

Abstract
Background and Purpose
Since endothelin (ET) may act as pro-fibrotic mediator, expression and release of ET isoforms, their receptors and potential pro-fibrotic ET effects were studied in human lung fibroblasts.
Experimental Approach
MRC-5 and primary human lung fibroblasts (phLFb) were cultured. Expression of prepro-ET isoforms was determined by qPCR and release of ET-1 by elisa. ET receptor function was analysed by real-time measurement of dynamic mass redistribution (DMR). Incorporation of [3H]-thymidine was determined as measure of proliferation and that of [3H]-proline for collagen synthesis. Phospho-p42/44 MAP kinase was determined by Western blot.
Key Results
ET-1 is the predominant ET in human lung fibroblasts (hLF), and TGF-β caused a further, selective and sustained up-regulation of ET-1 resulting in increased extracellular ET-1 accumulation. hLFb express mRNA encoding ET-A and ET-B receptors. Expression of both receptors was confirmed at protein level. ET-1 induced marked DMR signals, an effect that involved ET-A and ET-B receptors. Stimulatory effects of ET-1 on hLFb proliferation and collagen synthesis were mediated exclusively via ET-A receptors. ET-1, again via ET-A receptors, induced rapid activation of ERK MAPK, shown to be a crucial cellular signal in ET-1-induced collagen synthesis. ET-1-induced activation of ERK and collagen synthesis was, in contrast to corresponding effect of a muscarinic agonist, largely insensitive to pertussis toxin.
Conclusions and Implications
hLFb are endowed with all elements necessary to build a functional autocrine/paracrine endothelinergic system, which appears to drive pro-fibrotic airway and lung remodelling processes, effects for which only ET-A, but not ET-B receptors appear to be of significance.
Keywords: endothelin, ET-1, ET-A and ET-B receptor, collagen synthesis, lung fibroblasts, airway remodelling, myofibroblast, ERK MAPK, TGF-β, pertussis toxin, bosentan, dynamic mass redistribution (DMR)
Introduction
Endothelins are a family of small peptides of 21 amino acids (ET-1, -2 and -3). They are synthesized as large precursor proteins (prepro-ET) that are processed by a cascade of different proteases resulting in big-ET and finally the active ET (for review, see, e.g. Polikepahad et al., 2006; Khimji and Rockey, 2010). The last step is mediated by endothelin-converting enzyme (ECE), of which several isoenzymes have been described, some of them located intracellularly and some appear to act as ectoenzymes on the extracellular cell surface (for review, see, e.g. Battistini and Jeng, 2001; Corder, 2001). ET-1 is one of the most potent constrictors of vascular smooth muscle, and its role in the pathophysiology of pulmonary hypertension has extensively been studied (for review, see Shao et al., 2011). In addition, ET-1 is a potent constrictor of airway smooth muscle and may exert various pro-fibrotic effects. Therefore, it is also considered to participate in the pathogenesis of pulmonary fibrosis and obstructive airway disease (e.g. Chalmers et al., 1997; Goldie and Henry, 1999; Polikepahad et al., 2006; Ross et al., 2010, Swigris and Brown, 2010). There is evidence that ET-1 is synthesized and released by different pulmonary cells, among them pulmonary fibroblasts and airway epithelial cells, and increased levels of ET-1 were observed in asthmatic patients (Pégorier et al., 2007). Moreover, the levels of ET-1 in exhaled breath condensate appear to correlate with the severity of asthma (Zietkowski et al., 2008). In different experimental models, ET-1 was shown to promote pulmonary inflammatory reactions, particularly cellular infiltration (e.g. Bhavsar et al., 2008a,b; Landgraf and Jancar, 2008) and the release of cytokines (e.g. Gallelli et al., 2005; Iwata et al., 2009) and prostanoids (Juergens et al., 2008). Furthermore, as ET-1 acts also as proliferative stimulus in airway smooth muscle (Panettieri et al., 1996) and fibroblasts (Gallelli et al., 2005), it may significantly contribute to airway and lung remodelling processes. Although there are several reports describing pro-fibrotics effects of ET-1 in rat and human pulmonary fibroblasts (e.g. Gallelli et al., 2005; Préfontaine et al., 2008), a detailed analysis of the expression of ET isoforms and ET receptors and their potential regulation in human pulmonary fibroblasts as well as a detailed pharmacological characterization of their functions are still missing.
Therefore, the present study aimed to explore systematically the expression and release of ET isoforms, their receptors, as well as potential functional effects of ET and ET-activated cellular signals in human lung fibroblasts. The present data show that human lung fibroblasts are endowed with all elements necessary to build a functional autocrine/paracrine endothelinergic system that appears to be involved in the regulation pro-fibrotic features.
Methods
Culture of lung fibroblasts
MCR-5 human lung fibroblasts (CCL-171, ATCC, Manassas, VT) were grown in Eagle's minimal essential medium (MEM) supplemented with 10% FCS, 2 mM l-glutamine, Earle's BBS adjusted to contain 2.2 g·L−1 sodium bicarbonate, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. Cells were grown in a humidified incubator at 37°C and 5% CO2, and passaged by trypsinization at nearly confluence.
Primary human lung fibroblasts (phLFb) were established from normal areas of surgically resected lung tissue, which was obtained from lung cancer patients after thoracotomy (Matthiesen et al., 2006). Anonymous lung tissue of male Caucasians was obtained from patients who gave informed consent. The protocol for obtaining human tissue was approved by the local ethical review board for human studies (Ethics Committee, Medical Faculty, University of Bonn, Germany). Tissue consisting of lung parenchyma and small airways was cut into small pieces, treated with pronase (1 mg·mL−1, Calbiochem Novabiochem, San Diego, CA) at 37°C for 30 min, placed in cell culture plates and incubated in Eagle's MEM supplemented as described above, with FCS increased to 15%. After 2 weeks, fibroblasts had grown out from the tissue and were passaged by standard trypsinization. For experiments described here, cells of passage 3–11 were used.
Extraction of RNA and real-time reverse transcription-PCR
Total RNA was isolated by help of silica-gel-based membranes according to manufacturer's instructions including an additional DNase digestion protocol to beware any contamination by genomic DNA (Qiagen, Hilden, Germany). First strand cDNA was synthesized using Omniscript reverse transcriptase (Qiagen).
Quantitative PCR was performed by monitoring the fluorescence of SYBR Green dye on a Statagene Mx3000P real time PCR system. Applied primer pairs (based on human EMBL sequences) were specific for prepro-ET-1, 5′-TTATCAGCAGTTAGTGAGAGG-3′ and 5′-GAAGGTCTGTCACCAATGTG-3′; prepro-ET-2 5′-TGAGGGACATTTCCACAGTCAAG-3′ and 5′-CGGTTGCTCCTGGTTTGTAGC-3′, prepro-ET-3 5′-TATCGGCCTGGTGTCTATAC-3′ and 5′-AGTGGACTCCAAGCTAACTC-3′, ET-A receptor, 5′-GCTCTTTGCTGGTTCCCTGTT-3′ and 5′-GGTCATCAGACTTTTGGACTGG-3; ET-B receptor, 5′-TCTTTTGCCTGGTCCTTGTCT-3′ and 5′-GCAGTTTTTGAATCTTTTGCTCAC-3′; collagen I-α1, 5′-TGCTGGTCCCAAAGGTGCTGATG-3′ and 5′-GACCAGGCTCACCACGGTCT-3′; and the housekeeping gene GAPDH, 5′-CTGCACCACCAACTGCTTAGC-3′ and 5′-GGCATGGACTGTGGTCATGAG-3′, which was used for normalization. The cycling conditions were 10 min polymerase activation at 95°C and 40 cycles at 95°C for 30 s, 58°C for 30 s and 72°C for 30 s. The threshold was automatically set by the software. The crossing point of the amplification curve with the threshold represents the ‘Ct’.
Fluorescence data from each sample were analysed with the 2−[ΔΔCt] method: fold induction = 2−[ΔΔCt], where ΔΔCt = [Ct GI (unknown sample) − Ct GAPDH (unknown sample)] − [Ct GI (calibrator sample) − Ct GAPDH (calibrator sample)], GI is the gene of interest. Furthermore, ΔCt = 35 was set as detection limit, and the fold expression over detection limit (235−ΔCt) was used as a measure of the expression levels.
Endothelin elisa
MRC-5 cells were cultured for 24 h in presence of 10% FCS followed by 48 h in presence of 1% FCS and absence or presence of TGF-β. Thereafter, medium changed and cells incubated for further 1 to 8 h. Then the supernatant were collected for ET-1 determination by a highly sensitive (detection limit 0.41 pg·mL−1) and specific (cross-reactivity for ET-1100%, ET-2 21%, ET-3 3.6%, human big-ET-1 < 0.1%), commercially available elisa (Stressgen, Enzo Life Sciences GmbH, Lörrach, Germany).
Dynamic mass redistribution (DMR)
DMR measurements were carried out using the beta version of the Epic® device (Corning, NY). A detailed protocol of this technique has recently been published (Schröder et al., 2011). MRC-5 were seeded in a density of 15 000 cells per well in 384-well Epic® fibronectin-coated microplates with 40 μL of growth medium (Eagle's MEM, 10% FCS, 2 mM l-glutamine and 1% penicillin/streptomycin) and cultured at 37°C in an atmosphere of 5% CO2 for 20 h to achieve confluent cell layers. After cell culture medium had been removed and the cells had been washed twice with 50 μL of assay buffer per well (HBSS with 20 mM HEPES, pH 7.0), cells were allowed to rest in 30 μL of assay buffer for 2 h in the Epic® reader at a constant temperature of 28°C. After addition of 10 μL of test compound, dissolved in assay buffer, DMR responses were monitored for up to 3 h as described previously (Kebig et al., 2009; Schröder et al., 2010; Lamyel et al., 2011). In case of antagonist pretreatment, compounds were added to the assay buffer in the appropriate concentration directly after the washing procedure.
