

Abstract
Mitochondrial medicine is one of the few areas of genetic disease where germ-line transfer is being actively pursued as a treatment option. All of the germ-line transfer methods currently under development involve some carry-over of the maternal mitochondrial DNA (mtDNA) heteroplasmy, potentially delivering the pathogenic mutation to the offspring. Rapid changes in mtDNA heteroplasmy have been observed within a single generation, and so any ‘leakage’ of mutant mtDNA could lead to mtDNA disease in future generations, compromising the reproductive health of the first generation, and leading to repeated interventions in subsequent generations. To determine whether this is a real concern, we developed a model of mtDNA heteroplasmy inheritance by studying 87 mother–child pairs, and predicted the likely outcome of different levels of ‘mutant mtDNA leakage’ on subsequent maternal generations. This showed that, for a clinical threshold of 60%, reducing the proportion of mutant mtDNA to <5% dramatically reduces the chance of disease recurrence in subsequent generations, but transmitting >5% mutant mtDNA was associated with a significant chance of disease recurrence. Mutations with a lower clinical threshold were associated with a higher risk of recurrence. Our findings provide reassurance that, at least from an mtDNA perspective, methods currently under development have the potential to effectively eradicate pathogenic mtDNA mutations from subsequent generations.
Keywords: mitochondria, mtDNA, gene therapy, prenatal diagnosis, germ line
The importance of preventing mitochondrial DNA disease
Pathogenic mitochondrial DNA (mtDNA) mutations are found in 0.5% of the population (Elliott et al., 2008), and are a frequent cause of maternally inherited human disease affecting at least 1 in 6500 of the population (Schaefer et al., 2008). Many of these mutations are heteroplasmic, with a mixture of mutated and wild-type mtDNA present in varying proportions within cells of the same individual. High percentage levels of mutated mtDNA are associated with severe multi-system diseases that often affect the nervous system. In addition, some homoplasmic mutations can also cause disease (DiMauro and Schon, 2003). There are currently no treatments for these diseases (Pfeffer et al., 2012), so preventing maternal transmission is a high priority (Brown et al., 2006; Poulton et al., 2009). The medical ethics of modifying the germ-line mitochondrial genome have been discussed in detail (Bredenoord et al., 2011) and the UK Human Fertilisation and Embryology Authority is moving forwards with developing a legal framework for these methods (Callaway, 2012). Concerns include unexpected genetic and epigenetic consequences of the procedure itself. Although animal studies may provide some reassurance, including work in non-human primates (Tachibana et al., 2009), any complications of the procedure may be subtle and take time to emerge. Thus, despite extensive pre-clinical evaluation, there will inevitably be concerns about safety when the procedure is first used in humans, coupled to the ethical issue of generating a child harbouring genetic material from ‘three parents’.
Preventing mtDNA diseases: the challenges
Several unique features of mitochondrial genetics, including the physical separation of the mitochondrial and nuclear genome in the cell, heteroplasmy of some pathogenic mtDNA mutations and the generally high level of mutant mtDNA required to cause serious pathogenic effects, all contribute to the practicality of developing effective germ-line transfer in diseases due to pathogenic mtDNA mutations. Several techniques are currently under development to prevent the transmission of pathogenic mtDNA mutations. Prenatal diagnosis (PND) with amniocentesis or chorionic villus biopsy, and preimplantation genetic diagnosis (PGD) are currently offered in a few centres, based on the accurate measurement of mtDNA heteroplasmy in tissue samples from pre- or post-implantation embryos (Thorburn and Dahl, 2001; Jacobs et al., 2005; Steffann et al., 2007). However, these approaches cannot be used to prevent the transmission of homoplasmic mutations. For heteroplasmic mutations, interpreting the measured level of heteroplasmy is challenging, particularly if there is an intermediate level (20–80%), if the mutation is rare or unique to that particular family, or if the mutation level is known to change over time (as is the case for the most common pathogenic heteroplasmic mtDNA mutation, m.3243A>G) (Craven et al., 2011). Often both the clinician and the prospective heteroplasmic mother will use PND, or more so PGD, to identify embryos harbouring absolutely none of the pathogenic mtDNA mutation. It is also possible to select for fetuses and embryos, and thus prevent disease in the next generation and subsequent transmission. However, these approaches can result in termination in the context of PND, and will also reduce the number of embryos available for implantation in the context of PGD. As a result, several new techniques are in pre-clinical development, which can reduce the risk of transmission still further, and be broadly applicable to all mtDNA diseases. Early approaches involved cytoplasmic transfer (Meirelles and Smith, 1998), but more effective recent developments include spindle transfer (Tachibana et al., 2009; Tachibana et al., 2012) and pronuclear transfer (Brown et al., 2006; Craven et al., 2010). However, none of the current and proposed approaches assure the complete removal of mutant mtDNA. This raises concerns about the long-term consequences of ‘mutant mtDNA carry-over’ on future generations down the female line (Brown et al., 2006; Poulton et al., 2009). Although low levels (<20%) are unlikely to cause disease in the immediate offspring, the amount of the pathogenic mtDNA mutation could change on future transmission down the female line, limiting the effectiveness of the treatment in the longer term. Likewise, for mothers with a homoplasmic mutation, any approach that reduces the mutation load, but does not completely eliminate the mutation, will also introduce the possibility of recurrence in future generations, even if the level of mutation in the next generation is <20%. Why should this be the case?
