

Background: Apart from its mitochondrial localization, mechanistic details of STAT3 import and assembly in mitochondria remain elusive.
Results: Using an in vitro import assay, we show that STAT3 associates with the mitochondrial inner membrane in a GRIM-19-dependent manner.
Conclusion: GRIM-19 chaperones the recruitment of STAT3 into mitochondrial inner membrane complexes.
Significance: This study identifies a novel function of GRIM-19 and a mechanism for STAT3 import into mitochondria.
Keywords: Cell Death, Mitochondria, Mitochondrial Transport, Molecular Chaperone, Protein translocation, ROS
Abstract
The signal transducer and activator of transcription 3 (STAT3), a nuclear transcription factor, is also present in mitochondria and regulates cellular respiration in a transcriptional-independent manner. The mechanism of STAT3 import into mitochondria remains obscure. In this report we show that mitochondrial-localized STAT3 resides in the inner mitochondrial membrane. In vitro import studies show that the gene associated with retinoid interferon induced cell mortality 19 (GRIM-19), a complex I subunit that acts as a chaperone to recruit STAT3 into mitochondria. In addition, GRIM-19 enhances the integration of STAT3 into complex I. A S727A mutation in STAT3 reduces its import and assembly even in the presence of GRIM-19. Together, our studies unveil a novel chaperone function for GRIM-19 in the recruitment of STAT3 into mitochondria.
Introduction
Mitochondria are essential organelles involved in many cellular processes including energy transduction, apoptosis, and metabolism of lipids and amino acids. Even though they possess their own genome, most mitochondrial proteins are encoded by the nuclear genome and are imported into different sub-compartments of mitochondria by multisubunit protein import receptors (1–3). Understanding the mechanisms by which proteins are imported into the mitochondria will provide insights concerning the role of these proteins in mitochondrial respiration, biogenesis, and apoptosis (4).
The molecular mechanisms by which Stat transcription factors regulate nuclear gene expression have been actively pursued for decades. Recent reports suggested non-canonical functions for STAT3 and other nuclear transcription factors in mitochondria that involve regulation of energy management by the mitochondria. For instance, researchers have identified STAT3 (5), estrogen receptors (6, 7), myocyte enhancer factor 2D (8), glucocorticoid receptors (9, 10), p53 (11), NF-κB (12), and CREB (cAMP-responsive element binding protein) (13–15) in mitochondria regulating mitochondrial functions in a manner that is dependent or independent of transcription (16). These findings suggest a paradigm shift in the functions of these transcription factors to target and modulate mitochondrial driven cellular functions.
STAT3 is a key transcription factor that is phosphorylated on tyrosine 705 and serine 727 in response to cytokines and growth factors. Phosphorylated STAT3 translocates into nucleus and regulates expression of genes associated with various cellular processes. We and others have recently shown that STAT3 is also localized to mitochondria (5, 17–20). Mitochondrial STAT3 increases activity of complex I and II of the electron transport chain in a transcriptional-independent manner (5). In addition, Ras-mediated cellular transformation is shown to be dependent on mitochondrial STAT3 (17). Interestingly, phosphorylation of STAT3 on Ser-727, but not Tyr-705, seems to be integral for its mitochondrial activity (5, 17). Recent reports also suggest involvement of mitochondrial STAT3 in cardioprotection during ischemia and reperfusion possibly by preventing leakage of electrons from complex I (18, 21). The regulatory mechanisms involved in STAT3 import, integration into complex I, and its role in respiration remain obscure.
GRIM-19 was identified as a principal mediator of IFN-β/Retinoic acid-induced cell death (22). Subsequently, GRIM-19 was identified as a component of complex I in the electron transport chain. Small amounts of GRIM-19 have also been detected in nuclei (22–24) where it has been reported to be a negative transcriptional regulator of STAT3 (25–28). Phosphorylation of STAT3 on Ser-727 seems to be essential for its interaction with GRIM-19 (28). The requirement of Ser-727 phosphorylation for its mitochondrial functions and its interaction with GRIM-19 suggested that STAT3-GRIM-19 interaction may influence each other's localization or function.
In the present study we find that STAT3 anchors to the inner mitochondrial membrane. Using an in vitro import system, we demonstrate the involvement of GRIM-19 in the recruitment of STAT3 to mitochondria and its integration into complex I. Import of STAT3 requires phosphorylation at Ser-727 site as removal of the C terminus of STAT3 or mutation of Ser-727 reduces integration of STAT3 into the inner membrane of mitochondria. Together, our results disclosed a novel role of GRIM-19 as a chaperone in STAT3 localization into the mitochondria.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies used in this study were STAT3C-20, TOM20, and ND1 (Santa Cruz Biotechnology), Cyt c (Cell Signaling Technology), GRIM-19 (Mitosciences), aconitase 2 (Novus Biologicals), and Mia40 (in-house-generated and purified using CNBr-Sepharose). All chemicals were obtained from Sigma Aldrich and Amersco.
Plasmid Constructs
Full-length GRIM-19, TIM23, and RBM3 cDNA were amplified by polymerase chain reaction from total HeLa cell RNA using gene specific primers and cloned into a myc-tagged mammalian expression vector, pCDNA 3.1/myc(A). All STAT3 clones in pCDNA 3.1 (5) and pGEMT-SP6-Su9-DHFR4 have been described earlier (29). Full-length STAT3 was subcloned into Pet28 (a+) vector. The protein was expressed in bacteria and purified to homogeneity by using Ni-NTA column. His6-DHFR was amplified using Su9-DHFR as a template and cloned into pGEMT vector. pCDNA-ER-α was a kind gift from Dr. Bramanandam Manavathi (University of Hyderabad, Hyderabad, India).