[3H]-Thymidine incorporation
Cells were trypsinized, harvested and seeded into 12-well dishes at a density of 4 × 104 cells per well. In most experiments, cells were first cultured for 24 h in presence of 10% FCS, followed by 18 h under FCS-free conditions. Thereafter, test drugs were added and present for 30 h under FCS-free conditions, and [3H]-Thymidine (37 kBq) was present during the last 24 h. In one series of experiment, cells were seeded at density of 2*105 per well and cultured from the onset under FCS-free conditions in absence or presence of ET-1 for 30 h. [3H]-thymidine was present during the last 24 h. At the end of incubation, cells were washed in ice-cold PBS, and radioactivity was incorporated into DNA was extracted as described previously (Freitag et al., 1996; Matthiesen et al., 2006). Briefly, cells were denatured in TCA (5%) for 10 min, washed in ice-cold PBS and DNA was extracted during incubation for 1 h in 0.1 mol·L−1 NaOH at 37°C; 300 μL portions of the supernatant solution were neutralized, combined with scintillation cocktail (PerkinElmer, Rodgau–Jügesheim, Germany), and radioactivity was determined by liquid scintillation spectrometry in a Packard 2100 TR liquid scintillation analyser. External standardization was used to correct for counting efficiency. [3H]-thymidine incorporation was expressed either in absolute terms (d.p.m) or as percentage of the mean of the control group of each series of experiments.
Cell count
Cells were grown in serum-free medium in absence or presence of test substances for 28 or 48 h and trypsinized at the end of the incubation period. Cells were resuspended in medium, and 200 μL of cell suspension were diluted into 10 mL of a ready-to-use isotonic saline solution (CASYton®). This dilution was counted immediately in a CASY®CellCounter+Analyzer System (Innovatis, Bielefeld, Germany).
[3H]-proline incorporation
Collagen synthesis and deposition into the extracellular matrix were assessed by [3H]-proline incorporation assays originally developed by Peterkofsky and Diegelmann (1971) and subsequently established also in our laboratory (Haag et al., 2008). Cells were trypsinized, harvested and seeded into 12-well dishes at a density of 105 cells per well. Cells were first cultured for 24 h in the presence of 10% FCS, followed by an additional 18–24 h under FCS-free conditions. Thereafter, [3H]-proline (37 kBq) was added alone or in combination with test drugs, and cells were cultured for further 24 h. At the end, culture medium was removed, and cells were washed twice with 4°C cold PBS followed by 1–2 h incubation in 1 mL 20% trichloroacetic acid (TCA) at 4°C. Denaturated cells were scraped off, transferred into a reaction tube and centrifuged at 15 000× g for 10 min. The pellet was washed with 1 mL 10% TCA and centrifuged again at 15 000× g for 5 min dissolved in 300 μL 0.2 M NaOH followed by neutralization with 300 μL 0.2 M HCl; 300 μL portions were combined with scintillation cocktail (PerkinElmer), and radioactivity was determined by liquid scintillation spectrometry in a Packard 2100 TR liquid scintillation analyser. External standardization was used to correct for counting efficiency. [3H]-Proline incorporation was expressed either in absolute terms (d.p.m.) or as percentage of the mean of the control group of each series of experiments.
In previous experiments, it was confirmed by collagenase digestion that total radioactivity incorporated into proteins largely reflects de novo synthesis of collagen (Haag et al., 2008).
QuickZyme collagen assay
Collagen synthesis was additionally assessed by QuickZyme Soluble Collagen Assay (QuickZyme BioSciences, Leiden, the Netherlands). Cells were cultured for 24 h in presence of 10% FCS, followed by an additional 24 h under FCS-free conditions in absence or presence of test substances. At the end, culture medium was removed, and cells were washed with 4°C cold PBS followed by overnight incubation in 500 μL 0.5 M acetic acid at 4°C. Denaturated cells were scraped off, and the extract centrifuged at 3000× g for 10 min. The supernatant was analysed according to the manufacturer's instructions.
Western blot analysis
Cellular proteins were extracted in RIPA buffer [50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5% sodium deoxycholat, 1% Nonidet P-40, 0.1% (w/v) SDS, 2 mM EDTA pH 8.0] or NP-40 buffer [100 mM Tris–HCl pH 7.5, 100 mM NaCl,10 mM MgCl2, 100%(v/v) Nonidet P-40] containing the protease inhibitors PMSF (1 mM), pepstatin A (0.7 μg·mL−1) and leupeptin (0.5 μg·mL−1); 10–20 μg protein-equivalents were mixed with reducing protein loading buffer (Roti-Load 1, Roth, Karlsruhe, Germany) and boiled for 10 min at 70°C. Samples were separated on 4–12% acrylamide Tris–glycine precast gels (Invitrogen, Karlsruhe, Germany) using the precast MOPS-Buffer-System (Invitrogen) and transferred (20% methanol, 25 mM Tris, 14.4% glycine) to PVDF membranes (Millipore, Billerica, MA). Blots were blocked in 5% dried milk protein TBST (150 mM NaCl, 50 mM Tris, 0.05% Tween 20), and proteins were detected by use of a mouse monoclonal antibody against α-smooth muscle actin (clone 1A4, Sigma-Aldrich, München, Germany) or rabbit polyclonal anti-human phospho-p44/42 MAPK (Cell Signaling Technology, Beverly, MA), rabbit polyclonal anti-rat ERK2 (C-14) (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-ET-A receptor (Santa Cruz Biotechnology, H-60) and rabbit polyclonal anti-ET-B receptor (Santa Cruz Biotechnology, H-74) antibodies. The ‘housekeeping’ protein α-tubulin was detected with a mouse monoclonal anti-human α-tubulin antibody (Cedar Lane, Hornby, Canada). Bands were visualized by peroxidase-conjugated secondary goat anti-rabbit IgG-HRP (Biorad, Hercules, CA) or anti-mouse (Santa Cruz Biotechnology) antibodies employing BM chemoluminescence blotting substrate POD (Roche, Mannheim, Germany). Then blots were exposed to Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ).
Statistical analysis
All values are means with SEM of n experiments. Statistical significance of differences was evaluated by anova followed by Dunnett's or Bonferroni's test using GraphPad InStat (GraphPad Software, San Diego, CA). P < 0.05 was accepted as significant.
Drugs and materials
Bosentan was a gift from Actelion (Freiburg, Germany). All other drugs were purchased, BQ123 [Cyclo(d-Trp-d-Asp-Pro-d-Val-Leu)], BQ788 (N-cis-2,6-dimethylpiperidinocarbonyl-β-tBu-Ala-d-Trp(1-methoxycarbonyl)-d-Nle-OH) and big-endothelin-1 (1–31) from Bachem (Bubendorf, Switzerland); human endothelin-1, oxotremorine sesquifumarate, penicillin–streptomycin solution, pertussis toxin, TGF-β, trypsin were from Sigma (Deisenhofen, Germany); SIS3 (6,7-dimethoxy-2-((2E)-3-(1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl-prop-2-enoyl))-1,2,3,4-tetrahydroisoquinoline) was from Calbiochem (Darmstadt, Germany); desoxynucleotide mixture was from Fermentas (St. Leon-Rot, Germany); Eagle's MEM with Earl's salts and l-glutamine, non-essential amino acids from PAA (Cölbe, Germany); fetal calf serum (FCS) from Biochrom (Berlin, Germany); Taq DNA-polymerase from Invitrogen Omniscript reverse transcriptase, RNeasy Mini kit, QuantiTect™ SYBR Green PCR kit and RNase-free DNase set from Qiagen; [3H]-thymidine and [3H]-proline from PerkinElmer. Oligodesoxynucleotides for qPCR were obtained from Eurofins MWG Operon (Ebersberg, Germany).
Results
Expression of ET isoforms and ET receptor subtypes in human lung fibroblasts
As shown in Figure 1, phLFb as well as cells of the MRC-5 human lung fibroblast cell line clearly express mRNA encoding prepro-ET-1 and prepro-ET-2, but in both cell types the levels of prepro-ET-1 mRNA were about 100-fold higher than those of the ppET-2 (Figure 1A). mRNA for prepro-ET-3 was below detection limit in both, phLFb and MRC-5 cells. Both, phLFb and MRC-5 human lung fibroblasts express also mRNA for ET-A and ET-B receptors. Whereas MRC-5 cells expressed similar levels of mRNA for ET-A and ET-B receptor, in phLFb, the levels of mRNA for ET-A receptors were significantly lower than those for the ET-B receptor (Figure 1B). The expression of ET-A and ET-B receptor proteins could be confirmed by Western blot analysis using specific, commercially available polyclonal antibodies (Figure 1C). The immunoblot for ET-B receptor showed one band at about 70 kDa, which is in the range of the expected molecular weight, and an additional band of lower molecular weight, which may reflect a cellular degradation product still recognized by the antibody. The immunoblot for ET-A receptor showed also two bands, both with molecular weights lower than the expected molecular weight. Nonetheless, the larger band may reflect an ET-A receptor signal as it corresponds to a specific band of about 45 kDa seen in ET-A receptor transfected cells (antibody reference sheet of Santa Cruz).
Figure 1.
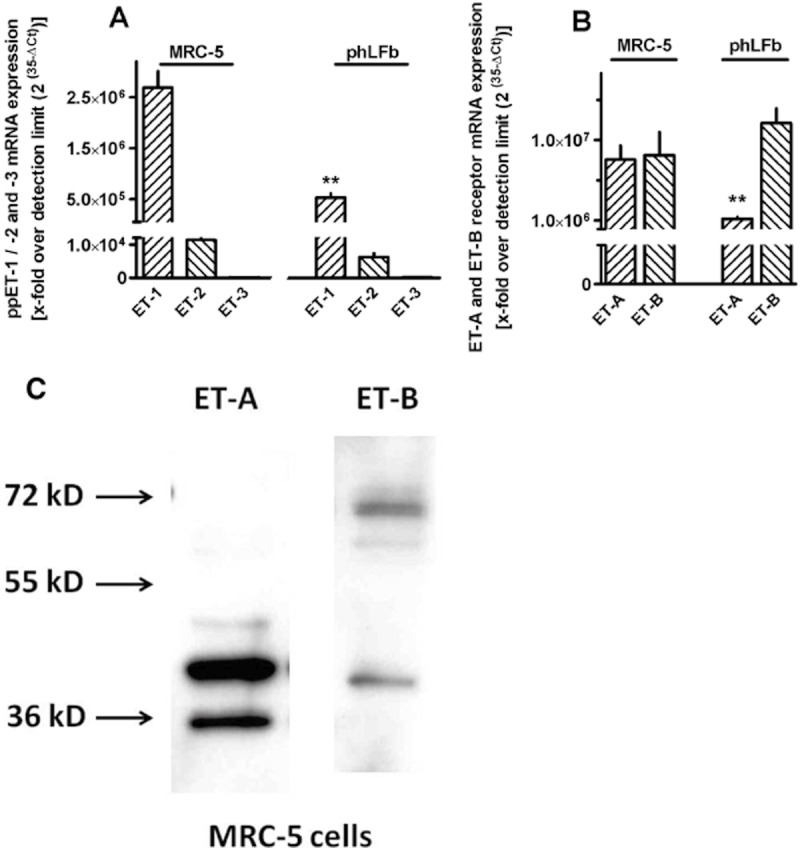
Expression of prepro-ET-1 (ppET-1), -2 and -3 (A) and ET-A and ET-B receptors (B) in MRC-5 and primary (phLFb) human lung fibroblasts. Cells were grown in presence of 10% FCS in 35 mm culture dishes to confluency. Either total RNA was isolated, treated with DNase and used for quantitative real-time PCR with primers specific for the respective human sequences or proteins were extracted and used for immunoblot analysis using commercially available antibodies against ET-A and ET-B receptor protein (C). (A and B) Height of columns: mRNA expression as n-fold over detection limit (2(35−ΔCt)), given are means ± SEM of n ≥ 6. Significance of differences: **P < 0.01 versus respective value in MRC-5 cells. (C) Representative samples of SDS-PAGE of four similar experiments.