Large changes in the percentage level of mutated mtDNA are observed in small human pedigrees transmitting heteroplasmic mtDNA mutations (Chinnery et al., 2000), and so even low levels of mutant mtDNA carry-over could lead to maternal descendants with high mutation levels, causing severe multi-system disease (DiMauro and Schon, 2003). The shifts in mutation level that have been observed are sufficiently large to cause disease recurrence even within a single generation after producing effectively treated offspring. Should this be a cause for concern, and how low must the maternal mtDNA carry-over be in order to consider these therapies successful? These questions will be difficult to address experimentally because current animal models of mtDNA disease do not closely resemble human disorders (Nakada and Hayashi, 2011) and there may be significant differences in the mechanism of mtDNA transmission (i.e. the mtDNA bottleneck) between humans and mice (Wonnapinij et al., 2010). This means that the results of animal studies could be either falsely reassuring, or overly pessimistic. Primate work may provide further reassurance that there are no catastrophic unwanted side effects of the treatment (Tachibana et al., 2009), but short-term experiments will not detect late-onset complications related to epigenetic reprogramming. Given the theoretical risks and uncertainties, experimental treatment in humans transmitting non-pathogenic, polymorphic variants are likely to be considered unethical. It is therefore likely that the first-in-human studies will actually be to offer mitochondrial gene replacement as a potential treatment. A means of estimating the risks is essential to enable patients to make informed decisions at that stage.
Modelling the inheritance of mtDNA heteroplasmy
mtDNA heteroplasmy levels are well described by the Kimura distribution, which is based on neutral genetic drift theory, although at this time rigorous testing of this theory in humans is limited to the common pathogenic variation m.3243A>G (Wonnapinij et al., 2008). This model has two parameters, p0 and b. The parameter p0 was set as the amount of mutation carry-over from the mother. The parameter b is the bottleneck parameter determining the width of the heteroplasmy distribution in the offspring. The bottleneck parameter can be set from the heteroplasmy variance of a number of offspring from a single mother, or by pooling the heteroplasmy values from offspring from mothers with similar heteroplasmy levels. For human data, only the latter choice is practical. Mothers with heteroplasmy in the range of 40–60% were used, since in this range the variation in offspring heteroplasmy from the mother's heteroplasmy level is minimal. Siblings were not included.
The key parameters of the Kimura distribution during transmission were determined by studying 87 human mother–offspring pairs transmitting a known pathogenic mtDNA mutation (Wonnapinij et al., 2010). This parameter value was set at b = 0.66 based on 87 human mother–offspring pairs (Lott et al., 1990; Ciafaloni et al., 1992; Larsson et al., 1992; Martinuzzi et al., 1992; Tatuch et al., 1992; Zhu et al., 1992; Hammans et al., 1993, 1995; Piccolo et al., 1993; Howell et al., 1994; Santorelli et al., 1994; Harding et al., 1995; Houstek et al., 1995; Makelabengs et al., 1995; Black et al., 1996; Mak et al., 1996; Carelli et al., 1997; Uziel et al., 1997; Olsson et al., 1998; Onishi et al., 1998; Tanaka et al., 1998; Chinnery et al., 1999; White et al., 1999; Lien et al., 2001; Porto et al., 2001; Hurvitz et al., 2002; Wong et al., 2002; Kaplanova et al., 2004; Enns et al., 2006; Phasukkijwatana et al., 2006), including the following mutations: m.3243A>G (15 pairs), m.83446A>G (10 pairs), m.11778G>A (23 pairs), m.3460G>A (15 pairs), m.9883T>C (10 pairs) and m.8993T>G (14 pairs). Data from the A3243G mutation taken from blood samples were adjusted to correct for the known decrease in the A3243G mutation level in blood with age (Rajasimha et al., 2008). Of course, these pathogenic variants cause a range of different phenotypes, which could in principle affect the inheritance of that variant. With the limited data currently available, the best that can be done is to average the data from all of the different pathogenic variants, to provide a general inheritance model. As more data become available in the future, specific inheritance models for each pathogenic mutation could be developed.