Cell-free Synthesis of Proteins
Full-length STAT3 and mutants, GRIM-19, Su9-DHFR, RBM3, ER-α, TIM23, and DHFR proteins were synthesized using T7 or Sp6 in vitro coupled transcription and translation system (Promega) according to the manufacturer's instructions. Each translation mix contains 20 μCi of 35S-labeled methionine (1170 Ci/mmol, BARC). Translated products were analyzed using phosphorimaging and scintillation counting.
Isolation of Mitochondria
Mitochondria were isolated from rat heart using differential centrifugation (30, 31). Briefly, excised tissues were minced in 0.9% saline and then homogenized in cold homogenization buffer (H medium: 220 mm mannitol, 70 mm sucrose, 0.2 mm EDTA, 2 mm HEPES, pH 7.2, and added 0.36 mg/ml BSA before use). Homogenates were centrifuged at 2000 rpm for 10 min. Supernatants were centrifuged at 10,000 rpm for 10 min. The pellet was washed in H-medium twice and suspended in import buffer (0.25 m sucrose, 1.5 mm MgCl2, 2.5 mg/ml BSA, and 10 mm HEPES, pH 7.2). To obtain a highly purified mitochondrial fraction, the crude mitochondrial suspension was layered on top of a 2.5 m sucrose-Percoll gradient and centrifuged at 46,000 × g at 4 °C for 45 min, and mitochondria were isolated as described (5).
Separation of Inner Mitochondrial Membrane (IMM) and Matrix Fraction of Mitochondria
IMM and matrix fractions were generated from mitoplasts as described (32). Mitochondria were resuspended in 450 μl of hypotonic buffer (5 mm Tris-HCl and 1 mm EDTA, pH 7.4) and incubated on ice for 15 min to generate mitoplasts. The solution was centrifuged at 20,000 × g for 10 min at 4 °C to pellet mitoplasts. The resulting mitoplasts were resuspended in 450 μl of hypotonic buffer and sonicated for 2 min (30 s off and 30 s on at 150 watts, Branson Sonifer) on ice. The solution was then spun at 100,000 × g for 40 min. The resultant pellet contains the IMM-enriched fraction, whereas the supernatant contains matrix-enriched fraction. For high salt treatment, mitoplasts were incubated with 400 mm KCl on ice for 10 min followed by centrifugation (15,000 rpm for 15 min). For high pH treatment, mitoplasts were incubated with 200 mm Na2CO3, pH 11.5, for 10 min followed by centrifugation.
In Vitro Import Assay
35S-Labeled proteins of GRIM-19 (6,000 cpm), STAT3 (12,000 cpm), STAT31–470 (12,000 cpm), STAT3S727A (12,000 cpm), Su9-DHFR (10,000 cpm), His6-DHFR (10,000 cpm), RBM3 (10,000 cpm), ERα (10,000 cpm), and TIM23 (10,000 cpm) were used in import assays unless otherwise described in the legends. Labeled proteins were incubated with 200 μg of isolated mitochondria in import buffer (0.25 m sucrose, 1.5 mm MgCl2, 2.5 mg/ml BSA, and 10 mm HEPES, pH 7.2) supplemented with 2 mm ATP, 2 mm GTP, 5 mm Mg(OAC)2, 20 mm KCl, and 2 mm succinate at 37 °C for 60 min. After import, one-half of each sample was directly used to assess the mitochondrial association, and the other half was treated with proteinase K on ice for 15 min to remove the non-imported protein. After inhibiting proteinase K with PMSF (1 mm), mitochondria were re-isolated by passing through a sucrose cushion (0.8 m sucrose in 10 mm HEPES, pH 7.2) at 12,000 rpm for 10 min. Mitochondria were separated on SDS-PAGE, and the import was analyzed by using phosphorimaging. For mitoplasts preparation, after import, mitochondria were resuspended in hypotonic buffer (20 mm KCl, 10 mm HEPES, pH 7.2) or 0.1% digitonin and incubated on ice for 20 min. The resulting mitoplasts were reisolated by centrifugation at 12,000 rpm for 10 min and processed as mentioned above.
Immunoblotting
For Western blot analysis, mitochondria were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.2, 150 mm NaCl, 1% deoxycholic acid, 1% Triton X-100, 0.1% SDS, and 0.25 mm EDTA) with the protease inhibitor mixture (Roche Applied Science) and centrifuged to remove the debris. Protein concentrations were measured using Bradford reagent (Amersco). Then lysates were resolved on SDS-PAGE and transferred to polyvinylidene difluoride or nitrocellulose membranes and probed with specific antibodies. After incubation with HRP-conjugated secondary antibodies, the blots were developed using the Bio-Rad Versa doc imaging system.
In Vitro Phosphorylation Assay
Recombinant purified STAT3 protein was incubated with rabbit reticulocyte lysate in phosphorylation buffer (20 mm Tris-Cl, pH 7.5, 1 mm DTT, 25 μm ATP, 10 mm MgCl2, 1 mm NaF, 1 mm Na3VO4, 40 mm KCl) containing γ-32P-labeled ATP at 35 °C for 30 min. Reactions were quenched by adding SDS, and recombinant protein was pulled down using Ni-NTA resin and separated on SDS-PAGE and analyzed using a phosphorimaging system.
Two-dimensional BN-PAGE
For the first dimension BN-PAGE, mitochondrial pellets were resuspended in solubilization buffer (1% dodecyl maltoside, 0.75 m aminocaproic acid, and 50 mm Bis-Tris, pH 7.2). Mitochondrial suspensions were cleared at 14,000 rpm for 30 min at 4 °C and mixed with 5% serva blue G dye. Complexes were resolved on native 6–13% acrylamide gradient gels as described (33). For the second dimension, excised bands from BN-PAGE were soaked in 1% mercaptoethanol and 1% SDS for 1 h at room temperature. Gel pieces were layered on top of a 12% Tricine-SDS gel, and electrophoresis was performed at room temperature at 100 V (34).