Effects of TGF-β and ET receptor antagonists on ET expression and release
TGF-β caused in a concentration-dependent manner a strong, sustained and selective increase in the expression of prepro-ET-1, in both, MRC-5 and phLFb (Figure 2). Concentrations of TGF-β higher than those shown in Figure 2 (5 ng·mL−1) did not cause stronger effects (n = 3; data not shown). In both, MRC-5 and phLFb, the low levels of prepro-ET-2 mRNA further decreased (Figure 2B), and prepro-ET-3 mRNA remained below detection limit (n = 9; data not shown) after 24 h exposure to 1 ng·mL−1 TGF-β. The marked stimulatory effect of TGF-β on prepro-ET-1 was already seen within 1 h (Figures 3 and 4), persisted after 72 h (Figure 2B) and also after one week (data no shown) of exposure.
Figure 2.
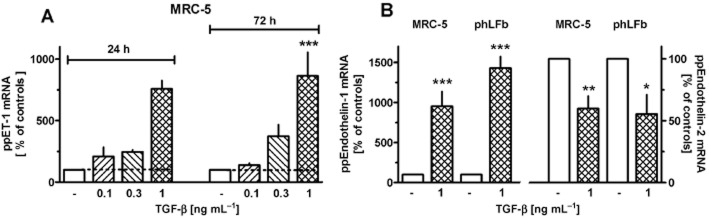
Effects of TGF-β on expression of prepro-ET-1 (ppET-1) and prepro-ET-2 (ppET-2) in MRC-5 and primary (phLFb) human lung fibroblasts. After dissemination, cells were cultured for 24 h in presence of 10% FCS followed by 24 or 72 h (A, as indicated) or 24 h (B) in presence of 1% FCS and absence or presence of TGF-β (0.1–1 ng·mL−1). Thereafter, total RNA was isolated, treated with DNase and used for quantitative real time PCR. Height of columns: ppET-1 or -2 mRNA (ΔCt over GAPDH) is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 6. Significance of differences: *P < 0.05; **P < 0.01; ***P < 0.001 versus respective value in absence of TGF-β.
Figure 3.
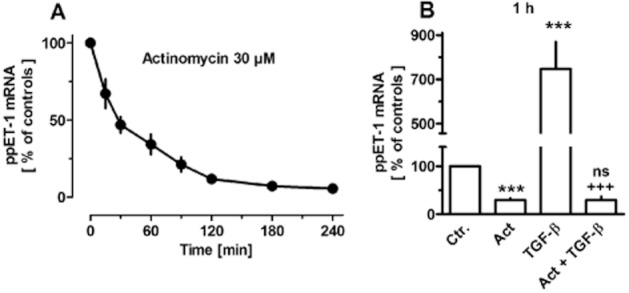
Time-dependent effects of actinomycin D (30 μM, A) and effect TGF-β (1 ng·mL−1, 1 h) in absence and presence of actinomycin D (Act, present 15 min before TGF-β, B) on expression of prepro-ET-1 (ppET-1) in MRC-5 human lung fibroblasts. After dissemination, cells were cultured for 24 h in presence of 10% FCS followed by up to 4 h in presence of 1% FCS and absence or presence test drugs. Thereafter, total RNA was isolated, treated with DNase and used for quantitative real time PCR. Ordinate (A) and height of columns (B): ppET-1 mRNA (ΔCt over GAPDH) is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 4. Significance of differences: ***P < 0.001 versus Ctr.; +++P < 0.001 versus value in presence of TGF-β alone; ns, not significant versus Act alone.
Figure 4.
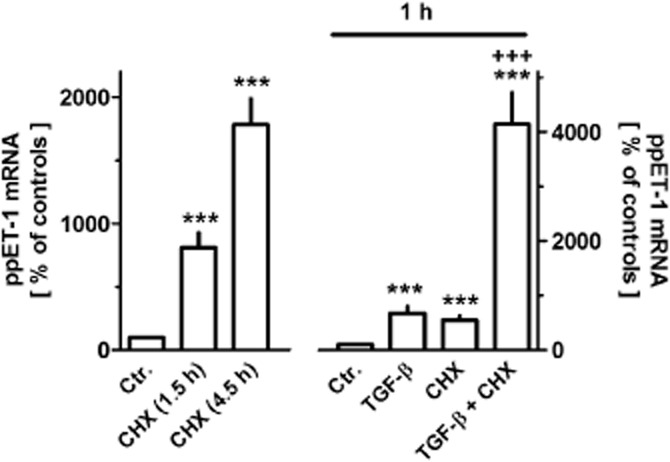
Effects of cycloheximide (CHX, 30 μM, left-hand ordinate) or TGF-β in absence or presence of 30 μM CHX (right-hand ordinate) on expression of prepro-ET-1 (ppET-1) in MRC-5 human lung fibroblasts. After dissemination, cells were cultured for 24 h in presence of 10% FCS followed by 1.5 or 4.5 h in presence of 1% FCS and absence or presence of cycloheximide (30 μM, left-hand ordinate) or 1 h TGF-β (1 ng·mL−1) and/or cycloheximide (30 μM, 1.5 h, i.e. present 30 min before TGF-β). Thereafter, total RNA was isolated, treated with DNase and used for quantitative real time PCR. Height of columns: ppET-1 mRNA (ΔCt over GAPDH) is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 6. Significance of differences: ***P < 0.001 versus respective Ctr.; +++P < 0.001 versus respective value in presence of TGF-β or CHX alone.
In MRC-5 cells, it was further demonstrated that after inhibition of de novo RNA synthesis by actinomycin D, prepro-ET-1 mRNA showed a rapid decline, with a half-life of about 30 min (Figure 3A). In presence of actinomycin D, the stimulatory effect of TGF-β was prevented (Figure 3B). On the other hand, inhibition of protein synthesis by cycloheximide resulted in a rapid, marked increase in prepro-ET-1 mRNA, about eightfold within 1.5 h and about 18-fold after 4.5 h (Figure 4). Strikingly, in presence of cycloheximide, the stimulatory effect of TGF-β was markedly augmented, resulting in a more that 40-fold increase in prepro-ET-1 mRNA after 1 h exposure to TGF-β and cycloheximide (Figure 4).
Bosentan, which alone had no effect of the expression of prepro-ET-1 mRNA, prevented the up-regulation of prepro-ET-1 mRNA caused by a sub-maximally effective concentration of TGF-β (0.3 ng·mL−1, Figure 5). Likewise, the ET-A receptor selective antagonist BQ123 (1 μM) also prevented the increase in prepro-ET-1 mRNA expression caused by 24 h exposure to 0.3 ng·mL−1 TGF-β (102 ± 13% vs. controls, n = 4), whereas the ET-B receptor selective antagonist BQ788 (0.1 μM) did not affect TGF-β (0.3 ng·mL−1)-induced up-regulation of prepro-ET-1 mRNA (n = 3; data not shown). The increase in prepro-ET-1 mRNA expression caused by 1 ng·mL−1 TGF-β (838 ± 125%, n = 11) was neither affected by the ET-A receptor selective antagonist BQ123 (1 μM, 877 ± 204%, n = 6) nor the ET-B receptor selective antagonist BQ788 (0.1 μM, 877 ± 174%, n = 7).
Figure 5.
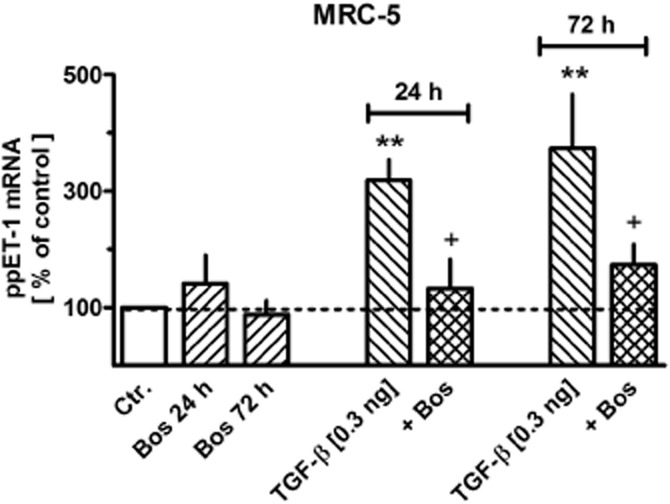
Effects of TGF-β and/or bosentan (Bos) on expression of prepro-ET-1 (ppET-1) in MRC-5 and primary (phLFb) human lung fibroblasts. After dissemination, cells were cultured for 24 h in presence of 10% FCS followed by 24 or 72 h in presence of 1% FCS and absence or presence of 0.3 ng mL−1 TGF-β and/or 10 μM Bos. Thereafter, total RNA was isolated, treated with DNase and used for quantitative real time PCR. Height of columns: ppET-1 (ΔCt over GAPDH) is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 6. Significance of differences: **P < 0.01 versus Ctr.; +P < 0.05 versus respective value in absence of Bos.