From this data-based model of heteroplasmy transmission in humans, we calculated the effect of low levels of mutant mtDNA carry-over on subsequent maternal generations for a range of different clinical heteroplasmy threshold values. Our analysis began with the initial percentage level of mutated mtDNA ‘carried over’ in the treated embryo that formed the F1 generation. We then calculated heteroplasmy levels based on a Kimura distribution (Wonnapinij et al., 2008) in 20 000 offspring in the next generation (F2, or ‘grandchildren’ of the original mother) and used these values to calculate heteroplasmy levels in the subsequent generation (F3, or ‘great-grandchildren’). Although there is still the theoretical possibility that low levels of heteroplasmy might segregate to higher levels in different tissues as the embryo develops, the heteroplasmy level appears to be uniformly distributed in both pre- and early post-implantation human embryos (Harding et al., 1992; Matthews et al., 1995; Steffann et al., 2007), and extreme differences have not been observed within neonates following the transmission of mtDNA heteroplasmy. Thus, we estimated the recurrence risks using a range of clinical thresholds of mutant mtDNA, showing a disease threshold of >60% mutant as an example (Fig. 1), and other clinical threshold values (Fig. 2).
Figure 1.

The effects of a carry-over of mutated mtDNA from the mother into the embryo on later generations. Triangles = grandchildren of the original mother seeking treatment (F2), and circles = the great-grandchildren (F3). This graph shows the recurrence risk in subsequent generations down the maternal line. (A) The probability that a descendant along the female line would develop an mtDNA mutation heteroplasmy level above 60% as a function of the amount of mutant mtDNA remaining in the treated embryo. This was chosen as a conservative threshold since levels below 60% are unlikely to cause disease. (B) The probability of fixing on the wild type for descendants along the female line. Having had more time to segregate, the chance of inheriting only wild-type mtDNA was greater in the great-grandchildren. (C) The upper 90% confidence interval on the mutant mtDNA level in the immediate descendants along the female line. (D) A measure of the impact of the transfer on later generations calculated by dividing the probability of a descendant having >60% mutation after the transfer by the probability without the transfer, for a mother with 60% mtDNA mutation threshhold. Similar trends were seen for mutations with a lower clinical threshold, although the recurrence risk was greater and the impact of the gene transfer techniques was less. Conversely, for mutations with a higher percentage clinical threshold, the recurrence risk for subsequent generations was lower, and the impact of the gene transfer procedure was greater (Fig. 3). Horizontal arrows denoting the reported range of mtDNA carry-over for the spindle transfer and pronuclear transfer techniques, as well as for PGD and PND are given at the bottom of the figure. It should be noted that the higher range of levels of carry-over with PND and PGD are based on a decision made by the clinician and the prospective mother, and could be zero if so required. The range indicated in the figure shows the heteroplasmy values determined by PND and PGD in pregnancies that were allowed to continue because the perceived risk of a child being affected with this level of mutation is low.
Figure 2.
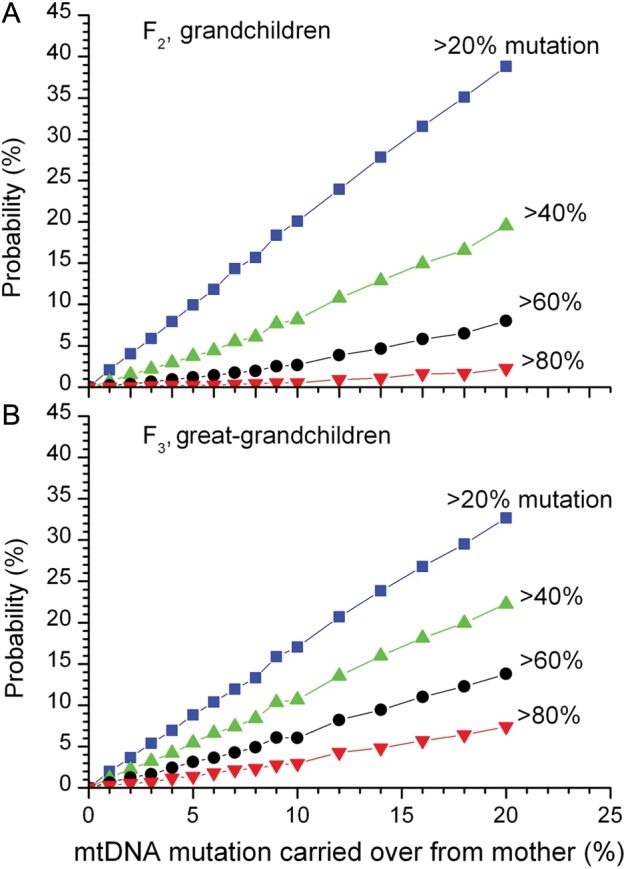
Simulation results for the probabilities that grandchildren (A) or great-grandchildren (B) would have mtDNA mutation levels greater than given thresholds as a function of the mutation level carried over from the mother. Results were calculated for 20% mutation, 40% mutation, 60% mutation (the same as in Fig. 1A and B) and 80% mutation threshholds.