RESULTS
Mitochondrial STAT3 Resides in the IMM of Mitochondria
Though STAT3 was shown to be associated with mitochondria, the exact submitochondrial localization remains ambiguous. To determine submitochondrial localization of STAT3, highly purified rat heart mitochondria were subjected to increasing concentrations of trypsin treatment (Fig. 1A). Mitochondrial fractions were separated on SDS-PAGE, Western-transferred, and probed with antibodies specific for each mitochondrial subcompartment (Fig. 1A). As shown in the Fig. 1A (top panel), STAT3 is resistant to low concentrations of protease treatment like matrix-localized protein, aconitase (bottom panel), whereas the outer membrane protein, TOM20 (Fig. 1A, lane 2), and the intermembrane space protein, Mia40, were completely digested (Fig. 1A, lane 3). A further increase in the trypsin concentration to 100 μg/ml reduced the mitochondrial associated STAT3 by 50% when compared with aconitase levels (Fig. 1A, lane 4). However, solubilization of mitochondrial membranes by Triton X-100 before protease treatment completely degraded the STAT3 and aconitase (Fig. 1A, lane 6). A similar observation was made when mitochondria was subjected to proteinase K treatment (not shown). These results show that a significant fraction of endogenous STAT3 is localized in the protease-inaccessible compartment of mitochondria.
FIGURE 1.

STAT3 is localized to inner mitochondrial membranes. A, purified rat heart mitochondria were subjected to different concentrations of trypsin (25–200 μg/ml, lanes 2–6) either in the absence (lanes 2–5) or presence (lane 6) of Triton X-100 for 20 min on ice. After inhibiting the trypsin by the addition of trypsin inhibitor, mitochondrial fractions were re-isolated by centrifugation and separated on SDS-PAGE followed by immunoblotting with antibodies specific for STAT3, TOM20 (outer membrane marker), Mia40 (inter membrane space marker), and aconitase (matrix marker). B, STAT3 sub-compartmental localization is shown. Mitochondrial outer membranes were solubilized with digitonin (0.1%) for 15 min on ice, and the resulting mitoplasts were re-isolated by centrifugation at 20,000 × g for 10 min. Both soluble (supernatant (S)) and insoluble fractions (pellet (P)) were collected and untreated or treated with proteinase K (25 μg/ml). After inhibition of proteinase K with PMSF (1 mm), all the fractions were separated on SDS-PAGE and probed with antibodies as specified in the figure. C, rat heart mitochondria (M) were subfractionated into mitoplasts (MP), IMM, and matrix (Mat) fractionations as described under “Experimental Procedures” and separated on SDS-PAGE and probed with specific markers as indicated in the figure. D, to show the tight association of STAT3 with membranes, isolated mitoplasts were subjected to high salt (400 mm KCl) and high pH (200 mm Na2CO3, pH 11.5) treatment for 15 min on ice. Mitoplasts were re-isolated, and equivalent fractions were resolved on SDS-PAGE and analyzed by Western blot. E, mitochondria (lanes 1 and 2) and mitoplasts (lanes 3 and 4) were treated without (1 and 3) or with 25 μg/ml proteinase K (lanes 2 and 4). After inhibiting proteinase K with PMSF, samples were solubilized in 1% dodecyl maltoside buffer as described under “Experimental Procedures.” The samples were subjected to BN-PAGE and probed for STAT3 and ND1.
To further investigate the submitochondrial localization of STAT3, mitochondrial outer membranes were solubilized with digitonin (Fig. 1B). The resulting mitoplasts were spun at high speed to separate insoluble mitoplasts and soluble intermembrane space fraction. 40% Mia40, an intermembrane space marker, was detected in the supernatant fraction but not ND1, an inner membrane marker (Fig. 1B, lane 2). Like ND1, most of the STAT3 was associated with mitoplast fraction (Fig. 1B, lane 3) and also partially resistant to protease treatment (Fig. 1B, lane 5). These results suggest that STAT3, like ND1, is not associated with soluble intermembrane space fraction; rather, it is likely to be associated with the inner membrane or matrix fraction.
To ascertain the exact localization of STAT3, we further separated mitoplasts into IMM and matrix fractions as described under “Experimental Procedures.” Separation of these fractions was confirmed by immunoblotting against specific marker proteins (Fig. 1C). Immunoblotting of these fractions with STAT3-specific antibody revealed enrichment of STAT3 in IMM fraction (Fig. 1C, lane 3). Treatment of mitoplasts with high salt (400 mm KCl) failed to dislodge STAT3 from the membrane fraction-like GRIM-19 (an integral membrane protein), whereas cytochrome c, a loosely associated inner membrane protein, was released into soluble fraction (Fig. 1D, compare lanes 3 and 5). However, high pH treatment (200 mm Na2CO3, pH 11.5) releases STAT3 into a soluble fraction (Fig. 1D, lane 4), indicating that STAT3 is tightly associated but not integrated into the membrane. Because STAT3 is known to increase the activities of complex I and II (5), we further evaluated the presence of STAT3 in the mitochondrial electron transport chain complexes. Mitochondria and mitoplast fractions were subjected to blue native gel electrophoresis and probed for STAT3 and a complex I subunit, ND1 (Fig. 1E). As shown in Fig. 1E, STAT3 was found to be associated with a high molecular mass complex above 700 kDa like a known complex I subunit, ND1. Protease treatment of the mitoplast fraction before to BN-PAGE reduced STAT3 association with this high molecular mass complex by 50%. In contrast, the levels of ND1 did not change significantly (Fig. 1E, lanes 3 and 4). Separation of this high molecular mass complex in second dimension showed the presence of STAT3 and GRIM-19, a known complex I subunit (supplemental Fig. S1). Taken together, our results indicate that STAT3 tightly associates with inner mitochondrial membrane complexes, likely to complex I, in a protease-inaccessible manner.