ET-1 accumulation in the culture medium of MRC-5 cells could also be measured. Under control conditions, it amounted to 6.8 ± 0.3 pg·mL−1 (≍3 nM) at the end of a 6 h incubation period, corresponding to 44.8 ± 2.6 pg mg−1 protein (n = 74). TGF-β caused in a concentration-dependent manner also a strong increase in the accumulation of ET-1 (Figure 6A), resulting in a maximal accumulation of 34 ± 7 pg·mL−1 ET-1 (≍15 nM) in presence of 0.3 ng·mL−1 TGF-β (n = 17). Interestingly, to note that the concentrations of TGF-β that caused maximal increase in ET-1 accumulation (0.1–0.3 ng·mL−1) were lower than those required for maximal effect on prepro-ET-1 mRNA expression (1 ng·mL−1, Figure 2), in other words, the further substantial increase in mRNA seen at 1 ng mg−1 TGF-β did not result in a corresponding further increase in ET-1 accumulation. Nonetheless, in line with observations on mRNA levels, the up-regulation of ET-1 accumulation by a sub-maximally effective concentration of TGF-β was also significantly attenuated by bosentan (Figure 6B).
Figure 6.
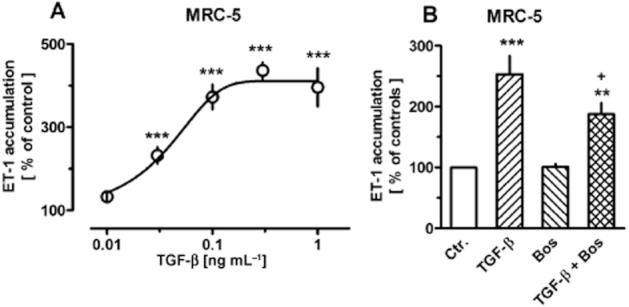
Concentration-dependent effects of TGF-β (A) and effects of 0.03 ng·mL−1 TGF-β in absence and presence of bosentan (Bos, 10 μM) (B) on release of ET-1 by MRC-5 human lung fibroblasts. After dissemination, cells were cultured for 24 h in presence of 10% FCS followed by 48 h in presence of 1% FCS and absence or presence of test substances as indicated. The last medium change was performed 6 h before the supernatant were collected for ET-1 determination by elisa. ET-1 accumulation is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 12. Significance of differences: **P < 0.01; ***P < 0.001 versus Ctr.; +P < 0.05 versus respective value in absence of Bos.
In order to test whether ET converting enzyme activity could have been a limiting factor for the accumulation of ET-1 under the present culture conditions, the conversion of big-ET-1 into ET-1 was studied. Addition of big-ET-1 to confluent cell cultures resulted in concentration- and time-dependent accumulation of ET-1 in the supernatant. Maximal rate of conversion appears to occur at a concentration of 100 nM big-ET-1, and steady-state conditions (where formation and degradation might be balanced) appear to be reached after about 6 h (Figure 7). Notably, the ET-1 concentrations during 6 h incubation with 100 nM big-ET-1 were about 40 times higher than those observed during a 6 h collection period after exposure to maximally stimulating concentrations of TGF-β. Pretreatment of the cells with TGF-β (0.3 ng·mL−1 for 48 h) did not affect accumulation of ET-1 during 3 or 6 h incubation with 1, 10 or 100 nM big-ET-1 (data not shown; each point n ≥ 6).
Figure 7.
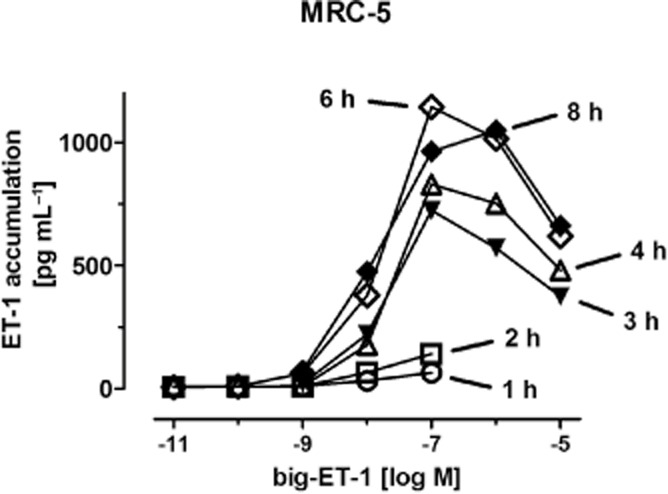
Time- and concentration-dependent conversion of big-ET-1 into ET-1 by MRC-5 human lung fibroblasts in culture. Big-ET-1 at the concentrations given was added to nearly confluent cultures in absence of FCS for 1 to 8 h, as indicated, followed by determination of ET-1 accumulation in the supernatant. Given are means of n ≥ 6, for clarity of the figure without SEM, which were between 5% and 15% of the respective mean value.
TGF-β causes selective down-regulation of ET-B receptor expression
In both, MRC-5 cells and phLFb, TGF-β caused in a concentration-dependent manner a marked and persistent down-regulation of the expression of ET-B receptor mRNA, whereas the expression of the ET-A receptor was not significantly affected by TGF-β in MRC-5 cells and only slightly reduced in phLFb after prolonged (72 h) exposure (Figure 8).
Figure 8.
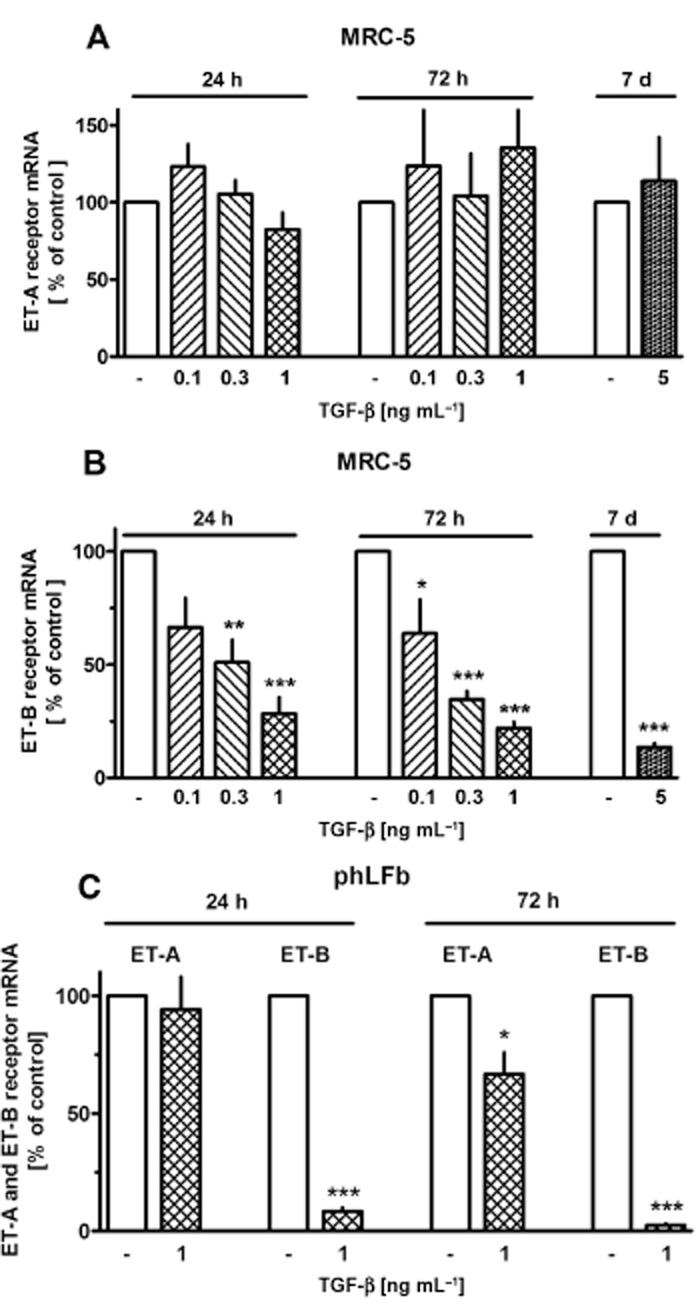
Effects of TGF-β on expression of ET-A and ET-B receptors in MRC-5 and primary (phLFb) human lung fibroblasts. After dissemination cells were cultured for 24 h in presence of 10% FCS followed by 24, 72 h or 7 days in presence of 1% FCS and absence or presence of TGF-β at the concentrations indicated. Thereafter, total RNA was isolated, treated with DNase and used for quantitative real time PCR. Height of columns: ET-A or ET-B mRNA (ΔCt over GAPDH) is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 3. Significance of differences: ***P < 0.001; **P < 0.01; *P < 0.05 versus respective controls.
ET-1-mediated changes in DMR signals
In order to check whether ET receptors in MRC-5 human lung fibroblasts are functional, effects of ET-1 on cellular DMR were measured. DMR allows label-free real-time analysis of cellular responses to GPCR activation in live cells (e.g. Schröder et al., 2010). In a previous study, it was observed that Gs-signalling in MRC-5 cells causes a negative DMR signal (Lamyel et al., 2011). In the present study, ET-1 caused in a concentration dependent manner a marked positive DMR signal, with an EC50 of 56 nM (Figure 9A and B). The effect of 0.3 μM ET-1 was inhibited in a concentration-dependent manner by the non-selective ET receptor antagonist bosentan (Figure 9C and D). The ET-A receptor selective antagonist BQ123 (1 μM) inhibited the effect of 0.3 μM ET-1 by only about 35% (Figure 9E and F), and increasing the concentration of BQ123 to 10 μM did not consistently cause a further reduction (data not shown). The ET-B receptor selective antagonists BQ788 (0.1 μM) alone attenuated only marginally the effect of 0.3 μM ET-1 but caused a marked further inhibition in presence of 1 μM BQ123, resulting in an almost complete suppression of the signal in the presence of both antagonists (Figure 9E and F).
Figure 9.