The fate of low-level heteroplasmy on future generations
As expected, a higher mutant mtDNA carry-over from the mother increased the chance that later maternal descendants would inherit high levels of the mutation, and thus be at risk for recurrence of the mitochondrial disease (Fig. 1A) (Poulton and Turnbull, 2000). Likewise, a higher mutant mtDNA carry-over reduced the chance of grandchildren and great-grandchildren fixing on the wild-type mtDNA (Fig. 1B). The same trends were observed for other clinical threshold heteroplasmy levels (Fig. 2). Fixation on wild-type mtDNA would protect all subsequent maternal generations from developing the disease, which is the ultimate goal of germ-line therapies. For the >60% clinical threshold, decreasing the mutation carry-over below 3% dramatically reduced the probability of subsequent generations inheriting high levels of mutant mtDNA (Fig. 1C), and thus the impact of the treatment was greatly increased (Fig. 1D). In contrast, even relatively modest levels of maternal mtDNA carry-over of >5% were associated with a strong possibility of inheriting high levels of mutant mtDNA in later generations (Fig. 1C), leading to the re-emergence of the disease within the family, and thus a poor outcome (Fig. 1D). These calculations illustrate the importance of limiting the carry-over of the mutant mtDNA to very low levels of <3% for the success of the germ-line transfer in future generations.
Similar trends were observed for other clinical threshold heteroplasmy levels (Fig. 3), with lower clinical thresholds associated with a greater risk of recurrence in subsequent generations, and less impact of the gene transfer procedure. Conversely, a higher clinical threshold was associated with a reduced risk of recurrence in subsequent generations, and a greater impact of the gene transfer procedure.
Figure 3.
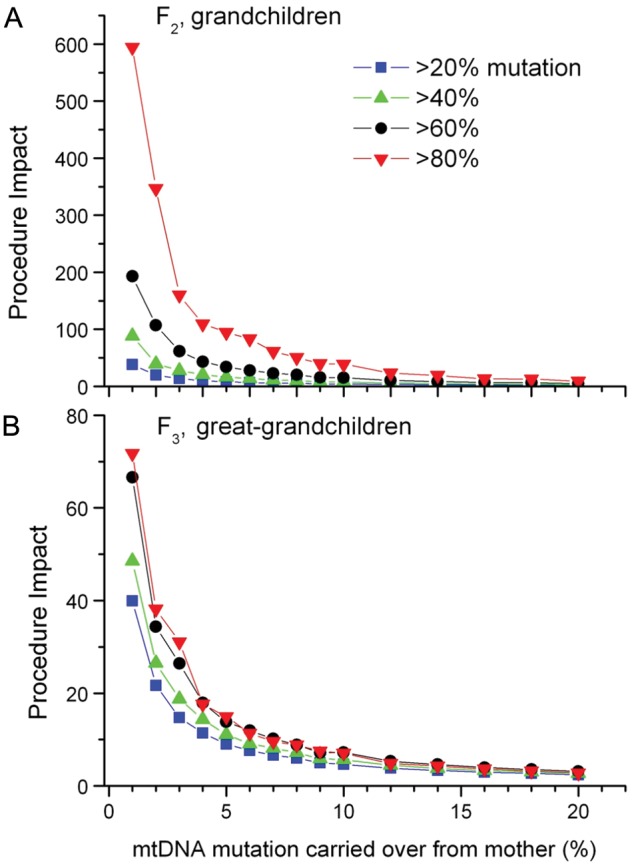
Simulation results for a measure of the impact of the procedure on grandchildren (A) or great-grandchildren (B). Impact is defined as in Fig. 1D, as the ratio of the probability maternal line descendants have greater than the given threshold without the procedure divided by the probability with the procedure, for a mother with 50% mtDNA mutation. A value of 1 indicates no impact of the procedure while higher values represent a larger positive impact. Results were calculated for 20% mutation, 40% mutation, 60% mutation (the same as in Fig. 1D) and 80% mutation thresholds.