In Vitro Import of STAT3 into Isolated Mitochondria
To investigate the translocation of STAT3 across mitochondrial membranes, in vitro translated and 35S-labeled STAT3 was incubated with isolated mitochondria. Import of STAT3 was confirmed by its resistance to externally added proteinase K. As shown in the Fig. 2A, STAT3 was resistant to proteinase K (top panel), which indicates the presence of STAT3 within mitochondria. Import of 35S-labeled Su9-DHFR (bottom panel) and His6-DHFR proteins (Fig. 2C) was used as the positive and negative control, respectively. Upon import, Su9-DHFR, which has a presequence of subunit 9 of F1F0 ATPase (Fig. 2A, bottom panel) was protected from the protease treatment, whereas His6-DHFR protein, having no mitotargeting sequence, was susceptible to protease treatment (Fig. 2C). Most of the imported fragments were completely digested when the mitochondrial membranes were solubilized with Triton X-100 (Fig. 2A, lane 6). Primary criteria for translocation of proteins into mitochondria are the requirement of energy in the form of ATP and GTP (35–37). In addition, most of the matrix-targeted proteins and some of the inner membrane-targeted proteins require membrane potential to cross the inner membrane (1, 2, 38). To analyze whether STAT3 translocation requires membrane potential and energy, the import was performed in the absence of membrane potential, and external energy source and immunoblots were probed with aconitase antibody to show equal amount of protein in all samples (Fig. 2B). When membrane potential was dissipated with valinomycin, import of both Su9-DHFR (Fig. 2B, bottom panel, lane 2) and STAT3 (Fig. 2B, top panel, lane 5) was abolished, whereas import under energy deficit conditions resulted in 20–30% reduction of import (Fig. 2B, top panel, lane 4, and bottom panel, lane 3). The persistent translocation of STAT3 in the absence of external energy could be due to ATP leaking out of the mitochondria (39) or mitochondria respiring on endogenous substrates. These results indicate that STAT3 import and translocation across mitochondrial membrane requires membrane potential and likely requires energy.
FIGURE 2.

In vitro import of STAT3 into isolated mitochondria. A, in vitro import of STAT3 and Su9-DHFR was performed for 60 min as described under “Experimental Procedures.” After import, samples were treated with increasing concentrations of proteinase K (lanes 2–6) either in the absence (2–5) or presence of Triton X-100 (lane 6). After inhibition of proteinase K with PMSF (1 mm), samples were re-isolated and resolved on SDS-PAGE (P, precursor; M, mature). B, import of STAT3 (top panel) and Su9-DHFR (bottom panel) was examined in the absence of membrane potential by incubating mitochondria with valinomycin (5 μm) for 5 min (top panel, lane 5; bottom panel, lane 2) or by excluding the external energy source in the form ATP, GTP, and succinate (top panel, lane 4; bottom panel, lane 3) in the import reaction. C, import of His6-DHFR was carried out as described under “Experimental Procedures,” and the samples were treated with proteinase K (25 μg/ml) and analyzed by phosphorimaging.
GRIM-19 Regulates STAT3 Translocation into Mitochondria
Because GRIM-19 is a negative transcriptional regulator of STAT3, we investigated whether GRIM-19 also regulates STAT3 import into mitochondria. Co-import of 35S-labeled STAT3 and GRIM-19 was carried out as described under “Experimental Procedures.” Interestingly, STAT3 import was steadily enhanced up to certain concentrations of GRIM-19, and the stimulatory effect was more profound at lower concentrations (Fig. 3A). To rule out the possibility of interference from reticulocyte lysate components, we performed an in vitro import with increasing concentrations of lysate but found no difference in STAT3 recruitment (supplemental Fig. S2). To show specificity, co-import was performed with another transcription factor, ERα, known to be associated with mitochondria. Most of mitochondrial-localized ERα is accessible to externally added protease (40). However, in our in vitro import studies, ERα neither increased its association with mitochondria (not shown) nor was it protected from externally added protease even in the presence of GRIM-19 (Fig. 3B). Most importantly, GRIM-19 does not have any effect on the import of a mitochondrial inner membrane protein, TIM23 (Fig. 3C). Additionally, as a negative control, we also performed co-import of GRIM-19 with RBM3, a RNA binding motif protein localized mostly in the nucleus and cytosol (data not shown). As shown in the Fig. 3D, RBM3 failed to be internalized into mitochondria, and GRIM-19 does not affect the import of this protein. In addition, we also performed co-import of STAT3 with increasing concentrations of another mitochondrial-targeted protein, Su9-DHFR, and found no significant difference in STAT3 recruitment to mitochondria (Fig. 3E). Together these results demonstrate the specific role of GRIM-19 in STAT3 recruitment to mitochondria.
FIGURE 3.

GRIM-19 stimulates the import of STAT3 into mitochondria. A, in vitro co-import of STAT3 (6 × 103 cpm) was performed with increasing concentrations (1 × 103, 1. 5 × 103, 2 × 103, 2.5 × 103, 3 × 103, 4 × 103, and 6 × 103) of 35S-labeled GRIM-19 for lanes 2–8 for 60 min at 37 °C. After import, outer membrane-associated proteins were digested with proteinase K (25 μg/ml). Then samples were resolved on SDS-PAGE, and imported proteins were analyzed by phosphorimaging. As a negative control, co-import was also performed with ER α (B), TIM23 (C), and RBM3 (D) with increasing concentrations of GRIM-19. In another control experiment, STAT3 (6 × 103 cpm) was co-imported with increasing concentrations of Su9-DHFR (3 × 103, 5 × 103, 8 × 103, and 10 × 103 cpm of Su9-DHFR for lanes 2–6 of panel E as described above.