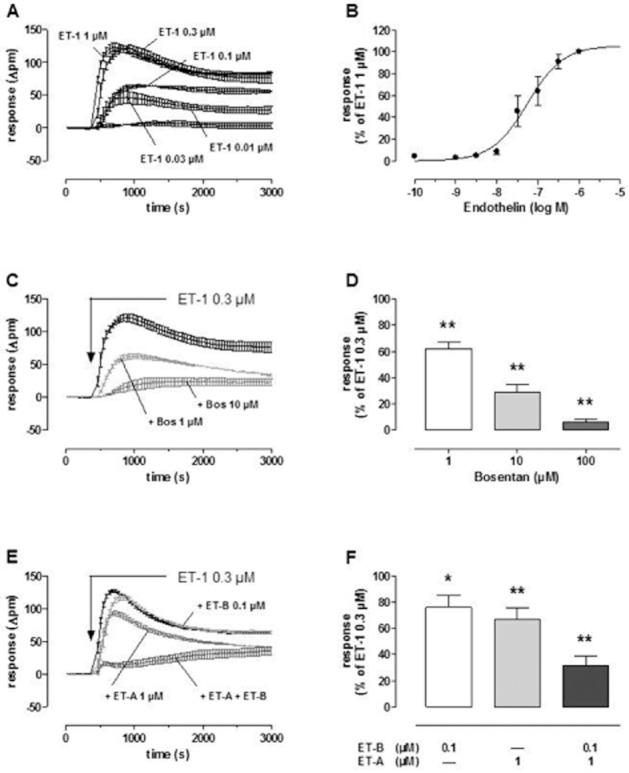
ET-1-induced DMR in MRC-5 human lung fibroblasts in absence and presence of bosentan (Bos) or the ET-A (BQ123) or ET-B (BQ788) receptor selective antagonist. Cells were seeded in a density of 15 000 cells per well in 384-well Epic fibronectin-coated microplates with 40 μL of growth medium cultured at 37°C for 20 h to achieve confluent cell layers. Thereafter, cell culture medium was replaced by 30 μL assay buffer per well (HBSS with 20 mM HEPES, pH 7.0), and cells were allowed to rest in assay buffer in absence or presence of antagonists for 2 h in the Epic® reader at 28°C. After addition of 10 μL of ET-1 (final concentrations 0.01–1 μM), DMR responses were monitored for 1 h. Indicated are real-time recordings of ET-1-induced DMR-signals from representative experiments (A, C and E, means ± SEM of three to four measurements run in parallel). ET-1-induced concentration–effect curve (B) resulted from the maximal DMR signals within 1800 s: log IC50 is 7.25 ± 0.11 (ET-1 1 μM, bottom and slope factor are fixed to 100 %, 0% and 1, respectively; n = 5). Height of columns (D and F): maximal DMR signal evoked by 0.3 μM ET-1 in presence of antagonists at the concentrations given, expressed as % of the ET-1 response in absence of antagonists, means ± SEM, n = 3–5. Significance of differences: *P < 0.05; **P < 0.01 versus ET-1 response in absence of antagonists.
Endothelinergic stimulation of human lung fibroblast proliferation
In order to investigate whether fibroblast proliferation is regulated by endothelinergic mechanisms, the effects of ET-1 and its receptor antagonists on [3H]-thymidine incorporation were studied. As shown in Figure 10A, in MRC-5 fibroblasts bosentan and the ET-A receptor selective antagonist BQ123 caused in concentration-dependent manner a reduction in [3H]-thymidine incorporation, maximally by about 45%, whereas the ET-B selective antagonist BQ788 showed no significant effects. ET-1 alone caused only a minor increase in [3H]-thymidine incorporation, maximally by about 15%. However, ET-1 was able to surmount in a concentration-dependent manner the inhibitory effect of bosentan (Figure 10B). In phLFb, bosentan caused also a reduction in [3H]-thymidine incorporation, but the maximum effect (an inhibition by about 25%) was smaller than that observed in MRC-5 cells. Correspondingly, the stimulatory effect of ET-1 on its own was somewhat larger in phLFb than in MRC-5 cells (Figure 10B and C). With intention to reduce the influence of a possible endogenous endothelinergic tone, one series of experiments was performed in which ET-1 was added immediately after cell dissemination, and cells were incubated for 30 h with [3H]-thymidine present for the last 24 h. Under these conditions, consistently a stronger proliferative effect (increases in [3H]-thymidine incorporation by about 48%) was observed in MRC cells (Figure 10D). However, also under these conditions, the proliferative effect in phLFb was stronger than in MRC-5 cells, with increases in [3H]-thymidine incorporation by about 100%. Noteworthy, for both cells types, the maximum effect appears already reached at lower concentrations of ET-1 than under conditions described in Figure 10A–C.
Figure 10.

Effects of bosentan, the ET-A (BQ123) and ET-B (BQ788) receptor selective antagonist and ET-1 in absence or presence of bosentan (Bos) on [3H]-thymidine incorporation (A–D) or cell count (E) in MRC-5 and primary (phLFb) human lung fibroblasts. A–C: 4 × 104 cells were seeded in 12-well dishes and first cultured for 24 h in presence of 10% FCS, followed by additional 24 h under FCS-free conditions. After a 24 h, FCS-free period test drugs were added as indicated for 30 h. [3H]-Thymidine (37 kBq) was present for the last 24 h (i.e. it was added 6 h after addition of test drugs). (D) 7.5 × 104 cells were seeded in 12-well dishes and cultured from the onset under FCS-free conditions for 30 h. ET-1 was present from the beginning, and [3H]-thymidine during the last 24 h. Cellular radioactivity is expressed as % of the mean value of the controls of each cell preparation. (E) 7.5 × 104 cells were seeded in 12-well dishes and cultured from the onset under FCS-free conditions for 28 or 48 h in absence or presence of test substance. Cell count is expressed as % of the mean value of the controls of each cell preparation Given are means ± SEM of n ≥ 9 (A–D) and n = 18–22 (E) experiments. Significance of differences: *P < 0.05; **P < 0.01; ***P < 0.001 versus respective controls; +P < 0.01 versus respective value in presence of bosentan alone; #P < 0.01; ##P < 0.001 versus respective value in absence of bosentan.
The growth stimulatory effect of ET-1 was also demonstrated in MRC-5 cells by an increased cell count. When cells were cultured under FCS-free condition for 28 and 48 h (conditions comparable with those in Figure 10D), the number of cells under control conditions was 45 000 ± 5300 (at 28 h, n = 22) and 63 500 ± 3850 (at 48 h, n = 18, P < 0.05 vs. 24 h). Exposure to ET-1 (10 nM) caused an increase in the number of cells by 70% and 80% respectively (Figure 10E). For comparison addition of 1% FCS for 28 h caused an increase by 70%. In presence of bosentan (10 μM) alone, cell count tended to be reduced after 28 h and was significantly reduced by 35% after 48 h (Figure 10E). In addition, bosentan prevented the stimulatory effect of ET-1 (Figure 10E).
Endothelinergic control of myofibroblast differentiation
In confirmation of previous observations (Lamyel et al., 2011), MRC-5 cells cultured under standard conditions showed expression of α-smooth muscle actin, a marker of myofibroblast differentiation, and after 24 h exposure to bosentan a clear reduction in α-smooth muscle actin protein levels by about 50% was observed (Figure 11), whereas ET-1 10 nM (Figure 11) or 100 nM (data not shown) induced only a minor increase by about 20%. In presence of TGF-β (0.3 ng·mL−1, 24 h), which alone caused an increase in α-smooth muscle actin by about 120%, bosentan had no effect (Figure 11).
Figure 11.
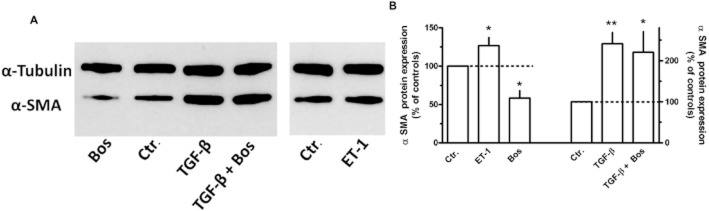
Effects of the ET-1 (10 nM), TGF-β (0.3 ng·mL−1) and/or bosentan (Bos, 10 μM) on α-smooth muscle actin (α-SMA) expression in MRC-5 human lung fibroblasts. 3 × 105 cells were seeded in 35 mm dishes and cultured for 24 h in presence of 10% FCS. After a 24 h FCS-free period, cells were cultured for additional 24 h in FCS-free medium in absence or presence of test substances. Cellular proteins were extracted and Western blot analysis for α-SMA and α-tubulin was performed. (A) Samples of original Western blots. (B) Densitometric analysis of a series of Western blots, ratio α-SMA/α-tubulin, expressed as % of controls. Given are means ± SEM of n ≥ 7, significance of differences versus Ctr.: *P < 0.05, **P < 0.01.
Endothelinergic stimulation of human lung fibroblast collagen synthesis
In MRC-5 cells, collagen synthesis (determined by [3H]-proline incorporation) was increased by ET-1 in a concentration-dependent manner, maximally by about 75% (Figure 12A). Bosentan and the ET-A receptor selective antagonist BQ123 caused a reduction in [3H]-proline incorporation by about 20% (Figure 12A), whereas the ET-B receptor selective antagonist BQ788 in a concentration of 100 nM (Figure 12A) had no significant effect. The effect of ET-1 was effectively, but in a surmountable manner, antagonized by bosentan and BQ123, whereas BQ788 had no effects (Figure 12A). The above-described DMR experiments suggested that blockade of ET-A receptors could unmask an ET-B-receptor-mediated effect. Therefore, BQ788 was also studied in presence of BQ123 either alone or in combination with 10 or 100 nM ET-1, but again failed to affect [3H]-proline incorporation (data not show; each n = 6–9).
Figure 12.
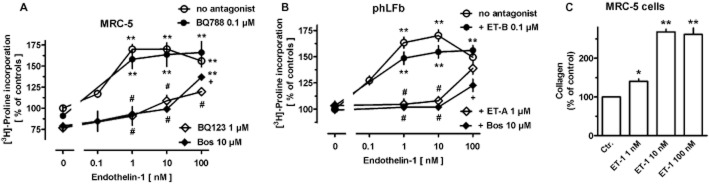
Concentration-dependent effects of ET-1 in absence and presence or bosentan (Bos) or the ET-A (BQ123, 1 μM) or ET-B (BQ788, 100 nM) receptor-selective antagonist on [3H]-proline incorporation (A, B) and collagen content (C) in MRC-5 (A, C) and primary (B, phLFb) human lung fibroblasts. 105 cells were seeded in 12-well dishes and first cultured for 24 h in presence of 10% FCS, followed by additional 24 h under FCS-free conditions. Thereafter (A, B), [3H]-proline (37 kBq) was added alone or in combination with test drugs and cells were cultured for further 24 h. Radioactivity incorporated in cellular and extracellular protein was determined. (C) Cells were cultured for 24 h in absence of presence of ET-1 and collagen content of cell extracts determined by QuickZyme collagen assay. Data are expressed as % of the mean value of the controls of each cell preparation. Given are means ± SEM of n ≥ 9 (A, B) or n = 4–5 (C) experiments. Significance of differences: *P < 0.05; **P < 0.001 versus controls (absence of drugs); +P < 0.05; #P < 0.01 versus respective value in absence of antagonists.
