

Abstract
Aberrant JAK-STAT signaling is a hallmark of myeloproliferative neoplasms (MPNs). These hyperproliferative disorders are classically associated with activating mutations in tyrosine kinases such as JAK2 and the thrombopoietin (TPO) receptor MPL. Activation of JAK-STAT signaling and responses to JAK2 inhibitors have been observed in MPN patients lacking JAK2 or MPL mutations, suggesting that other regulatory elements in the JAK-STAT pathway are altered. However, the molecular basis for this observation has been unclear. Recently, the role of inhibitory regulators of JAK-STAT signaling in MPN pathogenesis has been increasingly recognized. LNK is an adaptor protein that forms a negative feedback loop by binding to MPL and JAK2 and inhibiting downstream STAT activation. Murine models indicate that loss of LNK function can promote the development of a MPN phenotype. Several recent studies have identified novel LNK mutations in MPNs, thus validating this notion in humans. These findings represent a novel genetic paradigm of loss of negative feedback regulation of JAK-STAT activation in MPNs and have implications for the future development of targeted therapies in MPNs.
Keywords: essential thrombocythemia, JAK2 V617F, JAK-STAT, LNK, myeloproliferative neoplasms, polycythemia vera, primary myelofibrosis
Introduction
Myeloproliferative neoplasms (MPNs) are clonal hematologic malignancies derived from hematopoietic stem/progenitor cells and are characterized by unrestrained proliferation leading to overproduction of one or more myeloid lineages. The three classic chronic myeloproliferative neoplasms (MPNs) polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) have an estimated aggregate incidence of 5/100,000 persons, or approximately 15,000 new cases in the United States annually [Mesa et al. 1999; Ania et al. 1994]. Although phlebotomy and/or cytoreductive therapies can often effectively manage elevated blood counts, patients with PV and ET may experience significant morbidity associated with thrombosis and bleeding. In addition, these patients carry a risk of transformation to myelofibrosis, for which current treatment options are limited. All three of these disorders exhibit a propensity for transformation to acute myeloid leukemia (AML), for which the prognosis is poor.
Dysregulated JAK-STAT signaling via activated tyrosine kinases in MPNs
JAK2 is a cytoplasmic tyrosine kinase that associates with a variety of cytokine receptors (e.g. EPOR, MPL, IL-3R, G-CSFR, GM-CSFR) and becomes activated upon ligand binding (Figure 1a) [Ihle and Gilliland, 2007]. JAK2 knockout mice are embryonic lethal due to an absence of effective erythropoiesis [Neubauer et al. 1998; Parganas et al. 1998]. Fetal liver myeloid progenitors from JAK2−/− mice fail to respond to erythropoietin (EPO), thrombopoietin (TPO), interleukin-3 (IL-3), or granulocyte macrophage-colony stimulating factor (GM-CSF), indicating that JAK2 is essential for transmitting signals via these cytokines [Parganas et al. 1998]. Upon receptor-ligand binding, JAK2 autophosphorylation occurs, subsequently leading to activation of downstream signaling via recruitment of Src homology 2 (SH2)-domain-containing proteins such as STAT3 and STAT5. After phosphorylation by JAK2, the STAT proteins dimerize and translocate to the nucleus, where they activate transcription of target genes involved in regulating a variety of cellular processes, including proliferation, differentiation, and apoptosis [Valentino and Pierre, 2006; Kisseleva et al. 2002].
Figure 1.

Schematic of JAK-STAT signaling and activating tyrosine kinase gene mutations in myeloproliferative neoplasms (MPNs).
(a) Cytokine-receptor binding (e.g. TPO/MPL) leads to JAK2 activation, followed by recruitment and phosphorylation of STAT proteins, which then translocate to the nucleus and activate transcription of genes involved in proliferation and survival.
(b) Activating mutations in tyrosine kinases (e.g. MPL, JAK2) lead to JAK-STAT activation in the absence of cytokine stimulation.