GRIM-19 Enhances Integration of STAT3 into Complex I
As GRIM-19 was found to be a positive regulator of STAT3 import into mitochondria, we further evaluated whether GRIM-19 could influence STAT3 topology and its association with membranes or complex I. After in vitro import of 35S-labeled STAT3 in the presence or absence of GRIM-19 (Fig. 4A), mitochondria were subjected to hypotonic shock to generate mitoplasts. As expected, imported STAT3 was protected from protease treatment of mitochondria (Fig. 4A, lane 2). However, most of the imported fraction is susceptible to protease treatment in mitoplasts (Fig. 4A, right panel, lane 3), indicating STAT3 is translocated across the outer membrane and is probably loosely associated with the inner membrane. Surprisingly, upon its co-import with GRIM-19, a significant fraction of STAT3 was protected from externally added protease even in mitoplasts (Fig. 4A, right panel, lane 5), indicating GRIM-19-dependent integration of STAT3 into a protease-inaccessible fraction that is likely a matrix or inner mitochondrial membrane. Protection of in vitro imported GRIM-19 and Su9-DHFR in mitoplasts indicates the intactness of mitochondrial inner membrane and matrix (supplemental Fig. S3). Immunoblots were also performed to confirm the formation of mitoplasts by protease susceptibility of Mia40, an intermembrane space marker, but not ND1, an inner membrane marker (supplemental Fig. S3).
FIGURE 4.
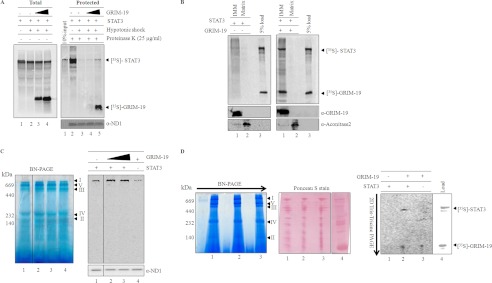
GRIM-19-mediated association of STAT3 with complex I. A, labeled STAT3 was imported into mitochondria for 60 min in the absence (left panel lanes 1 and 2; right panel, lanes 2 and 3) or presence of GRIM-19 (left panel, lanes 3 and 4; right panel, lanes 4 and 5), and mitoplasts were generated by hypotonic treatment of mitochondria. Mitochondria (left panel, lane 1; right panel, lane 2) and the resulting mitoplasts (left panel, lanes 2–4; right panel, lanes 3–5) were either left untreated (left panel) or treated with proteinase K (right panel). After inhibiting the protease, mitochondria and mitoplasts fractions were re-isolated and separated on SDS-PAGE and analyzed by phosphor imager. As a loading control, the right panel fractions were probed with antibodies specific for ND1. B, to demonstrate the association of co-imported STAT3 (with GRIM-19) with IMM, mitoplasts were further separated into IMM and matrix. The equivalent fractions of IMM and matrix were separated on SDS-PAGE and analyzed by phosphorimaging. Left and right panels represent the IMM associated STAT3 in the absence and presence of GRIM-19 respectively. To confirm the separation of IMM and matrix fractions, these fractions were also probed with inner membrane-specific antibody, GRIM-19, and matrix-specific antibody, aconitase (bottom panels). C, to analyze complex I association of STAT3, import of radiolabeled STAT3 was performed without (lane 1) or with increasing concentrations of unlabeled GRIM-19 (lanes 2 and 3). Import of labeled GRIM-19 (lane 4) was used as a positive control. After import, mitochondria were re-isolated and solubilized in 1% dodecyl maltoside buffer, and electron transport chain components were separated by BN-PAGE. Association of STAT3 with the components of the electron transport chain was detected by using phosphorimaging (C, right panel). Immunoblot, with ND1-specific antibody, was performed to show the equal loading of protein in all the fractions. D, to provide direct evidence for STAT3 association with complex I, in vitro import of labeled STAT3 (lane 1) or STAT3/GRIM-19 (lane 2) or GRIM-19 (lane 3) was performed, and these imported samples were protease-treated (25 μg/ml). After inhibiting the protease, the mitochondria samples were solubilized in 1% dodecyl buffer and separated on BN-PAGE (left panel) in the first dimension. For the second dimension, complex I bands were excised and separated on Tris-Tricine PAGE (right panel) and analyzed by phosphorimaging. The Ponceau S stained blot shows equal amount of protein in all lanes (middle panel). Lane 4 represents the in vitro translated 35S-labeled products of STAT3 and GRIM-19 to identify the STAT3 and GRIM-19 on two-dimensional-PAGE.
