., Burris L. L., Wang Y., Busby S. A., Pascal B. D., Garcia-Ordonez R. D., Bruning J. B., Istrate M. A., Kojetin D. J., Dodge J. A., Burris T. P., Griffin P. R. (2011) DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat. Struct. Mol. Biol. 18, 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts V. A., Pique M. E., Hsu S., Li S., Slupphaug G., Rambo R. P., Jamison J. W., Liu T., Lee J. H., Tainer J. A., Ten Eyck L. F., Woods V. L., Jr. (2012) Combining H/D exchange mass spectroscopy and computational docking reveals extended DNA-binding surface on uracil-DNA glycosylase. Nucleic Acids Res. 40, 6070–6081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chung K. Y., Rasmussen S. G., Liu T., Li S., DeVree B. T., Chae P. S., Calinski D., Kobilka B. K., Woods V. L., Jr., Sunahara R. K. (2011) Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature 477, 611–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iacob R. E., Engen J. R. (2012) Hydrogen Exchange Mass Spectrometry: Are We Out of the Quicksand? J. Am. Soc. Mass Spectrom. 23, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan J., Han J., Borchers C. H., Konermann L. (2009) Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J. Am. Chem. Soc. 131, 12801–12808 [DOI] [PubMed] [Google Scholar]
- 10. Rand K. D., Zehl M., Jensen O. N., Jørgensen T. J. (2009) Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal. Chem. 81, 5577–5584 [DOI] [PubMed] [Google Scholar]
- 11. Garcia B. A. (2010) What Does the Future Hold for Top Down Mass Spectrometry? J. Am. Soc. Mass Spectrom. 21, 193–202 [DOI] [PubMed] [Google Scholar]
- 12. Fajer P. G., Bou-Assaf G. M., Marshall A. G. (2012) Improved Sequence Resolution by Global Analysis of Overlapped Peptides in Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 23, 1202–1208 [DOI] [PubMed] [Google Scholar]
- 13. Mayne L., Kan Z. Y., Chetty P. S., Ricciuti A., Walters B. T., Englander S. W. (2011) Many Overlapping Peptides for Protein Hydrogen Exchange Experiments by the Fragment Separation-Mass Spectrometry Method. J. Am. Soc. Mass Spectrom. 22, 1898–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Landgraf R. R., Chalmers M. J., Griffin P. R. (2012) Automated Hydrogen/Deuterium Exchange Electron Transfer Dissociation High Resolution Mass Spectrometry Measured at Single-Amide Resolution. J. Am. Soc. Mass Spectrom. 23, 301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L., Pan H., Smith D. L. (2002) Hydrogen exchange-mass spectrometry - Optimization of digestion conditions. Mol. Cell. Proteomics 1, 132–138 [DOI] [PubMed] [Google Scholar]
- 16. Cravello L., Lascoux D., Forest E. (2003) Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. 17, 2387–2393 [DOI] [PubMed] [Google Scholar]
- 17. Mazon H., Marcillat O., Forest E., Vial C. (2005) Local dynamics measured by hydrogen/deuterium exchange and mass spectrometry of creatine kinase digested by two proteases. Biochimie 87, 1101–1110 [DOI] [PubMed] [Google Scholar]
- 18. Zhang H. M., Kazazic S., Schaub T. M., Tipton J. D., Emmett M. R., Marshall A. G. (2008) Enhanced Digestion Efficiency, Peptide Ionization Efficiency, and Sequence Resolution for Protein Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 80, 9034–9041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Y., Kaveti S., Engen J. R. (2006) Extensive deuterium back-exchange in certain immobilized pepsin columns used for H/D exchange mass spectrometry. Anal. Chem. 78, 1719–1723 [DOI] [PubMed] [Google Scholar]
- 20. Zhang Q., Willison L. N., Tripathi P., Sathe S. K., Roux K. H., Emmett M. R., Blakney G. T., Zhang H. M., Marshall A. G. (2011) Epitope Mapping of a 95 kDa Antigen in Complex with Antibody by Solution-Phase Amide Backbone Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 83, 7129–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruan C. Q., Chi Y. J., Zhang R. D. (2010) Kinetics of hydrolysis of egg white protein by pepsin. Czech J. Food Sci. 28, 355–363 [Google Scholar]
- 22. Sachdev G. P., Fruton J. S. (1975) Kinetics of Action of Pepsin on Fluorescent Peptide Substrates. Proc. Natl. Acad. Sci. U.S.A. 72, 3424–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Slysz G. W., Schriemer D. C. (2005) Blending protein separation and peptide analysis through real-time proteolytic digestion. Anal. Chem. 77, 1572–1579 [DOI] [PubMed] [Google Scholar]
- 24. Marcoux J., Thierry E., Vivès C., Signor L., Fieschi F., Forest E. (2010) Investigating Alternative Acidic Proteases for H/D Exchange Coupled to Mass Spectrometry: Plasmepsin 2 but not Plasmepsin 4 Is Active Under Quenching Conditions. J. Am. Soc. Mass Spectrom. 21, 76–79 [DOI] [PubMed] [Google Scholar]
- 25. Vines S. H. (1876) On the digestive ferment of nepenthes. J. Anat. Physiol. 11, 124–127 [PMC free article] [PubMed] [Google Scholar]
- 26. Amagase S., Nakayama S., Tsugita A. (1969) Acid protease in nepenthes . 2. Study on specificity of nepenthesin. J. Biochem. 66, 431–439 [DOI] [PubMed] [Google Scholar]