In 2005, multiple groups identified the JAK2 V617F mutation in patients with PV, ET, and PMF (Figure 1b) [Baxter et al. 2005; James et al. 2005; Kralovics et al. 2005; Levine et al. 2005]. Multiple subsequent studies have confirmed these findings, indicating that >95% of PV patients are JAK2 V617F-positive, while approximately 50-60% of ET and PMF patients carry the mutation [Tefferi, 2008; Levine and Wernig, 2006]. The V617F mutation localizes to the autoinhibitory pseudokinase domain of JAK2 and results in constitutive activation of JAK2 tyrosine kinase activity [Baxter et al. 2005; James et al. 2005; Kralovics et al. 2005; Levine et al. 2005]. Overexpression of JAK2 V617F in cell lines leads to phosphorylation of JAK2 and STAT5 in the absence of cytokine stimulation, and cells expressing the mutant allele exhibit cytokine independence and/or hypersensitivity [Baxter et al. 2005; James et al. 2005; Kralovics et al. 2005]. Based on modeling studies, it has been postulated that the V617F mutation causes a structural change in the pseudokinase domain, relieving inhibition of JAK2 kinase activity, thereby leading to activated downstream signaling.
The identification of the JAK2 V617F mutation in PV, ET, and PMF raises the question of how a single mutation can result in such phenotypic diversity. In murine retroviral transplant models, high levels of JAK2 V617F expression lead to a PV-like phenotype, with marked erythrocytosis as the predominant feature [Bumm et al. 2006; Lacout et al. 2006]. In contrast, transgenic models with more physiologic levels of JAK2 V617F expression result in phenotypes resembling ET and PMF [Shide et al. 2008; Tiedt et al. 2008; Xing et al. 2008]. These findings are somewhat corroborated by human studies, in which the highest JAK2 V617F allele burden is typically seen in PV patients, and the lowest in ET patients [Antonioli et al. 2008; Tiedt et al. 2008; Vannucchi et al. 2008].
In addition to the V617F mutation, a variety of missense, insertion, and deletion mutations in exon 12 of JAK2 have been identified in a subset of patients with idiopathic erythrocytosis (Figure 1b) [Scott et al. 2007]. The mutations are located just 5′ of the pseudokinase domain, and like V617F, are speculated to cause a structural change resulting in JAK2 activation. This is supported by cell line experiments, in which the mutant alleles confer IL-3 independent growth and constitutive phosphorylation of JAK2. In addition, in murine retroviral transplant models, one of the mutant alleles (K539L) causes erythrocytosis. In contrast to the JAK2 V617F mutation, exon 12 mutations are restricted to PV [Pietra et al. 2008; Wang et al 2008; Pardanani et al. 2007; Scott et al. 2007]. These findings suggest that the specific type of mutation in JAK2 may confer disparate clinical phenotypes, perhaps mediating differential effects on downstream signaling.
Activating mutations in the transmembrane domain of the TPO receptor MPL (W515L/K) have been reported in small numbers of ET (1%) and PMF (5%) patients (Figure 1b) [Pardanani et al. 2006; Pikman et al. 2006]. Overexpression of the mutant MPL W515L allele in cell lines leads to cytokine-independent growth, TPO hypersensitivity, and activated JAK-STAT signaling. In retroviral transplant models, the W515L allele results in marked thrombocytosis, splenomegaly, and reticulin fibrosis, but not erythrocytosis. MPL W515L/K mutations have not been found in PV, or other myeloid disorders such as myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML), or AML [Pardanani et al. 2006].
Taken together, these findings demonstrate that aberrant JAK-STAT signaling due to activating mutations in tyrosine kinases is a recurrent biologic feature of MPNs. In addition, activation of JAK-STAT signaling has been demonstrated in some MPN patients lacking JAK2 or MPL mutations, suggesting that alterations in other regulatory members of the JAK-STAT axis may be responsible for driving the pathogenesis of these MPNs. However, the molecular basis of these MPN cases has largely been unexplained. While several studies have identified additional genes mutated in MPNs (e.g. TET2, IDH1/2, ASXL1, 1KZF1, CBL) [Tefferi, 2010] considerable recent attention has focused on the role of the adaptor protein LNK in MPN pathogenesis.