In phLFb, none of the antagonists showed a significant effect (Figure 12B), whereas ET-1 caused a concentration-dependent increase in [3H]-proline incorporation comparable with that observed in MRC-5 cells (Figure 12B). Like in MRC-5 cells, the effect of ET-1 was effectively, but in a surmountable manner, antagonized by bosentan and the ET-A selective antagonist BQ123, whereas the ET-B-selective antagonist BQ788 did not significantly affect the ET-1 response (Figure 12B).
The stimulatory effect of ET-1 on collagen synthesis was also demonstrated using the QuickZyme collagen assay which is based on the specific binding of the dye Sirius Red to collagen. The amount of collagen detected in extracts of control cultures was 5.1 ± 0.7 μg (n = 5). Exposure to ET-1 for 24 h caused in concentration-dependent manner a marked increase, maximally by 170% at 10 nM (Figure 12C).
Finally, it could be demonstrated that ET-1 enhances also the mRNA expression of collagen I-α1, a major collagen type in lung fibroblasts. As shown in Figure 13, after 24 h exposure to ET-1 (10 and 100 nM), collagen I-α1 mRNA was enhanced by about 60%, an effect blocked by bosentan, which alone had no effect (Figure 13).
Figure 13.
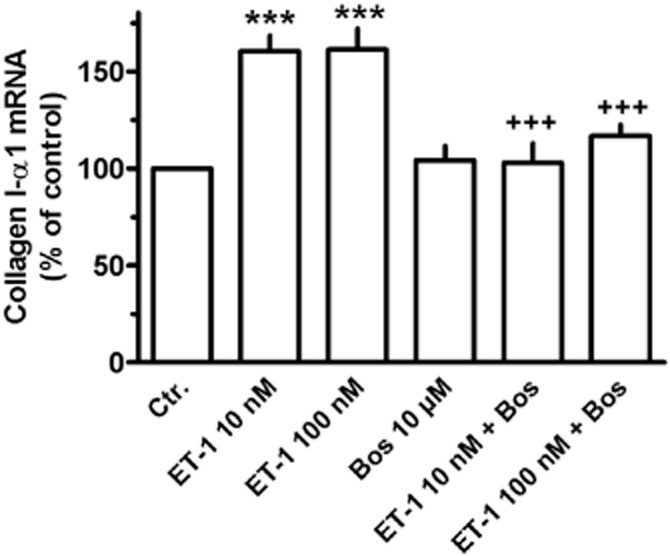
Effect of ET-1 in absence or presence of bosentan (Bos) on collagen I-α1 mRNA expression in MRC-5 human lung fibroblasts; 105 cells were seeded in 12-well dishes and first cultured for 24 h in presence of 10% FCS, followed by additional 24 h under FCS-free conditions in absence or presence of test substances at the concentrations given. Thereafter, total RNA was isolated, treated with DNase and used for quantitative real time PCR. Height of columns: collagen I-α1 mRNA (ΔCt over GAPDH) is expressed as % of the control of the individual cell preparation, given are means ± SEM of n ≥ 6. Significance of differences: ***P < 0.01 versus control (Ctr.); +++P < 0.001 versus respective value in absence of Bos.
Since TGF-β enhanced prepro-ET-1 expression and ET-1 release, it was further studied whether a putative stimulatory effect of TGF-β on collagen synthesis involves ET-1. TGF-β in the relatively low concentrations of 0.3 and 1.0 ng·mL−1 enhanced [3H]-proline incorporation by 60% and 73% respectively (Figure 14A). Presence of bosentan under control conditions resulted (like in the preceding experiments, Figure 12) in a reduction of [3H]-proline incorporation by about 20%. Of the enhanced [3H]-proline incorporation in presence of TGF-β, a significantly larger fraction was sensitive to ET receptor blockade by bosentan compared with control conditions (Figure 14B). The ET-A-receptor-selective antagonist BQ123 (1 μM) caused a similar reduction in [3H]-proline incorporation, whereas the ET-B-receptor-selective antagonist BQ788 (100 nM) was again without effect (data not shown; each n = 6).
Figure 14.
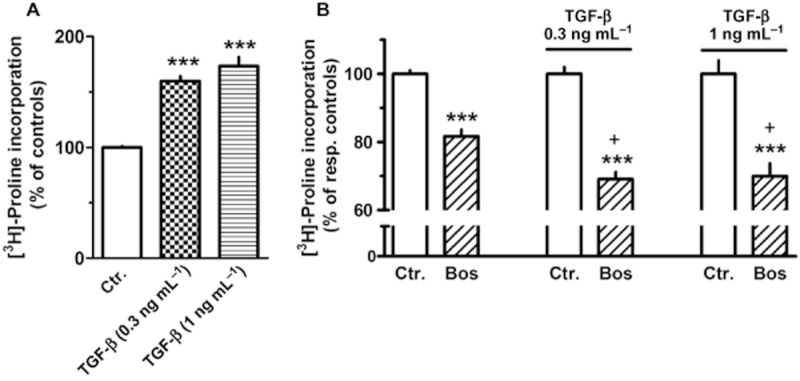
Effects of TGF-β and of bosentan (Bos, 10 μM) in absence and presence of TGF-β on [3H]-proline incorporation in MRC-5 (A); 105 cells were seeded in 12-well dishes and first cultured for 24 h in presence of 10% FCS, followed by additional 24 h under FCS-free conditions. Thereafter, [3H]-proline (37 kBq) was added alone or in combination with test drugs, and cells were cultured for further 24 h. Radioactivity incorporated in cellular and extracellular protein was determined and is expressed as % of the mean value of the respective controls (in absence or presence of TGF-β) of each cell preparation. Given are means ± SEM of n ≥ 8 experiments. Significance of differences: ***P < 0.001 versus respective Ctr.; +P < 0.05 versus respective value in absence of TGF-β.
ET-1-induced activation of ERK MAPK and its role in collagen synthesis
We previously reported that muscarine receptor mediated stimulation of proliferation and collagen synthesis in human lung fibroblasts was mediated by activation of the ERK MAPK pathway (Matthiesen et al., 2007; Haag et al., 2008). As shown in Figure 15A, ET-1, similar to the muscarinic agonist oxotremorine, induced a rapid and transient increase in phospho-p42/44, i.e. activation of the ERK MAP kinase pathway. The maximum effect of ET-1, an increase by 310% was seen at 100 nM and was somewhat larger than that of oxotremorine (10 μM, also a maximally effective concentration; Matthiesen et al., 2007; Haag et al., 2008). However, pre-treatment with pertussis toxin, which alone tended to reduce basal levels of phospho-p42/44, largely attenuated the increase in phospho-p42/44 induced by oxotremorine, but not that by ET-1 (Figure 15). When expressed in relation to untreated controls as shown in Figure 15, the ET-1-induced increase in phospho-p42/44 was somewhat reduced, but when expressed in relation to the respective (somewhat reduced) PTX value, an increase in phospho-p42/44 by more than 400% is seen. The ET-A receptor selective antagonist BQ123 caused a 30% reduction of the basal phospho-p42/44 levels and was able to oppose the stimulatory effect of ET-1 (Figure 15C), whereas the ET-B receptor selective antagonist BQ788 showed no effects, neither in absence nor presence of ET-1. It should be mentioned that the solvent DMSO alone (in the concentration present with BQ788) significantly attenuated the stimulatory effect of ET-1. Therefore, the data in Figure 15C show effects in relation to respective controls in absence of presence of the solvent.
Figure 15.

Western blot analysis of phospho-p42/44 (ERK1/2) MAPK in MRC-5 human lung fibroblasts. Cells were cultured in 55 mm dishes to nearly confluence, serum-starved for 24 h and exposed to 100 nM ET-1, 10 μM oxotremorine (Oxo), or vehicle (Ctr.) for 5–30 min (as indicated, A) or 5 min (B and C). In the respective experiments, pertussis toxin (PTX, 50 ng·mg−1) was present 6 h and the ET-A (BQ123, 1 μM) or ET-B (BQ788, 100 nM) receptor antagonist 30 min prior to exposure to ET-1. In controls, cells were treated with solvent at the respective time points (i.e. 5 min and additionally 30 min or 6 h before cell lysis respectively). Cell lysates were prepared, and 25 μg was separated in 4–10% acrylamide Tris–glycine gels, immobilized to PVDF membranes and detected by antibodies specific for phosphorylated and unphosphorylated ERK. Densitometrical quantification of the p44/42 bands was performed, and values (arbitrary units, normalized over unphosphorylated ERK) are expressed as % of the respective control, in absence or presence of ET-1 and absence or presence of the solvent DMSO (which was required to dissolve BQ788). Given are means ± SEM of n = 4–14. Significance of differences: *P < 0.05, **P < 0.01, ***P < 0.001 versus respective Ctr.; #P < 0.01 versus respective value in absence of PTX; ++P < 0.05 versus respective value in presence of PTX alone.
In correspondence to the above observation, the stimulatory effect of oxotremorine (in confirmation of previous data; Haag et al., 2008), but not that of ET-1 on collagen synthesis was prevented by pertussis toxin (Figure 16A). Nonetheless, the activation of the ERK–MAPK pathway appears to be crucially involved also in the ET-1-induced collagen synthesis, as PD 098059 (a specific inhibitor of the MAPK-activating enzyme, MEK) (Dudley et al., 1995) prevented the ET-1-mediated stimulation of collagen synthesis (Figure 16B).
Figure 16.
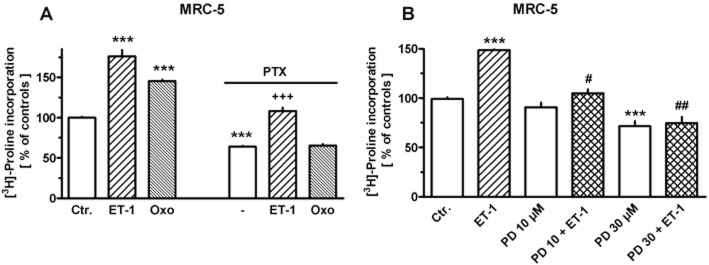
Effects of ET-1 (10 nM), oxotremorine (Oxo, 10 μM) and/or pertussis toxin (PTX, 50 ng·mL−1) (A) and/or PD 098059 (PD, at the concentrations given) (B) on [3H]-proline incorporation in MRC-5 human lung fibroblasts; 105 cells were seeded in 12-well dishes. Cells were first cultured for 24 h in presence of 10% FCS, followed by additional 24 h under FCS-free conditions. Thereafter, [3H]-proline (37 kBq) was added alone or in combination with test drugs, and cells were cultured for further 24 h. In the respective experiments, PTX was present 6 h and PD 30 min prior to the exposure to ET-1 or Oxo. Radioactivity incorporated in cellular and extracellular protein was determined and is expressed as % of the mean value of the controls of each cell preparation. Given are means ± SEM of n = 9 experiments. Significance of differences: ***P < 0.001 versus Ctr.; +++P < 0.001 versus respective value in presence PTX or ET-1 alone; #P < 0.01; ##P < 0.001 versus respective value in presence of ET-1 alone.