To precisely determine the localization of protease-protected 35S-labeled STAT3 in mitoplasts, we carried out the import of STAT3 in the presence of GRIM-19. Protease-treated mitoplasts were separated into IMM and matrix fractions (Fig. 4B). Separation of these fractions was demonstrated by immunoblotting with known marker proteins such as GRIM-19 (inner membrane) and aconitase (matrix) (Fig. 4B, bottom panels). When STAT3 was imported alone, a small fraction of STAT3 was found to be associated with IMM and matrix fraction (Fig. 4B, left panel, lanes 1 and 2). However, upon its co-import with GRIM-19, most of the STAT3 was recruited to IMM (Fig. 4B, right panel, lane 1). These results demonstrate that GRIM-19 integrates STAT3 into the inner membrane of mitochondria. To further assess whether the co-import of STAT3 with GRIM-19 enhances the association of STAT3 with respiratory complexes, in particular complex I, in vitro co-imported samples of 35S-labeled STAT3 with unlabeled GRIM-19 (cold) were separated by BN-PAGE, and STAT3 association was monitored by phosphor imager. As shown in the Fig. 4C, STAT3 association with complex I was increased in the presence of GRIM-19 (Fig. 4C, compare lanes 1 and 2), and any further increase in GRIM-19 had no effect on STAT3 association with complex I (Fig. 4C, compare lanes 2 and 3). 35S-Labeled STAT3 or 35S-labeled GRIM-19 when imported alone was associated with the complex I (Fig. 4C, lanes 1 and 4). However, 35S-labeled STAT3 that was associated with complex I was loosely bound (Fig. 4D) and susceptible to protease treatment (supplemental Fig. S5, right panel, lane 1). This was further confirmed by separating labeled 35S-labeled STAT3 alone (Fig. 4D, right panel, lane 1) or co-imported 35S-labeled STAT3 and 35S-labeled GRIM-19 (Fig. 4D, lane 2) or 35S-labeled GRIM-19 alone (Fig. 4D, lane 3) by BN-PAGE (Fig. 4D, left panel) followed by two-dimensional Tris-Tricine gels (Fig. 4D, right panel). The association was monitored by phosphorimaging of two-dimensional gels. STAT3 was found only in co-imported fraction (Fig. 4D, right panel, lane 2) but not in STAT3 alone fraction (Fig. 4D, right panel, lane 1). GRIM-19, a known complex I subunit, served as a positive control (Fig. 4D, right panel, lane 3). A Ponceau S-stained two-dimensional gel showed equal amounts of complex I proteins in all samples, and MALDI analysis confirms the presence of NDUFS1, a subunit of complex I (supplemental Fig. S4). Taken together these results demonstrate that GRIM-19 probably alters topology of mitochondrial STAT3 and also promotes its integration into complex I.
Ser-727 of STAT3 Is Required for GRIM-19-dependent Import and Assembly
The transcriptional activation domain lies in the C terminus of STAT3. To test whether the C terminus is required for the protein to be imported into the mitochondria, we generated a STAT3 truncation mutant, STAT3ΔC (STAT31–470). We carried out an in vitro import of labeled STAT3ΔC in the presence and absence of GRIM-19 (Fig. 5). Import of STAT3ΔC was absent even in the presence of GRIM-19 (Fig. 5A, lanes 2–5). Furthermore, the protein devoid of C-terminal domain failed to assemble into complex I even in the presence of GRIM-19, whereas full-length STAT3 was significantly present in complex I (supplemental Fig. S5 panel). This clearly indicates that the C terminus of STAT3 is required for GRIM-19-dependent import and integration of STAT3 into complex I. Ser-727 of STAT3 is located in the C terminus region of the protein and is known to be essential for STAT3 functions in mitochondria (5, 17) and is also a prerequisite for GRIM-19/STAT3 interaction (28). We evaluated the role of Ser-727 on mitochondrial import of STAT3 in the presence of GRIM-19. Interestingly, mutation of serine 727 to alanine reduced the import of STAT3 into mitochondria even in the presence of GRIM-19 (Fig. 5B, compare lane 1 with 2–4). This study indicates a requirement of the Ser-727 residue in the C terminus of STAT3 for its GRIM-19-dependent import into mitochondria. To further clarify whether STAT3 was phosphorylated before import, we performed an in vitro phosphorylation assay using purified recombinant STAT3 and reticulocyte lysate (used as a kinase source). STAT3 was found to be phosphorylated by the reticulocyte lysate in vitro (Fig. 5C, lane 4). In addition, alkaline phosphatase treatment of reticulocyte lysate containing labeled STAT3 before import reduces the import of STAT3, whereas no such inhibition was observed with Su9-DHFR (supplemental Fig. S6 comparelanes 1 with2 and 3). These results likely indicate that phosphorylation at Ser-727 of STAT3 may be required for its import into mitochondria.
FIGURE 5.

Requirement of Ser-727 of STAT3 for GRIM-19-dependent import of STAT3. A and B, in vitro import of STAT3ΔC (STAT31–470) and STAT3S727A with increasing concentrations of GRIM-19 was performed. After proteinase K treatment, samples were resolved on SDS-PAGE and analyzed by phosphorimaging (A and B). C, to determine whether STAT3 is phosphorylated before import, in vitro phosphorylation was performed using rabbit reticulocyte lysate as the kinase source. Recombinant purified His-tagged STAT3 was incubated either in the presence (lane 2 and 4) or absence (lane 3) of rabbit reticulocyte lysate and in the presence of (lanes 3–5) or absence (lane 2) of [γ-32P]ATP as indicated under “Experimental Procedures.” Rabbit reticulocyte lysate incubated with [γ-32P]ATP serves as a negative control (lane 5). Ni-NTA pulldown products were analyzed by phosphorimaging and also probed with STAT3 antibodies to show equal amounts of protein.
DISCUSSION
Recent findings from us and others indicate that STAT3 is associated with mitochondria (5, 17–20), where it modulates cellular respiration (5) and supports cellular transformation induced by the Ras oncogene (17). The mechanism by which STAT3 is imported into mitochondria has not been addressed in detail. In this report we demonstrate that a distinct mechanism is employed by STAT3 for its import and assembly into mitochondria and GRIM-19 has a major influence on this process.