Conclusions and future prospects
The development of germ-line therapies for mtDNA diseases is now focused on limiting the carry-over of the mutant mtDNA as much as is practically possible. It has been shown that it is technically possible to transmit <3% maternal mtDNA with spindle–chromosomal complex transfer in non-human primates (Tachibana et al., 2009, 2012), and with pronuclear transfer in preimplantation human embryos (Craven et al., 2010). Our observations indicate that this level of mutant mtDNA carry-over is highly unlikely to cause mitochondrial disease in any maternal descendants, effectively (and quite likely completely) eradicating the disease for good. These predictions not only apply to mtDNA gene transfer techniques, but also for embryos screened by PGD and PND. At present, both PND and PGD remain the first port-of-call for the prevention of mtDNA disease. These techniques can also be used in subsequent generations if there is an ongoing perceived risk of recurrence. However, our findings support the clinical development of gene transfer techniques as a definitive approach to prevent mtDNA disease in these families for all subsequent generations.
Further pre-clinical work in animal models is required to provide further reassurance that these new approaches do not have catastrophic consequences in higher mammalian species, and the ethical and legal debate will need to progress in parallel. However, ultimately, the first successful mtDNA gene transfer in humans will probably be offered to a patient with severe, highly penetrant mtDNA disease, for whom the benefits would outweigh the perceived risks. The work we present here will hopefully assist the prospective mother in making this bold decision.
Authors' roles
The study was conceived by P.F.C. and D.C.S. D.C.S. supervised the modelling, which was carried out by P.W. P.F.C. and D.C.S. wrote the manuscript.
Funding
P.F.C. is an Honorary Consultant Neurologist at Newcastle upon Tyne Foundation Hospitals NHS Trust, is a Wellcome Trust Senior Fellow in Clinical Science (084980/Z/08/Z) and a UK NIHR Senior Investigator. P.F.C. receives additional support from the Wellcome Trust Centre for Mitochondrial Research (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease research, the Association Française contre les Myopathies, and EU FP7 TIRCON, and the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University. Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Conflict of interest
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
References
- Black GCM, Morten K, Laborde A, Poulton J. Leber's hereditary optic neuropathy: heteroplasmy is likely to be significant in the expression of LHON in families with the 3460 ND1 mutation. Br J Ophthalmol. 1996;80:915–917. doi: 10.1136/bjo.80.10.915. doi:10.1136/bjo.80.10.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenoord AL, Dondorp W, Pennings G, De Wert G. Ethics of modifying the mitochondrial genome. J Med Ethics. 2011;37:97–100. doi: 10.1136/jme.2010.037481. doi:10.1136/jme.2010.037481. [DOI] [PubMed] [Google Scholar]
- Brown DT, Herbert M, Lamb VK, Chinnery PF, Taylor RW, Lightowlers RN, Craven L, Cree L, Gardner JL, Turnbull DM. Transmission of mitochondrial DNA disorders: possibilities for the future. Lancet. 2006;368:87–89. doi: 10.1016/S0140-6736(06)68972-1. doi:10.1016/S0140-6736(06)68972-1. [DOI] [PubMed] [Google Scholar]
- Callaway E. UK sets sights on gene therapy in eggs. Nature. 2012;481:419. doi: 10.1038/481419a. doi:10.1038/481419a. [DOI] [PubMed] [Google Scholar]
- Carelli V, Ghelli A, Ratta M, Bacchilega E, Sangiorgi S, Mancini R, Leuzzi V, Cortelli P, Montagna P, Lugaresi E, et al. Leber's hereditary optic neuropathy: biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology. 1997;48:1623–1632. doi: 10.1212/wnl.48.6.1623. doi:10.1212/WNL.48.6.1623. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Zwijnenburg PJG, Walker M, Howell N, Taylor RW, Lightowlers RN, Bindoff L, Turnbull DM. Nonrandom tissue distribution of mutant mtDNA. Am J Med Genet. 1999;85:498–501. doi:10.1002/(SICI)1096-8628(19990827)85:5<498::AID-AJMG13>3.0.CO;2-8. [PubMed] [Google Scholar]