Discussion
The present study shows that human lung fibroblasts are endowed with all elements of a functional autocrine/paracrine endothelinergic system. They express prepro-ET and are able to process it to the active mediator. They also express functional ET receptors that appear to regulate ‘pro-fibrotic’ features.
MRC-5 cells and phLFb express mRNA for prepro-ET-1 and prepro-ET-2, but no transcript for prepro-ET-3. However, in both MRC-5 cells and primary cells, mRNA levels for prepro-ET-1 largely exceeded those for prepro-ET-2, indicating that ET-1 is the predominant ET in human lung fibroblasts. Under standard culture conditions, mRNA levels for prepro-ET-1 and prepro-ET-2 were somewhat higher in MRC-5 cells than in phLFb. However, the expression of prepro-ET-1 mRNA is highly up-regulated by TGF-β, up to about ninefold in MRC-5 and more than 15-fold in phLFb, resulting in nearly similar expression levels in both cell types. The stimulatory effect of TGF-β was rapid in onset and did not show any fatigue during prolonged exposure suggesting that pathological conditions with prolonged elevation of TGF-β might be accompanied by a sustained up-regulation of ET-1 expression.
The stimulatory effect of TGF-β on ET expression is confined to ET-1, as TGF-β did not induce the expression of prepro-ET-3 and caused even a reduction of the already low levels of prepro-ET-2 (Figure 2B).
Expression of ET-1 mRNA in human lung fibroblasts appears to be highly regulated. As a consequence of the short half-life (30 min), prepro-ET-1 mRNA levels are expected to reflect immediately changes in prepro-ET-1 gene transcription. In fact, prepro-ET-1 mRNA levels were increased about sevenfold already after 1 h exposure to TGF-β (Figures 3 and 4).
TGF-β-induced up-regulation of prepro-ET-1 mRNA was prevented by actinomycin D, indicating that it is caused by increased gene transcription. The rapid onset of the TGF-β effect and the observation that it occurred also in presence of cycloheximide suggest a direct regulation of prepro-ET-1 mRNA gene expression by TGF-β signalling pathways. That cycloheximide caused a rapid increase in prepro-ET-1 mRNA indicates that prepro-ET-1 gene expression is under inhibitory control of short-living regulatory peptides, which appear also to oppose the stimulatory effect of TGF-β, as the TGF-β response was markedly enhanced in presence of cycloheximide (Figure 4).
Accumulation of the biological active ET-1 in culture media could also be detected and exposure to TGF-β resulted in increased levels. Strikingly, the maximum effect of TGF-β on ET-1 accumulation was seen at concentrations that induced only sub-maximal increase in prepro-ET-1 mRNA (compare Figures 2A and 6A). A simple relationship between prepro-ET-1 mRNA levels and ET-1 accumulation may not be expected, since ET-1 accumulation is determined by a number of processes, including translation into the precursor protein, its processing to big-ET-1, release of big-ET-1, conversion to ET-1 and finally degradation of the active peptide. Since the concentrations of ET-1 accumulating in presence of exogenous big-ET-1 were about 40-fold higher (Figure 7) than those observed in media from cells stimulated with TGF-β (Figure 6), limited ET converting enzyme activity can be excluded. Limited translation and/or limited processing of the precursor protein may account for the non-linear relationship between mRNA levels and accumulation of the active peptide.
At present, two ET receptors, ET-A and ET-B, have been identified; and mRNA for both receptors was found to be expressed in MRC-5 and phLFb. ET-B receptor mRNA appears to be strongly regulated, as indicated by the marked down-regulation by TGF-β, whereas expression of ET-A receptors appears to be constitutive (Figure 8B and C). Constitutive expression of ET-A receptors, but strong regulation of ET-B receptor expression has also been described for other tissues, for example rat mesenteric artery (Uddman et al., 2002).
To check whether ET receptors in human lung fibroblasts are functional, ET-1-induced cellular DMR was measured in MRC-5 cells. DMR allows label-free real-time analysis of signalling pathway activation by GPCRs in living cells (Antony et al., 2009; Schröder et al., 2010). The direction of G-protein pathway mediated DMR depends on the cell-type (Schröder et al., 2010), and we recently showed that in MRC-5 cells β2-adrenoceptor agonists, like direct activation of AC by forskolin, induced negative DMR (Lamyel et al., 2011). In the present experiments, ET-1 induced in a concentration-dependent manner a positive DMR. The non-selective ET receptor antagonist bosentan inhibited ET-1-induced DMR in a manner suggesting a competitive interaction. Combination of the ET-A and ET–B receptor selective antagonists BQ123 and BQ788, in concentrations, in which they may selectively block the respective receptor (Ishikawa et al., 1994), inhibited the ET-1-induced DMR much more effectively than each antagonist alone. This indicates that in MRC-5 human lung fibroblasts both ET receptors are functional. BQ788 alone had only minor effects indicating that ET-A receptors alone are able to induce almost the full DMR response. However, after blockade of ET-A receptors, ET-B receptors appear to be able to maintain a substantial response.
Bosentan attenuated the up-regulation of prepro-ET-1 expression and ET-1 accumulation induced by sub-maximal concentrations of TGF-β (Figures 5 and 6B), indicating that ET-1 may contribute in a kind of auto-regulatory positive feedback to the regulation of its own expression and release. This effect appears to be mediated via ET-A receptors, since only BQ123, but not BQ788 mimicked the effect of bosentan. A similar ET-A receptor-mediated feedback mechanism was also described for the TNF-α-induced up-regulation of ET-1 in human airway smooth muscle cells (Knobloch et al., 2009).
In both, MRC-5 cells and phLFb, ET-1 exerted proliferative effects as measured by [3H]-thymidine incorporation. Under standard culture conditions (Figure 10A–C), ET-1 induced only a minor proliferative effect. However, bosentan caused a marked inhibition, indicating that endogenously released ET-1 exerts already a significant proliferative signal. The inhibitory effect of bosentan was surmountable by exogenous ET-1, indicating a competitive interaction between ET-1 and bosentan, and supporting that the effect of bosentan was caused by specific ET receptor blockade. As only BQ123, but not BQ788, mimicked the effect of bosentan, it is concluded that ET-A receptors mediate the proliferative stimulus. In phLFb, the inhibitory effect of bosentan was smaller, but on the other side ET-1 exerted a stronger proliferative effect. These functional observations are in line with lower expression levels of ET-1 in phLFb compared with MRC-5 cells. ET-1 mediated stronger proliferative effects, when applied already at the time of cell dissemination, presumably a condition with a lower endogenous ET-1 tone and in line with this conclusion, the potency of exogenous ET-1 was also higher. Under these conditions, a proliferative effect of ET-1 was confirmed by cell count. In contrast to present observations, Préfontaine et al. (2008) reported that ET-A and ET-B receptor were involved in ET-1-induced proliferation in rat lung fibroblasts, whereas Gallelli et al. (2005) showed in line with the present data that in phLFb only ET-A receptors mediate proliferative effects. Thus, species difference in the role of ET-A and ET-B receptors must be considered.
Endogenously released ET-1 appears to drive also myofibroblast differentiation as bosentan inhibited α-smooth muscle actin expression. However, ET-1, although up-regulated by exposure to TGF-β, appears not to be involved in TGF-β-induced myofibroblast differentiation, as bosentan did not affect TGF-β-induced up-regulation of α-smooth muscle actin expression (Figure 11).
Furthermore, ET-1 stimulated also collagen synthesis as measured by [3H]-proline incorporation in MRC-5 cells and phLFb. In agreement with the already discussed higher endogenous endothelinergic tone in MRC-5 cells compared to phLFb, bosentan and the ET-A receptor antagonist BQ123 caused a significant reduction of [3H]-proline incorporation in MRC-5 cells, but not in phLFb. Nonetheless, in both cell types, bosentan and BQ123 effectively antagonized the effect of ET-1 in a surmountable manner, whereas BQ788 was ineffective. Direct determination of collagen by the QuickZyme collagen assay confirmed the marked stimulatory effect of ET-1 on collagen synthesis. The observation, that ET-1 up-regulated the mRNA of collagen I-α1, a major collagen type in lung fibroblasts, indicates that enhanced collagen gene transcription may at least contribute to the up-regulation of collagen synthesis.
Activation of ERK–MAPK has been shown to be crucially involved in muscarinic receptor mediated stimulation of proliferation (Matthiesen et al., 2007) and collagen synthesis (Haag et al., 2008) in MRC-5 cell. In the present experiments, ET-1, like oxotremorine, induced a rapid, but transient activation of the ERK-MAPK (Figure 15A). In agreement with previous findings that muscarinic receptors in human lung fibroblasts are Gi coupled (Matthiesen et al., 2006; 2007; Haag et al., 2008), pertussis toxin prevented oxotremorine-induced ERK activation. It inhibited, however, only partially activation of ERK caused by ET-1 (Figure 15), indicating that ET-1-induced ERK activation may be mediated in part via Gi, but to a large extent also via pertussis-insensitive G-proteins, possible Gq or G11/12. In correspondence, pertussis toxin prevented muscarinic stimulation of collagen synthesis, but inhibited only partially ET-1-induced collagen synthesis (Figure 15A). Nonetheless, inhibition of ERK MAP kinase pathway by PD 098059 abolished ET-1-induced stimulation of collagen synthesis (Figure 16B), proving that this pathway is crucially involved not only in proliferative effects of ET-1 (Gallelli et al., 2005) but also in its effects on collagen synthesis.
In conclusion, human lung fibroblasts express a functional autocrine/paracrine endothelinergic system, which may, in interaction with other mediators such as TGF-β, drive pro-fibrotic features. Thus, ET-1 is highly up-regulated by TGF-β and can mediate stimulatory effects on proliferation, myofibroblast differentiation and collagen synthesis. Although ET-B receptors are expressed in human lung fibroblasts and their functional significance could be demonstrated by DMR, all other so far studied effects of ET-1 appear to be mediated exclusively by ET-A receptor activation.