LNK: A critical regulator of JAK-STAT signaling and hematopoiesis
LNK, APS, and SH2-B are members of a family of adaptor proteins that share several structural motifs, including a proline-rich N-terminal dimerization domain (Pro/DD), a pleckstrin homology (PH) domain, an SH2 domain, and a conserved tyrosine residue near the C-terminus (Figure 2a) [Rudd, 2001]. Early studies implicated LNK in the regulation of T-cell and B-cell signaling and development [Rudd, 2001; Li et al. 2000; Takaki et al. 2000, 1997; Huang et al. 1995].
Figure 2.
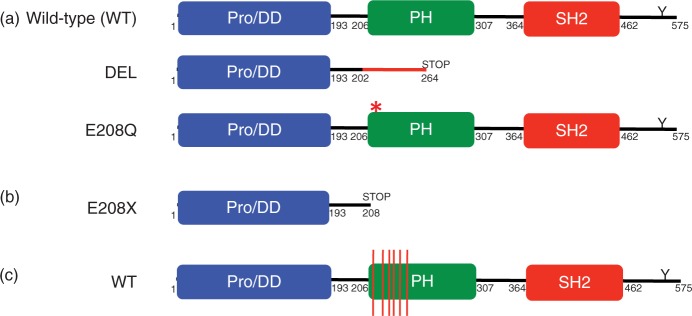
Schematic of LNK structure and mutations.
(a) Wild-type LNK is 575 amino acids and contains an N-terminal proline-rich dimerization domain (Pro/DD), a pleckstrin homology (PH) domain, a Src homology 2 (SH2) domain, and a C-terminal conserved tyrosine residue (Y). DEL is a five-base-pair deletion and missense mutation that leads to a frameshift and premature stop codon. E208Q is a point mutation at the beginning of the PH domain. (b) E208X is a point mutation leading to a stop codon. (c) Red lines depict the location of LNK PH domain missense mutations.
Subsequent studies revealed a more widespread role for LNK in myeloid development and hematopoiesis. For example, LNK−/− mice have an expanded hematopoietic stem cell (HSC) compartment.[Bersenev et al. 2008; Ema et al. 2005; Takaki et al. 2002; Velazquez et al. 2002] In addition, LNK−/− mice exhibit leukocytosis and thrombocytosis, as well as splenomegaly with marked fibrosis and extramedullary hematopoiesis [Tong and Lodish, 2004; Velazquez et al. 2002]. Megakaryocytic hyperplasia has also been observed in both the spleen and bone marrow from LNK−/− mice [Tong and Lodish, 2004; Velazquez et al. 2002]. These features are TPO-dependent, as LNK−/− mice crossed with TPO−/−mice do not exhibit increased HSC numbers, leukocytosis, or thrombocytosis [Buza-Vidas et al. 2006]. In addition, TPO-mediated activation of STAT3 and STAT5 is enhanced in LNF−/−mice [Tong and Lodish, 2004].
LNK also appears to play a role in erythroid development. Erythroid hyperplasia has been demonstrated in the spleen of LNK−/− mice [Velazquez et al. 2002]. Although hematocrit levels are not elevated, subtle differences in erythropoiesis may be masked by the large erythropoietic reserve activity observed in adult mice. In support of this notion, LNK−/− mice exhibit slightly higher numbers of erythroid progenitors in the bone marrow, and upon challenge with erythropoietic stress, superior recovery with an enhanced erythropoietic rate has been observed [Tong et al. 2005]. In addition, splenic erythroid progenitors derived from LNK−/− mice exhibit hypersensitivity to EPO, both in terms of colony formation as well as activation of JAK-STAT signaling [Tong et al. 2005].