- Chinnery PF, Thorburn DR, Samuels DC, White SL, Dahl HM, Turnbull DM, Lightowlers RN, Howell N. The inheritance of mitochondrial DNA heteroplasmy: random drift, selection or both? Trends Genet. 2000;16:500–505. doi: 10.1016/s0168-9525(00)02120-x. doi:10.1016/S0168-9525(00)02120-X. [DOI] [PubMed] [Google Scholar]
- Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, Simonetti S, Angelini C, Donati MA, Garcia C, et al. MELAS—clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31:391–398. doi: 10.1002/ana.410310408. doi:10.1002/ana.410310408. [DOI] [PubMed] [Google Scholar]
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. doi:10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven L, Elson JL, Irving L, Tuppen HA, Lister LM, Greggains GD, Byerley S, Murdoch AP, Herbert M, Turnbull D. Mitochondrial DNA disease: new options for prevention. Hum Mol Genet. 2011;20:R168–R174. doi: 10.1093/hmg/ddr373. doi:10.1093/hmg/ddr373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. doi:10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mtDNA mutations are common in the general population. Am J Hum Genet. 2008;80:254–260. doi: 10.1016/j.ajhg.2008.07.004. doi:10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns GM, Bai RK, Beck AE, Wong LJ. Molecular-clinical correlations in a family with variable tissue mitochondrial DNA T8993G mutant load. Mol Genet Metab. 2006;88:364–371. doi: 10.1016/j.ymgme.2006.02.001. doi:10.1016/j.ymgme.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Hammans SR, Sweeney MG, Brockington M, Lennox GG, Lawton NF, Kennedy CR, Morganhughes JA, Harding AE. The mitochondrial DNA transfer RNA (Lys) A>G(8344) mutation and the syndrome of myoclonic epilepsy with ragged-red fibers (MERRF). Relationship of clinical phenotype to proportion of mutant mitochondrial DNA. Brain. 1993;116:617–632. doi: 10.1093/brain/116.3.617. doi:10.1093/brain/116.3.617. [DOI] [PubMed] [Google Scholar]
- Hammans SR, Sweeney MG, Hanna MG, Brockington M, Morganhughes JA, Harding AE. The mitochondrial DNA transfer RNA(Leu(UUR)) A>G(3243) mutation. A clinical and genetic study. Brain. 1995;118:721–734. doi: 10.1093/brain/118.3.721. doi:10.1093/brain/118.3.721. [DOI] [PubMed] [Google Scholar]
- Harding A, Holt I, Sweeney M, Brockington M, Davis M. Prenatal diagnosis of mitochondrial DNA8993T>G disease. Am J Hum Genet. 1992;50:629–633. : [PMC free article] [PubMed] [Google Scholar]
- Harding AE, Sweeney MG, Govan GG, Riordaneva P. Pedigree analysis in Leber hereditary optic neuropathy families with pathogenic mtDNA mutation. Am J Hum Genet. 1995;57:77–86. [PMC free article] [PubMed] [Google Scholar]
- Houstek J, Klement P, Hermanska J, Houstkova H, Hansikova H, Vandenbogert C, Zeman J. Aletered properties of mitochondrial ATP synthase in patients with a T>G mutation in the ATPase 6 (subunit A) gene at position 8993 of mtDNA. Biochim Biophys Acta Mol Basis Dis. 1995;1271:349–357. doi: 10.1016/0925-4439(95)00063-a. doi:10.1016/0925-4439(95)00063-A. [DOI] [PubMed] [Google Scholar]
- Howell N, Xu M, Halvorson S, Bodiswollner I, Sherman J. Heteroplasmic LHON family—tissue distribution and transmission of the 11778 mutation. Am J Hum Genet. 1994;55:203–206. [PMC free article] [PubMed] [Google Scholar]
- Hurvitz H, Naveh Y, Shoseyov D, Klar A, Shaag A, Elpeleg O. Transmission of the mitochondrial t8993c mutation in a new family. Am J Med Genet. 2002;111:446–447. doi: 10.1002/ajmg.10613. doi:10.1002/ajmg.10613. [DOI] [PubMed] [Google Scholar]
- Jacobs LJ, de Coo IF, Nijland JG, Galjaard RJ, Los FJ, Schoonderwoerd K, Niermeijer MF, Geraedts JP, Scholte HR, Smeets HJ. Transmission and prenatal diagnosis of the T9176C mitochondrial DNA mutation. Mol Hum Reprod. 2005;11:223–228. doi: 10.1093/molehr/gah152. doi:10.1093/molehr/gah152. [DOI] [PubMed] [Google Scholar]
- Kaplanova V, Zeman J, Hansikova H, Cerna L, Houst'kova H, Misovicova N, Houstek J. Segregation pattern and biochemical effect of the G3460A mtDNA mutation in 27 members of LHON family. J Neurol Sci. 2004;223:149–155. doi: 10.1016/j.jns.2004.05.001. doi:10.1016/j.jns.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Tulinius MH, Holme E, Oldfors A, Andersen O, Wahlstrom J, Aasly J. Segregation and manifestations of the mtDNA trans RNALys A>G (8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am J Hum Genet. 1992;51:1201–1212. [PMC free article] [PubMed] [Google Scholar]