Acknowledgments
This work was supported by Research Grants from Actelion Pharmaceuticals Deutschland GmbH and Bonfor, Univ. Bonn. The paper contains part of the PhD thesis of ASA. We thank Mrs M Fuhrmann and Mr M Stöber for excellent technical assistance and Corning Inc. for their support on the Epic system. WKS is a member of the NRW International Graduate Research School BIOTECH-PHARMA.
Glossary
- BQ123
cyclo(d-Trp-d-Asp-Pro-d-Val-Leu)
- BQ788
N-cis-2,6-dimethylpiperidinocarbonyl-β-tBu-Ala-d-Trp(1-methoxycarbonyl)-d-Nle-OH
- d.p.m
disintegration per minute
- DMR
dynamic mass redistribution
- ET
endothelin
- FCS
fetal calf serum
- hLFb
human lung fibroblasts
- MEK
MAPK-activating enzymes
- phLFb
primary human lung fibroblasts
- qPCR
real-time reverse transcription-PCR
Conflict of interest
Authors declare that they have not any conflict of interest.
References
- Antony J, Kellershohn K, Mohr-Andrä M, Kebig A, Prilla S, Muth M, et al. Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 2009;23:442–450. doi: 10.1096/fj.08-114751. [DOI] [PubMed] [Google Scholar]
- Battistini B, Jeng AY. Endothelin-converting enzyme inhibitors and their effects. In: Warner TD, editor. Handbook of Experimental Pharmacology, Vol 152. Endothelin and Its Inhibitors. Berlin: Springer-Verlag; 2001. pp. 155–208. [Google Scholar]
- Bhavsar TM, Liu X, Cerreta JM, Liu M, Cantor JO. Endothelin-1 potentiates smoke-induced acute lung inflammation. Exp Lung Res. 2008a;34:707–716. doi: 10.1080/01902140802389701. [DOI] [PubMed] [Google Scholar]
- Bhavsar T, Liu XJ, Patel H, Stephani R, Cantor JO. Preferential recruitment of neutrophils by endothelin-1 in acute lung inflammation induced by lipopolysaccharide or cigarette smoke. Int J Chron Obstruct Pulmon Dis. 2008b;3:477–481. doi: 10.2147/copd.s2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers GW, Little SA, Patel KR, Thomson NC. Endothelin-1-induced bronchoconstriction in asthma. Am J Respir Crit Care Med. 1997;156:382–388. doi: 10.1164/ajrccm.156.2.9702066. [DOI] [PubMed] [Google Scholar]
- Corder R. Identity of endothelin-converting enzyme and other targets for the therapeutic regulation of endothelin biosynthesis. In: Warner TD, editor. Handbook of Experimental Pharmacology, Vol 152. Endothelin and Its Inhibitors. Berlin: Springer-Verlag; 2001. pp. 35–67. [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag A, Reimann A, Wessler I, Racké K. Effects of bacterial lipopolysaccharides (LPS) and tumor necrosis factor-α (TNF-α) on rat tracheal epithelial cells in culture: morphology, proliferation and induction of NO synthase. Pulm Pharmacol. 1996;9:149–156. doi: 10.1006/pulp.1996.0017. [DOI] [PubMed] [Google Scholar]
- Gallelli L, Pelaia G, D'Agostino B, Cuda G, Vatrella A, Fratto D, et al. Endothelin-1 induces proliferation of human lung fibroblasts and IL-11 secretion through an ET(A) receptor-dependent activation of MAP kinases. J Cell Biochem. 2005;96:858–868. doi: 10.1002/jcb.20608. [DOI] [PubMed] [Google Scholar]
- Goldie RG, Henry PJ. Endothelins and asthma. Life Sci. 1999;65:1–15. doi: 10.1016/s0024-3205(98)00614-6. [DOI] [PubMed] [Google Scholar]
- Haag S, Matthiesen S, Juergens UR, Racké K. Muscarinic receptors mediate stimulation of collagen synthesis in human lung fibroblasts. Eur Resp J. 2008;32:555–562. doi: 10.1183/09031936.00129307. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Ihara M, Noguchi K, Mase T, Mino N, Saeki T, et al. Biochemical and pharmacological profile of a potent and selective endothelin B-receptor antagonist, BQ-788. Proc Natl Acad Sci U S A. 1994;91:4892–4896. doi: 10.1073/pnas.91.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata S, Ito S, Iwaki M, Kondo M, Sashio T, Takeda N, et al. Regulation of endothelin-1-induced interleukin-6 production by Ca2+ influx in human airway smooth muscle cells. Eur J Pharmacol. 2009;605:15–22. doi: 10.1016/j.ejphar.2008.12.045. [DOI] [PubMed] [Google Scholar]
- Juergens UR, Racké K, Uen S, Haag S, Lamyel F, Stöber M, et al. Inflammatory responses after endothelin B (ETB) receptor activation in human monocytes: new evidence for beneficial anti-inflammatory potency of ETB receptor antagonism. Pulm Pharmcol Ther. 2008;21:533–539. doi: 10.1016/j.pupt.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Kebig A, Kostenis E, Mohr K, Mohr-Andrä M. An optical dynamic mass redistribution assay reveals biased signaling of dualsteric GPCR activators. J Recept Signal Transduct Res. 2009;29:140–145. doi: 10.1080/10799890903047437. [DOI] [PubMed] [Google Scholar]
- Khimji AK, Rockey DC. Endothelin – biology and disease. Cell Signal. 2010;22:1615–1625. doi: 10.1016/j.cellsig.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Knobloch J, Peters H, Jungck D, Müller K, Strauch J, Koch A. TNFalpha-induced GM-CSF release from human airway smooth muscle cells depends on activation of an ET-1 autoregulatory positive feedback mechanism. Thorax. 2009;64:1044–1052. doi: 10.1136/thx.2008.111047. [DOI] [PubMed] [Google Scholar]
- Lamyel F, Warnken-Uhlich M, Seemann WK, Mohr K, Kostenis E, Ahmedat AS, et al. The β2-subtype of adrenoceptors mediates inhibition of pro-fibrotic events in human lung fibroblasts. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:133–145. doi: 10.1007/s00210-011-0655-5. [DOI] [PubMed] [Google Scholar]
- Landgraf RG, Jancar S. Endothelin A receptor antagonist modulates lymphocyte and eosinophil infiltration, hyperreactivity and mucus in murine asthma. Int Immunopharmacol. 2008;8:1748–1753. doi: 10.1016/j.intimp.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Matthiesen S, Bahulayan A, Kempens S, Haag S, Fuhrmann M, Stichnote C, et al. Muscarinic receptor mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell Mol Biol. 2006;35:621–627. doi: 10.1165/rcmb.2005-0343RC. [DOI] [PubMed] [Google Scholar]
- Matthiesen S, Bahulayan A, Holz O, Racké K. MAPK pathway mediates muscarinic receptor-induced human lung fibroblast proliferation. Life Sci. 2007;80:2259–2262. doi: 10.1016/j.lfs.2007.02.027. [DOI] [PubMed] [Google Scholar]
- Panettieri RA, Jr, Goldie RG, Rigby PJ, Eszterhas AJ, Hay DW. Endothelin-1-induced potentiation of human airway smooth muscle proliferation: an ETA receptor-mediated phenomenon. Br J Pharmacol. 1996;118:191–197. doi: 10.1111/j.1476-5381.1996.tb15385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pégorier S, Arouche N, Dombret MC, Aubier M, Pretolani M. Augmented epithelial endothelin-1 expression in refractory asthma. J Allergy Clin Immunol. 2007;120:1301–1307. doi: 10.1016/j.jaci.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Peterkofsky B, Diegelmann R. Use of a mixture of proteinase-free collagenases for the specific assay of radioactive collagen in the presence of other proteins. Biochemistry. 1971;10:988–994. doi: 10.1021/bi00782a009. [DOI] [PubMed] [Google Scholar]
- Polikepahad S, Moore RM, Venugopal CS. Endothelins and airways – a short review. Res Commun Mol Pathol Pharmacol. 2006;119:3–51. [PubMed] [Google Scholar]
- Préfontaine A, Calderone A, Dupuis J. Role of endothelin receptors on basal and endothelin-1-stimulated lung myofibroblast proliferation. Can J Physiol Pharmacol. 2008;86:337–342. doi: 10.1139/Y08-024. [DOI] [PubMed] [Google Scholar]
- Ross B, D'Orléans-Juste P, Giaid A. Potential role of endothelin-1 in pulmonary fibrosis: from the bench to the clinic. Am J Respir Cell Mol Biol. 2010;42:16–20. doi: 10.1165/rcmb.2009-0175TR. [DOI] [PubMed] [Google Scholar]
- Schröder R, Janssen N, Schmidt J, Kebig A, Merten N, Hennen S, et al. Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotechnol. 2010;28:943–499. doi: 10.1038/nbt.1671. [DOI] [PubMed] [Google Scholar]
- Schröder R, Schmidt J, Blättermann S, Peters L, Janssen N, Grundmann M, et al. Applying label-free dynamic mass redistribution technology to frame signaling of G protein-coupled receptors noninvasively in living cells. Nat Protoc. 2011;6:1748–1760. doi: 10.1038/nprot.2011.386. [DOI] [PubMed] [Google Scholar]
- Shao D, Park JE, Wort SJ. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacol Res. 2011;63:504–511. doi: 10.1016/j.phrs.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Swigris JJ, Brown KK. The role of endothelin-1 in the pathogenesis of idiopathic pulmonary fibrosis. BioDrugs. 2010;24:49–54. doi: 10.2165/11319550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddman E, Adner M, Edvinsson L. Protein kinase C inhibitors decrease endothelin ETB receptor mRNA expression and contraction during organ culture of rat mesenteric artery. Eur J Pharmacol. 2002;452:215–222. doi: 10.1016/s0014-2999(02)02303-8. [DOI] [PubMed] [Google Scholar]
- Zietkowski Z, Skiepko R, Tomasiak MM, Bodzenta-Lukaszyk A. Endothelin-1 in exhaled breath condensate of stable and unstable asthma patients. Respir Med. 2008;102:470–474. doi: 10.1016/j.rmed.2007.10.013. [DOI] [PubMed] [Google Scholar]