A direct role for LNK in regulating signaling via MPL and JAK2 has also been demonstrated. Bersenev and colleagues showed that while LNK binds to MPL constitutively, it exhibits weak binding to JAK2 in the basal state [Bersenev et al. 2008]. Upon stimulation with TPO, however, JAK2 becomes phosphorylated and binds to LNK with much higher affinity, thus forming a critical negative feedback loop (Figure 3a). A kinase-inactive mutant of JAK2 results in decreased binding to LNK, suggesting that JAK2 phosphorylates LNK, which may lead to its enhanced binding. Gery and colleagues demonstrated that LNK binds to MPL and colocalizes with MPL at the plasma membrane [Gery et al. 2007]. The SH2 domain of LNK is essential for this interaction, as well as for the inhibition of downstream signaling. In addition, the PH domain is required for membrane localization. Interestingly, LNK can bind and inhibit JAK2 V617F as well as MPL W515L [Gery et al. 2009, 2007; Bersenev et al. 2008], suggesting that even in the presence of these activating mutations, additional mechanisms may need to be recruited in order to evade inhibition by LNK. In support of this notion, a recent study demonstrated that in mice expressing activated forms of JAK2 (either JAK2 V617F or the TEL-JAK2 fusion), loss of LNK function accelerates and exacerbates the development of an MPN-like disease [Bersenev et al. 2010].
Figure 3.

The adaptor protein LNK is a critical negative regulator of JAK-STAT signaling.
(a) The adaptor protein LNK negatively regulates JAK-STAT signaling by binding to MPL and JAK2, thereby inhibiting downstream STAT activation. (b) LNK mutations affecting the PH domain (depicted as a yellow line) may lead to mislocalization in the cytoplasm, thereby disrupting the ability of LNK to inhibit JAK-STAT signaling. As the dimerization domain is retained, mutant LNK forms may also sequester wild-type LNK, potentially resulting in a dominant negative effect. This research was originally published in Blood. Oh et al. (2010) Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 116: 988-992 © by the American Society of Hematology.
LNK may also play a role in mast cell development by inhibiting signaling mediated by stem cell factor (SCF) and its receptor KIT [Simon et al. 2008; Velazquez et al. 2002]. Hematopoietic progenitors from LNK−/− mice are hypersensitive to a variety of cytokines including SCF [Velazquez et al. 2002]. Furthermore, LNK has been shown to bind directly to KIT via its SH2 domain [Simon et al. 2008]. LNK has also been shown to inhibit SCF-induced signaling and proliferation in bone marrow mast cells isolated from LNK−/− mice [Simon et al. 2008]. These findings raise the question of whether LNK activity may be dysregulated in mast cell disorders such as systemic mastocytosis, although this has not been investigated directly.
Identification of novel LNK mutations in MPNs
The aforementioned studies clearly demonstrate that LNK plays a central role in the regulation of hematopoiesis, and that loss of its function in mice confers an MPN phenotype. We therefore hypothesized that LNK mutations could provide an alternative basis for JAK-STAT activation in MPN patients lacking JAK2 or MPL mutations. In a screen of 33 JAK2 V617F-negative ET and PMF patients, we identified two individuals with mutations in exon 2 of LNK (Figure 2a) [Oh et al. 2010]. One patient with PMF exhibited a five base-pair deletion and missense mutation (DEL) leading to a premature stop codon and loss of the pleckstrin homology (PH) and SH2 domains. A second patient with ET had a missense mutation (E208Q) in the PH domain. BaF3-MPL cells transduced with the LNK DEL mutant exhibited augmented and sustained TPO-dependent growth and signaling, consistent with a loss of LNK inhibitory function. The LNK E208Q mutant retained partial LNK inhibitory activity. Primary samples from these patients exhibited aberrant STAT3 5 activation in response to TPO and/ or G-CSF stimulation, and a similar phenotype was observed in MPN patients bearing the JAK2 V617F and MPL W515L mutations. These findings indicate that JAK-STAT activation due to loss of LNK negative feedback regulation can phenocopy disease related to activating mutations in tyrosine kinases such as JAK2 or MPL. In follow-up studies with an extended MPN cohort, we have identified additional patients with LNK exon 2 mutations [Oh and Gotlib, unpublished]. In addition, several recent reports have demonstrated that LNK mutations are present in a spectrum of myeloid disorders. In a screen of eight patients with JAK2 mutationnegative erythrocytosis, a LNK E208X stop codon mutation was identified in one patient (Figure 2b) [Lasho et al. 2010]. This finding is particularly notable given that it represents the first non-JAK2 gene mutation identified in this MPN. In a separate report, Pardanani and colleagues sequenced 61 blast phase MPN samples, identifying five patients with nonsynonymous LNK mutations at blast phase (Table 1) [Pardanani et al. 2010]. Three patients acquired LNK mutations only at blast phase, suggesting that LNK may play a role in transformation. Conversely, in two cases, LNK mutations were identified at chronic phase, but were no longer present at blast phase. This is reminiscent of some cases of JAK2 V617F-positive MPNs, in which the JAK2 V617F mutation can no longer be detected at transformation [Theocharides et al. 2007; Campbell et al. 2006]. Three of the patients with LNK mutations exhibited a concomitant JAK2 V617F mutation, and one patient was found to have an IDH2 R140Q mutation. However, no LNK mutations were indentified in a screen of 78 additional chronic phase MPN patients carrying one or more TET2, IDH, JAK2, or MPL mutations, indicating that LNK mutations infrequently coexist with other MPN-associated mutations. As LNK can still bind and inhibit JAK2 V617F and MPL W515L, these findings suggest that other mechanisms of abrogating LNK function may be invoked in these MPNs.