- 27. Hammel M., Yu Y., Fang S. J., Lees-Miller S. P., Tainer J. A. (2010) XLF Regulates Filament Architecture of the XRCC4.Ligase IV Complex. Structure 18, 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blonder J., Rodriguez-Galan M. C., Lucas D. A., Young H. A., Issaq H. J., Veenstra T. D., Conrads T. P. (2004) Proteomic investigation of natural killer cell microsomes using gas-phase fractionation by mass spectrometry. Biochim. Biophys. Acta 1698, 87–95 [DOI] [PubMed] [Google Scholar]
- 29. Slysz G. W., Baker C. A. H., Bozsa B. M., Dang A., Percy A. J., Bennett M., Schriemer D. C. (2009) Hydra: software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinf. 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bennett M. J., Barakat K., Huzil J. T., Tuszynski J., Schriemer D. C. (2010) Discovery and Characterization of the Laulimalide-Microtubule Binding Mode by Mass Shift Perturbation Mapping. Chem. Biol. 17, 725–734 [DOI] [PubMed] [Google Scholar]
- 31. Hatano N., Hamada T. (2008) Proteome analysis of pitcher fluid of the carnivorous plant Nepenthes alata. J. Proteome Res. 7, 809–816 [DOI] [PubMed] [Google Scholar]
- 32. Hatano N., Hamada T. (2012) Proteomic analysis of secreted protein induced by a component of prey in pitcher fluid of the carnivorous plant Nepenthes alata. J. Proteom. In press [DOI] [PubMed] [Google Scholar]
- 33. Tokes Z. A., Woon W. C., Chambers S. M. (1974) Digestive enzymes secreted by carnivorous plant Nepenthes-Macferlanei-L. Planta 119, 39–46 [DOI] [PubMed] [Google Scholar]
- 34. Hamuro Y., Coales S. J., Molnar K. S., Tuske S. J., Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046 [DOI] [PubMed] [Google Scholar]
- 35. Junop M. S., Modesti M., Guarne A., Ghirlando R., Gellert M., Yang W. (2000) Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. EMBO J. 19, 5962–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dunker A. K., Lawson J. D., Brown C. J., Williams R. M., Romero P., Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. R., Hipps K. W., Ausio J., Nissen M. S., Reeves R., Kang C. H., Kissinger C. R., Bailey R. W., Griswold M. D., Chiu M., Garner E. C., Obradovic Z. (2001) Intrinsically disordered protein. J. Mol. Graphics & Modelling 19, 26–59 [DOI] [PubMed] [Google Scholar]
- 37. Warwood S., Mohammed S., Cristea I. M., Evans C., Whetton A. D., Gaskell S. J. (2006) Guanidination chemistry for qualitative and quantitative proteomics. Rapid Commun. Mass Spectrom. 20, 3245–3256 [DOI] [PubMed] [Google Scholar]
- 38. Sibanda B. L., Critchlow S. E., Begun J., Pei X. Y., Jackson S. P., Blundell T. L., Pellegrini L. (2001) Crystal structure of an Xrcc4-DNA ligase IV complex. Nat. Struct. Biol. 8, 1015–1019 [DOI] [PubMed] [Google Scholar]
- 39. Wu P. Y., Frit P., Meesala S., Dauvillier S., Modesti M., Andres S. N., Huang Y., Sekiguchi J., Calsou P., Salles B., Junop M. S. (2009) Structural and Functional Interaction between the Human DNA Repair Proteins DNA Ligase IV and XRCC4. Mol. Cell. Biol. 29, 3163–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Athauda S. B., Matsumoto K., Rajapakshe S., Kuribayashi M., Kojima M., Kubomura-Yoshida N., Iwamatsu A., Shibata C., Inoue H., Takahashi K. (2004) Enzymic and structural characterization of nepenthesin, a unique member of a novel subfamily of aspartic proteinases. Biochem. J. 381, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Merbach M. A., Merbach D. J., Maschwitz U., Booth W. E., Fiala B., Zizka G. (2002) Mass march of termites into the deadly trap. Nature 415, 36–37 [DOI] [PubMed] [Google Scholar]
- 42. Wong T. S., Kang S. H., Tang S. K., Smythe E. J., Hatton B. D., Grinthal A., Aizenberg J. (2011) Bioinspired self-repairing slippery surfaces with pressure-stable omniphobicity. Nature 477, 443–447 [DOI] [PubMed] [Google Scholar]
- 43. Kubota K., Metoki Y., Athauda S. B., Shibata C., Takahashi K. (2010) Stability Profiles of Nepenthesin in Urea and Guanidine Hydrochloride: Comparison with Porcine Pepsin A. Biosci. Biotechnol. Biochem. 74, 2323–2326 [DOI] [PubMed] [Google Scholar]
- 44. Takahashi K., Athauda S. B. P., Matsumoto K., Rajapakshe S., Kuribayashi M., Kojima M., Kubomura-Yoshida N., Iwamatsu A., Shibata C., Inoue H. (2005) Nepenthesin, a unique member of a novel subfamily of aspartic proteinases: Enzymatic and structural characteristics. Curr. Prot. Pep. Sci. 6, 513–525 [DOI] [PubMed] [Google Scholar]
- 45. Fruton J. S. (2002) A history of pepsin and related enzymes. Q Rev. Biol. 77, 127–147 [DOI] [PubMed] [Google Scholar]
- 46. Koch C. A., Agyei R., Galicia S., Metalnikov P., O'Donnell P., Starostine A., Weinfeld M., Durocher D. (2004) Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 23, 3874–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mani R. S., Yu Y., Fang S., Lu M., Fanta M., Zolner A. E., Tahbaz N., Ramsden D. A., Litchfield D. W., Lees-Miller S. P., Weinfeld M. Dual modes of interaction between XRCC4 and polynucleotide kinase/phosphatase: implications for nonhomologous end joining. J. Biol. Chem. 285, 37619–37629 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.