Using an in vitro import system, we show that STAT3 efficiently traverses the mitochondrial membrane, although it does not have a canonical mito-targeting sequence. We further demonstrate that the mitochondrial import of this protein requires membrane potential and may require energy (Fig. 2). We also show that in vitro imported STAT3 is susceptible to proteases upon removal of the outer membrane by digitonin (Fig. 4A, lane 3). However, the endogenous mitochondrial STAT3 is partially protected from externally added protease (Fig. 1, B and C). In vitro imported STAT3, but not the endogenous protein, is accessible to protease treatment in mitoplasts, suggesting that an unknown factor(s) may be required for STAT3 transport in a protease-inaccessible mitochondrial compartment. This led us to evaluate whether GRIM-19 has any role in import of STAT3 into mitochondria given that both the proteins have been shown to interact and are present in mitochondria (24–27). Interestingly, we find that a low concentration of GRIM-19 facilitates transport of STAT3 into the mitochondria (Fig. 4). However, excessive GRIM-19 reduced internalization of STAT3 (Fig. 3A). We speculate that the decrease in recruitment of STAT3 at high concentrations of GRIM-19 may be due to competition for the same receptor, as both are devoid of cleavable mitochondrial targeting sequences, and proteins having similar sequences utilize the same receptor. In support of this notion, co-import of STAT3 with a pre-sequence-containing protein, Su9-DHFR, does not affect its mitochondrial recruitment, even at higher concentrations (Fig. 3E).
We also analyzed the role of GRIM-19 on subcompartmental localization of in vitro imported STAT3. In the absence of GRIM-19, most of the STAT3 is susceptible to protease upon opening of the outer mitochondrial membrane. Surprisingly, upon its co-import with GRIM-19, STAT3 is protease-resistant even in mitoplasts, indicating its association with a protease-inaccessible compartment of mitochondria (Fig. 4A, lane 5). We further demonstrate that this protease-inaccessible portion of imported STAT3 is associated with inner mitochondrial membranes by separating IMM and matrix fractions. The absence of STAT3 in the matrix fraction and resistance toward the protease in mitoplasts suggests that STAT3 might be anchored to the inner membrane facing toward the matrix, and this organization seems to require GRIM-19. In support of this idea, we find that a major portion of endogenous STAT3 also resides in inner mitochondrial membranes (Fig. 1). Similar to GRIM-19, high salt treatment failed to dislodge STAT3 from mitochondrial inner membranes, whereas high pH treatment releases most of the protein into the soluble fraction, which implies strong association with mitochondrial membranes. Based on these in vivo and in vitro import studies, we propose that STAT3 tightly associates with the inner mitochondrial membrane, and this association requires GRIM-19.
Studies indicate that 50% of mitochondrial proteins do not contain any canonical mitochondrial targeting sequences (41–43). Generally, proteins having no typical mitochondrial targeting sequence utilize the chaperone system (44) to translocate into the mitochondria, whereas some mitochondrial-targeted proteins contain an internal targeting sequence (45). It has been shown that phosphorylation of some proteins enhance their interaction with chaperones or exposes the cryptic mitochondrial targeting sequences to facilitate their mitochondrial recruitment (46, 47). Despite the absence of a mitochondrial targeting sequence, STAT3 efficiently traverses the mitochondrial membranes. Alkaline phosphatase treatment or S727A mutation diminishes the import of STAT3 into mitochondria, suggesting that serine 727 needs to be phosphorylated for efficient import of STAT3. We speculate that phosphorylation of STAT3 might be required for its interaction with GRIM-19 to facilitate its mitochondrial localization.
While our paper was in preparation Shulga and Pastorino (48) showed that phosphorylation of STAT3 on Ser-727 by RIPK1 enhances its interaction with GRIM-19 and thereby its mitochondrial localization.
Acknowledgments
We thank Department of Biotechnology - Centre for Research and Education in Biology and Biotechnology (DBT-CREBB) and University Grants Commission - Special Assistance Programme (UGC-SAP) for funding the School of Life Sciences and Department of Science and Technology - Funds for Improvement of S & T Infrastructure in Higher Educational Institutions for funding the Department of Biochemistry, University of Hyderabad, Hyderabad. We thank Dr. Bramanandam, Department of Biochemistry, University of Hyderabad, Hyderabad for kindly providing the pcDNA-ER-α clone. We also thank members of the Sepuri laboratory for suggestions.

This article contains supplemental Figs. 1–6.