Table 1.
Summary of patients with myeloproliferative neoplasms with nonsynonymous LNK mutations.
| LNK mutation | Domains affected | Diagnosis | Study |
|---|---|---|---|
| DEL | PH + SH2 | PMF | [Oh et al. 2010] |
| E208Q | PH | ET | [Oh et al. 2010] |
| E208X | PH + SH2 | JAK2-negative erythrocytosis | [Lasho et al. 2010] |
| A215V | PH | JAK2-negative erythrocytosis | [Lasho et al. 2010] |
| A215V | PH | Post-PMF AML1 | [Pardanani et al. 2010] |
| A215V | PH | Post-PMF AML1 | [Pardanani et al. 2010] |
| G220R | PH | PMF2 | [Pardanani et al. 2010] |
| G220V | PH | PMF2 | [Pardanani et al. 2010] |
| A223V D234N | PH | Post-PMF AML3 | [Pardanani et al. 2010] |
| G229S | PH | Post-PMF AML4 | [Pardanani et al. 2010] |
| 685–691_delGGCCCCG; 955_delA | PH + SH2; SH2 | PMF/AML5 PMF/AML5 | [Pardanani et al. 2010] |
Mutation not present at the chronic phase.
Mutation not detected at the blast phase.
Both mutations detected in the same patient and not present in the chronic phase.
Chronic phase samples not available for analysis.
Both deletion mutations detected in the same patient and present in both chronic and blast phases.
PH, pleckstrin homology; SH2, Src homology 2; PMF, primary myelofibrosis; ET, essential thrombocythemia; AML, acute myeloid leukemia.
The majority of the LNK mutations identified to date localize to exon 2, implicating this as a possible mutational hotspot. In addition, virtually all of the missense mutations affect highly conserved residues in the PH domain, indicating that plasma membrane localization of LNK may be disrupted (Figure 2c). Previous studies with synthetic LNK mutants have demonstrated that disruption of the PH domain results in loss of LNK inhibitory function [Bersenev et al. 2008; Gery et al. 2007; Tong and Lodish, 2004]. As most of the LNK mutations identified in MPNs appear to be heterozygous, an important biological question is whether loss of inhibition of JAK-STAT activation is related to haploinsufficiency of LNK or due to dominant negative effects of the mutant protein. In support of the former notion, LNK+/− mice exhibit an intermediate MPN phenotype, consistent with a haploinsufficiency model [Velazquez et al. 2002]. In addition, LNK mutations affecting the PH domain (but sparing the DD domain) could lead to sequestering of the wild-type protein, thus resulting in a dominant negative effect (Figure 3b).