- Lien LM, Lee HC, Wang KL, Chiu JC, Chiu HC, Wei YH. Involvement of nervous system in maternally inherited diabetes and deafness (MIDD) with the A3243G mutation of mitochondrial DNA. Acta Neurol Scand. 2001;103:159–165. doi: 10.1034/j.1600-0404.2001.103003159.x. doi:10.1034/j.1600-0404.2001.103003159.x. [DOI] [PubMed] [Google Scholar]
- Lott MT, Voljavec AS, Wallace DC. Variable genotype of Lebers hereditary optic neuropathy patients. Am J Ophthalmol. 1990;109:625–631. doi: 10.1016/s0002-9394(14)72429-8. [DOI] [PubMed] [Google Scholar]
- Mak SC, Chi CS, Liu CY, Pang CY, Wei YH. Leigh syndrome associated with mitochondrial DNA 8993 T->G mutation and ragged-red fibers. Pediatr Neurol. 1996;15:72–75. doi: 10.1016/0887-8994(96)00126-9. doi:10.1016/0887-8994(96)00126-9. [DOI] [PubMed] [Google Scholar]
- Makelabengs P, Suomalainen A, Majander A, Rapola J, Kalimo H, Nuutila A, Pihko H. Correlation between the clinical symptoms and the propertion of mitochondrial DNA carry the 8993 point mutation in the NARP syndrome. Pediatr Res. 1995;37:634–639. doi: 10.1203/00006450-199505000-00014. doi:10.1203/00006450-199505000-00014. [DOI] [PubMed] [Google Scholar]
- Martinuzzi A, Bartolomei L, Carrozzo R, Mostacciuolo M, Carbonin C, Toso V, Ciafaloni E, Shanske S, Dimauro S, Angelini C. Correlation between clinical and molecular features in 2 MELAS families. J Neurol Sci. 1992;113:222–229. doi: 10.1016/0022-510x(92)90250-o. doi:10.1016/0022-510X(92)90250-O. [DOI] [PubMed] [Google Scholar]
- Matthews PM, Brown RM, Morten K, Marchington D, Poulton J, Brown G. Intracellular heteroplasmy for disease-associated point mutations in mtDNA: implications for disease expression and evidence for mitotic segregation of heteroplasmic units of mtDNA. Hum Genet. 1995;96:261–268. doi: 10.1007/BF00210404. [DOI] [PubMed] [Google Scholar]
- Meirelles F, Smith LC. Mitochondrial genotype segregation during preimplantation development in mouse heteroplasmic embryos. Genetics. 1998;148:877–883. doi: 10.1093/genetics/148.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada K, Hayashi J. Transmitochondrial mice as models for mitochondrial DNA-based diseases. Exp Anim. 2011;60:421–431. doi: 10.1538/expanim.60.421. doi:10.1538/expanim.60.421. [DOI] [PubMed] [Google Scholar]
- Olsson C, Zethelius B, Lagerstrom-Fermer M, Asplund J, Berne C, Landegren U. Level of heteroplasmy for the mitochondrial mutation A3243G correlates with age at onset of diabetes and deafness. Hum Mutat. 1998;12:52–58. doi: 10.1002/(SICI)1098-1004(1998)12:1<52::AID-HUMU8>3.0.CO;2-K. doi:10.1002/(SICI)1098-1004(1998)12:1<52::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Onishi H, Hanihara T, Sugiyama N, Kawanishi C, Iseki E, Maruyama Y, Yamada Y, Kosaka K, Yagishita S, Sekihara H, et al. Pancreatic exocrine dysfunction associated with mitochondrial tRNA(Leu(UUR)) mutation. J Med Genet. 1998;35:255–257. doi: 10.1136/jmg.35.3.255. doi:10.1136/jmg.35.3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:CD004426. doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phasukkijwatana N, Chuenkongkaew WL, Suphavilai R, Luangtrakool K, Kunhapan B, Lertrit P. Transmission of heteroplasmic G11778A in extensive pedigrees of Thai Leber hereditary optic neuropathy. J Hum Genet. 2006;51:1110–1117. doi: 10.1007/s10038-006-0073-6. doi:10.1007/s10038-006-0073-6. [DOI] [PubMed] [Google Scholar]
- Piccolo G, Focher F, Verri A, Spadari S, Banfi P, Gerosa E, Mazzarello P. Myoclonus epolepsy and ragged-red fibers—blood miotochondrial DNA heteroplasmy in affected and symptomatic members of a family. Acta Neurol Scand. 1993;88:406–409. doi: 10.1111/j.1600-0404.1993.tb05368.x. doi:10.1111/j.1600-0404.1993.tb05368.x. [DOI] [PubMed] [Google Scholar]