Similar to the presence of JAK2 V617F in PV, ET, and PMF, the identification of LNK mutations in multiple MPN subtypes raises the question as to how these mutations may confer disease specificity. One possibility is that specific LNK mutations lead to differential effects on LNK inhibitory function and JAK-STAT signaling. In support of this notion, the LNK E208Q mutation results in a partial loss of function, and was identified in a patient with ET, whereas the LNK DEL mutation, which results in a complete loss of LNK inhibitory properties, was found in a patient with PMF [Oh et al. 2010]. As the DEL mutation leads to loss of both the PH and SH2 domains, this is likely to contribute to its more drastic effect on LNK function. This is supported by prior studies demonstrating that a synthetic PH domain mutation retains partial LNK activity, while mutations affecting the SH2 domain result in loss of binding to MPL and JAK2 and a more severe phenotype [Bersenev et al. 2008; Gery et al. 2007; Tong et al. 2005; Tong and Lodish, 2004]. However, the relationship between specific LNK mutations and MPN phenotype is likely to be complex, given that PH domain mutations were also identified in PMF and post-PMF AML [Pardanani et al. 2010], and that the E208X stop codon mutation was found in a patient with erythrocytosis [Lasho et al. 2010]. Further mechanistic studies will address how these LNK mutations contribute to MPN pathogenesis in vivo.
The impact of LNK mutations on disease phenotype may also be related to the specific cell type that acquires LNK mutations. In our initial report describing the LNK DEL and E208Q mutations, cytokine-responsive CD34+ early progenitors were abnormally abundant in both patients, and the LNK DEL mutation was specifically detected in this activated early progenitor compartment [Oh et al. 2010]. This suggests that LNK mutations can represent an early genetic event in MPN pathogenesis, akin to JAK2 V617F in PV [Jamieson et al. 2006]. In addition, as some MPN patients specifically acquire LNK mutations at transformation [Pardanani et al. 2010], the timing of these genetic events are likely to play a role in their pleiotropic effects on MPN pathogenesis.
Conclusions
Although activated signaling in MPNs has historically been linked to mutations in tyrosine kinases such as JAK2 and MPL, the role of negative feedback pathways in regulating JAK-STAT signaling has become evident in recent years. This has been highlighted by the identification of novel LNK mutations in a variety of MPN subtypes, as well as both chronic and blast phase MPNs. These findings indicate that specific LNK mutations may result in differential effects on LNK function and JAK-STAT signaling, and that their contribution to MPN pathogenesis may be further modulated by the context in which these mutations are acquired. As the majority of LNK mutations affect the PH domain, future studies should address the effects of these mutations on plasma membrane localization, and determine whether the mutant protein can exert a dominant negative effect. As some MPN patients lacking JAK2 or MPL mutations have exhibited responses to JAK2 inhibitors, the identification of LNK mutations may provide an explanation for at least a subset of these patients. These findings will aid in the future development of targeted therapies in MPNs.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
None declared.
References
- Ania B.J., Suman V.J., Sobell J.L., Codd M.B., Silverstein M.N., Melton L.J., III (1994) Trends in the incidence of polycythemia vera among Olmsted County, Minnesota residents, 1935-1989. Am J Hematol 47: 89–93 [DOI] [PubMed] [Google Scholar]
- Antonioli E., Guglielmelli P., Poli G., Bogani C., Pancrazzi A., Longo G., et al. (2008) Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica 93: 41–48 [DOI] [PubMed] [Google Scholar]
- Baxter E.J., Scott L.M., Campbell P.J., East C., Fourouclas N., Swanton S., et al. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365: 1054–1061 [DOI] [PubMed] [Google Scholar]