- Porto FBO, Mack G, Sterboul MP, Lewin P, Flament J, Sahel J, Dollfus H. Isolated late-onset cone-rod dystrophy revealing a familial neurogenic muscle weakness, ataxia, and retinitis pigmentosa syndrome with the T8993G mitochondrial mutation. Am J Ophthalmol. 2001;132:935–937. doi: 10.1016/s0002-9394(01)01187-4. doi:10.1016/S0002-9394(01)01187-4. [DOI] [PubMed] [Google Scholar]
- Poulton J, Turnbull DM. 74th ENMC international workshop: mitochondrial diseases. Neuromuscul Disord. 2000;10:460–462. doi: 10.1016/s0960-8966(00)00101-2. doi:10.1016/S0960-8966(00)00101-2. [DOI] [PubMed] [Google Scholar]
- Poulton J, Kennedy S, Oakeshott P, Wells D. Preventing transmission of maternally inherited mitochondrial DNA diseases. Br Med J. 2009;338:b94. doi: 10.1136/bmj.b94. doi:10.1136/bmj.b94. [DOI] [PubMed] [Google Scholar]
- Rajasimha HK, Chinnery PF, Samuels DC. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A -> G mutation in blood. Am J Hum Genet. 2008;82:333–343. doi: 10.1016/j.ajhg.2007.10.007. doi:10.1016/j.ajhg.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santorelli FM, Shanske S, Jain KD, Tick D, Schon EA, Dimauro S. A T- C mutation at nt-8993 of mitochondrial DNA in a child with Leign syndrome. Neurology. 1994;44:972–974. doi: 10.1212/wnl.44.5.972. doi:10.1212/WNL.44.5.972. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. doi:10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- Steffann J, Gigarel N, Corcos J, Bonniere M, Encha-Razavi F, Sinico M, Prevot S, Dumez Y, Yamgnane A, Frydman R, et al. Stability of the m.8993T->G mtDNA mutation load during human embryofetal development has implications for the feasibility of prenatal diagnosis in NARP syndrome. J Med Genet. 2007;44:664–669. doi: 10.1136/jmg.2006.048553. doi:10.1136/jmg.2006.048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. doi:10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2012 doi: 10.1038/nature11647. doi:10.1038/nature11647. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Kiyosawa M, Mashima Y, Tokoro T. A family with Leber's hereditary optic neuropathy with mitochondrial DNA heteroplasmy related to disease expression. J Neuroophthalmol. 1998;18:81–83. [PubMed] [Google Scholar]
- Tatuch Y, Christodoulou J, Feigenbaum A, Clarke JTR, Wherret J, Smith C, Rudd N, Petrovabenedict R, Robinson BH. Heteroplasmic mtDNA mutation (T) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet. 1992;50:852–858. [PMC free article] [PubMed] [Google Scholar]
- Thorburn DR, Dahl HHM. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet. 2001;106:102–114. doi: 10.1002/ajmg.1380. doi:10.1002/ajmg.1380. [DOI] [PubMed] [Google Scholar]
- Uziel G, Moroni I, Lamantea E, Fratta GM, Ciceri E, Carrara F, Zeviani M. Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. J Neurol Neurosurg Psychiatry. 1997;63:16–22. doi: 10.1136/jnnp.63.1.16. doi:10.1136/jnnp.63.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SL, Shanske S, McGill JJ, Mountain H, Geraghty MT, DiMauro S, Dahl HHM, Thorburn DR. Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue- or age-related variation. J Inherit Metab Dis. 1999;22:899–914. doi: 10.1023/a:1005639407166. doi:10.1023/A:1005639407166. [DOI] [PubMed] [Google Scholar]
- Wong LJC, Wong H, Liu AY. Intergenerational transmission of pathogenic heteroplasmic mitochondrial DNA. Genet Med. 2002;4:78–83. doi: 10.1097/00125817-200203000-00005. doi:10.1097/00125817-200203000-00005. [DOI] [PubMed] [Google Scholar]
- Wonnapinij P, Chinnery PF, Samuels DC. The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am J Hum Genet. 2008;83:582–593. doi: 10.1016/j.ajhg.2008.10.007. doi:10.1016/j.ajhg.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnapinij P, Chinnery PF, Samuels DC. Previous estimates of mitochondrial DNA mutation level variance did not account for sampling error: comparing the mtDNA genetic bottleneck in mice and humans. Am J Hum Genet. 2010;86:540–550. doi: 10.1016/j.ajhg.2010.02.023. doi:10.1016/j.ajhg.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu DP, Economou EP, Antonarakis SE, Maumenee IH. Mitochondrial DNA mutation and heteroplasmy in type I Leber hereditary optic neuropathy. Am J Med Genet. 1992;42:173–179. doi: 10.1002/ajmg.1320420208. doi:10.1002/ajmg.1320420208. [DOI] [PubMed] [Google Scholar]