- Bersenev A., Wu C., Balcerek J., Jing J., Kundu M., Blobel G.A., et al. (2010) LNK constrains myeloproliferative diseases in mice. J Clin Invest 120: 2058–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersenev A., Wu C., Balcerek J., Tong W. (2008) LNK controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest 118(8): 2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumm T., Elsea C., Corbin A., Loriaux M., Sherbenou D., Wood L., et al. (2006) Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 66: 11156–11165 [DOI] [PubMed] [Google Scholar]
- Buza-Vidas N., Antonchuk J., Qian H., Månsson R., Luc S., Zandi S., et al. (2006) Cytokines regulate postnatal hematopoietic stem cell expansion: Opposing roles of thrombopoietin and LNK. Genes Dev 20: 2018–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P., Baxter E., Beer P., Scott L., Bench A., Huntly B., et al. (2006) Mutation of JAK2 in the myeloproliferative disorders: Timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood 108: 3548–3555 [DOI] [PubMed] [Google Scholar]
- Ema H., Sudo K., Seita J., Matsubara A., Morita Y., Osawa M., et al. (2005) Quantification of self-renewal capacity in single hematopoietic stem cells from normal and LNK-deficient mice. Dev Cell 8: 907–914 [DOI] [PubMed] [Google Scholar]
- Gery S., Cao Q., Gueller S., Xing H., Tefferi A., Koeffler H.P. (2009) LNK inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J Leukoc Biol 85: 957–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gery S., Gueller S., Chumakova K., Kawamata N., Liu L., Koeffler H.P. (2007) Adaptor protein LNK negatively regulates the mutant MPL, MPLW515L associated with myeloproliferative disorders. Blood 110: 3360–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Li Y., Tanaka K., Moore K.G., Hayashi J.I. (1995) Cloning and characterization of LNK, a signal transduction protein that links T-cell receptor activation signal to phospholipase C gamma 1, Grb2, and phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 92: 11618–11622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle J., Gilliland D. (2007) Jak2: Normal function and role in hematopoietic disorders. Curr Opin Genet Dev 17: 8–14 [DOI] [PubMed] [Google Scholar]
- James C., Ugo V., Le Couedic J.P., Staerk J., Delhommeau F., Lacout C., et al. (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434: 1144–1148 [DOI] [PubMed] [Google Scholar]
- Jamieson C.H., Gotlib J., Durocher J.A., Chao M.P., Mariappan M.R., Lay M., et al. (2006) The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci USA 103: 6224–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T., Bhattacharya S., Braunstein J., Schindler C.W. (2002) Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285: 1–24 [DOI] [PubMed] [Google Scholar]
- Kralovics R., Passamonti F., Buser A.S., Teo S.S., Tiedt R., Passweg J.R., et al. (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352: 1779–1790 [DOI] [PubMed] [Google Scholar]
- Lacout C., Pisani D.F., Tulliez M., Gachelin F.M., Vainchenker W., Villeval J.L. (2006) JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 108: 1652–1660 [DOI] [PubMed] [Google Scholar]
- Lasho T.L., Pardanani A., Tefferi A. (2010) LNK mutations in JAK2 mutation-negative erythrocytosis. N Engl J Med 363: 1189–1190 [DOI] [PubMed] [Google Scholar]
- Levine R., Wadleigh M., Cools J., Ebert B., Wernig G., Huntly B.J., et al. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7: 387–397 [DOI] [PubMed] [Google Scholar]
- Levine R., Wernig G. (2006) Role of JAK-STAT signaling in the pathogenesis of myeloproliferative disorders. Hematology 510: 233–239 [DOI] [PubMed] [Google Scholar]
- Li Y., He X., Schembri-King J., Jakes S., Hayashi J. (2000) Cloning and characterization of human LNK, an adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J Immunol 164: 5199–5206 [DOI] [PubMed] [Google Scholar]
- Mesa R.A., Silverstein M.N., Jacobsen S.J., Wollan P.C., Tefferi A. (1999) Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: An Olmsted County Study, 1976-1995. Am J Hematol 61: 10–15 [DOI] [PubMed] [Google Scholar]
- Neubauer H., Cumano A., Muller M., Wu H., Huffstadt U., Pfeffer K. (1998) Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93: 397–409 [DOI] [PubMed] [Google Scholar]
- Oh S.T., Simonds E.F., Jones C., Hale M.B., Goltsev Y., Gibbs K.D., Jr, et al. (2010) Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 116: 988–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardanani A., Lasho T., Finke C., Hanson C., Tefferi A. (2007) Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 21: 1960–1963 [DOI] [PubMed] [Google Scholar]
- Pardanani A., Lasho T., Finke C., Oh S.T., Gotlib J., Tefferi A. (2010) LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia [ePub August 2010]. [DOI] [PubMed]
- Pardanani A., Levine R., Lasho T., Pikman Y., Mesa R., Wadleigh M., et al. (2006) MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 108(10): 3472–3476 [DOI] [PubMed] [Google Scholar]
- Parganas E., Wang D., Stravopodis D., Topham D.J., Marine J.C., Teglund S., et al. (1998) Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93: 385–395 [DOI] [PubMed] [Google Scholar]
- Pietra D., Li S., Brisci A., Passamonti F., Rumi E., Theocharides A., et al. (2008) Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 111: 1686–1689 [DOI] [PubMed] [Google Scholar]
- Pikman Y., Lee B.H., Mercher T., McDowell E., Ebert B., Gozo M., et al. (2006) MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 3: e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd C.E. (2001) LNK adaptor: Novel negative regulator of B cell lymphopoiesis. Sci STKE 85: PE1. [DOI] [PubMed] [Google Scholar]
- Scott L.M., Tong W., Levine R., Scott M.A., Beer P.A., Stratton M.R., et al. (2007) JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 356: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shide K., Shimoda H., Kumano T., Karube K., Kameda T., Takenaka K., et al. (2008) Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia 22: 87–95 [DOI] [PubMed] [Google Scholar]
- Simon C., Dondi E., Chaix A., De Sepulveda P., Kubiseski T., Varin-Blank N., et al. (2008) LNK adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood 112: 4039–4047 [DOI] [PubMed] [Google Scholar]
- Takaki S., Morita H., Tezuka Y., Takatsu K. (2002) Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, LNK. J Exp Med 195: 151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki S., Sauer K., Iritani B.M., Chien S., Ebihara Y., Tsuji K., et al. (2000) Control of B cell production by the adaptor protein LNK. Definition of a conserved family of signal-modulating proteins. Immunity 13: 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki S., Watts J.D., Forbush K.A., Nguyen N.T., Hayashi J., Alberola-Ila J., et al. (1997) Characterization of LNK. An adaptor protein expressed in lymphocytes. J Biol Chem 272: 14562–14570 [DOI] [PubMed] [Google Scholar]
- Tefferi A. (2008) JAK and MPL mutations in myeloid malignancies. Leukemia Lymphoma 49: 388–397 [DOI] [PubMed] [Google Scholar]
- Tefferi A. (2010) Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 24: 1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharides A., Boissinot M., Girodon F., Garand R., Teo S., Lippert E., et al. (2007) Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 110: 375–379 [DOI] [PubMed] [Google Scholar]
- Tiedt R., Hao-Shen H., Sobas M.A., Looser R., Dirnhofer S., Schwaller J., et al. (2008) Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 111: 3931–3940 [DOI] [PubMed] [Google Scholar]
- Tong W., Lodish H.F. (2004) LNK inhibits TPO-MPL signaling and TPO-mediated megakaryocytopoiesis. J Exp Med 200: 569–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W., Zhang J., Lodish H.F. (2005) LNK inhibits erythropoiesis and EPO-dependent JAK2 activation and downstream signaling pathways. Blood 105: 4604–4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino L., Pierre J. (2006) JAK/STAT signal transduction: Regulators and implication in hematological malignancies. Biochem Pharmacol 71: 713–721 [DOI] [PubMed] [Google Scholar]
- Vannucchi A., Antonioli E., Guglielmelli P., Pardanani A., Tefferi A. (2008) Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: A critical reappraisal. Leukemia 22: 1299–1307 [DOI] [PubMed] [Google Scholar]
- Velazquez L., Cheng A.M., Fleming H.E., Furlonger C., Vesely S., Bernstein A., et al. (2002) Cytokine signaling and hematopoietic homeostasis are disrupted in LNK-deficient mice. J Exp Med 195: 1599–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Vandris K., Jones A., Cross N., Christos P., Adriano F., et al. (2008) JAK2 Mutations are present in all cases of polycythemia vera. Leukemia 22: 1289. [DOI] [PubMed] [Google Scholar]
- Xing S., Wanting T., Zhao W., Ma J., Wang S., Xu X., et al. (2008) Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood 111: 5109–5117 [DOI] [PMC free article] [PubMed] [Google Scholar]