- DHFR
- dihydrofolate reductase
- Ni-NTA
- nickel-nitrilotriacetic acid
- ERα
- estrogen receptor α
- IMM
- inner mitochondrial membrane
- GRIM-19
- gene associated with retinoid-interferon induced mortality-19
- ND1
- NADH dehydrogenase 1
- RBM3
- RNA binding motif 3
- STAT3
- signal transducer and activator of transcription 3
- TIM23
- translocase of inner membrane 23
- TOM20
- translocase of outer membrane
- BN-PAGE
- blue native PAGE
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Schatz G., Dobberstein B. (1996) Common principles of protein translocation across membranes. Science 271, 1519–1526 [DOI] [PubMed] [Google Scholar]
- 2. Neupert W. (1997) Protein import into mitochondria. Annu. Rev. Biochem. 66, 863–917 [DOI] [PubMed] [Google Scholar]
- 3. Pfanner N., Craig E. A., Hönlinger A. (1997) Mitochondrial preprotein translocase. Annu. Rev. Cell Dev. Biol. 13, 25–51 [DOI] [PubMed] [Google Scholar]
- 4. Biswas T. K., Getz G. S. (2002) Import of yeast mitochondrial transcription factor (Mtf1p) via a nonconventional pathway, J. Biol. Chem. 277, 45704–45714 [DOI] [PubMed] [Google Scholar]
- 5. Wegrzyn J., Potla R., Chwae Y. J., Sepuri N. B., Zhang Q., Koeck T., Derecka M., Szczepanek K., Szelag M., Gornicka A., Moh A., Moghaddas S., Chen Q., Bobbili S., Cichy J., Dulak J., Baker D. P., Wolfman A., Stuehr D., Hassan M. O., Fu X. Y., Avadhani N., Drake J. I., Fawcett P., Lesnefsky E. J., Larner A. C. (2009) Function of mitochondrial Stat3 in cellular respiration, Science 323, 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen J. Q., Eshete M., Alworth W. L., Yager J. D. (2004) Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors α and β to human mitochondrial DNA estrogen response elements, J. Cell. Biochem. 93, 358–373 [DOI] [PubMed] [Google Scholar]
- 7. Yang S. H., Liu R., Perez E. J., Wen Y., Stevens S. M., Jr., Valencia T., Brun-Zinkernagel A. M., Prokai L., Will Y., Dykens J., Koulen P., Simpkins J. W. (2004) Mitochondrial localization of estrogen receptor β. Proc. Natl. Acad. Sci. U.S.A. 101, 4130–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. She H., Yang Q., Shepherd K., Smith Y., Miller G., Testa C., Mao Z. (2011) Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Invest. 121, 930–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demonacos C. V., Karayanni N., Hatzoglou E., Tsiriyiotis C., Spandidos D. A., Sekeris C. E. (1996) Mitochondrial genes as sites of primary action of steroid hormones. Steroids 61, 226–232 [DOI] [PubMed] [Google Scholar]
- 10. Ioannou I. M., Tsawdaroglou N., Sekeris C. E. (1988) Presence of glucocorticoid responsive elements in the mitochondrial genome. Anticancer Res. 8, 1405–1409 [PubMed] [Google Scholar]
- 11. Marchenko N. D., Zaika A., Moll U. M. (2000) Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 275, 16202–16212 [DOI] [PubMed] [Google Scholar]
- 12. Cogswell P. C., Kashatus D. F., Keifer J. A., Guttridge D. C., Reuther J. Y., Bristow C., Roy S., Nicholson D. W., Baldwin A. S., Jr. (2003) NF-κB and IκBα are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-κB. J. Biol. Chem. 278, 2963–2968 [DOI] [PubMed] [Google Scholar]
- 13. Cammarota M., Paratcha G., Bevilaqua L. R., Levi de Stein M., Lopez M., Pellegrino de Iraldi A., Izquierdo I., Medina J. H. (1999) Cyclic A-responsive element-binding protein in brain mitochondria. J. Neurochem. 72, 2272–2277 [DOI] [PubMed] [Google Scholar]
- 14. Lee J., Kim C. H., Simon D. K., Aminova L. R., Andreyev A. Y., Kushnareva Y. E., Murphy A. N., Lonze B. E., Kim K. S., Ginty D. D., Ferrante R. J., Ryu H., Ratan R. R. (2005) Mitochondrial cyclic A response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J. Biol. Chem. 280, 40398–40401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ryu H., Lee J., Impey S., Ratan R. R., Ferrante R. J. (2005) Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc. Natl. Acad. Sci. U.S.A. 102, 13915–13920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J., Sharma S., Kim J., Ferrante R. J., Ryu H. (2008) Mitochondrial nuclear receptors and transcription factors. Who's minding the cell? J. Neurosci. Res. 86, 961–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gough D. J., Corlett A., Schlessinger K., Wegrzyn J., Larner A. C., Levy D. E. (2009) Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324, 1713–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boengler K., Hilfiker-Kleiner D., Heusch G., Schulz R. (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 105, 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bernier M., Paul R. K., Martin-Montalvo A., Scheibye-Knudsen M., Song S., He H. J., Armour S. M., Hubbard B. P., Bohr V. A., Wang L., Zong Y., Sinclair D. A., de Cabo R. (2011) Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 286, 19270–19279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qiu H., Lizano P., Laure L., Sui X., Rashed E., Park J. Y., Hong C., Gao S., Holle E., Morin D., Dhar S. K., Wagner T., Berdeaux A., Tian B., Vatner S. F., Depre C. (2011) H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 124, 406–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szczepanek K., Chen Q., Derecka M., Salloum F. N., Zhang Q., Szelag M., Cichy J., Kukreja R. C., Dulak J., Lesnefsky E. J., Larner A. C. (2011) Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 286, 29610–29620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Angell J. E., Lindner D. J., Shapiro P. S., Hofmann E. R., Kalvakolanu D. V. (2000) Identification of GRIM-19, a novel cell death-regulatory gene induced by the interferon-β and retinoic acid combination, using a genetic approach. J. Biol. Chem. 275, 33416–33426 [DOI] [PubMed] [Google Scholar]
- 23. Fearnley I. M., Carroll J., Shannon R. J., Runswick M. J., Walker J. E., Hirst J. (2001) GRIM-19, a cell death regulatory gene product, is a subunit of bovine mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 276, 38345–38348 [DOI] [PubMed] [Google Scholar]
- 24. Huang G., Lu H., Hao A., Ng D. C., Ponniah S., Guo K., Lufei C., Zeng Q., Cao X. (2004) GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I, Mol. Cell. Biol. 24, 8447–8456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalakonda S., Nallar S. C., Gong P., Lindner D. J., Goldblum S. E., Reddy S. P., Kalvakolanu D. V. (2007) Tumor-suppressive protein gene associated with retinoid-interferon-induced mortality (GRIM)-19 inhibits src-induced oncogenic transformation at multiple levels. Am. J. Pathol. 171, 1352–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okamoto T., Inozume T., Mitsui H., Kanzaki M., Harada K., Shibagaki N., Shimada S. (2010) Overexpression of GRIM-19 in cancer cells suppresses STAT3-mediated signal transduction and cancer growth. Mol. Cancer Ther. 9, 2333–2343 [DOI] [PubMed] [Google Scholar]
- 27. Lufei C., Ma J., Huang G., Zhang T., Novotny-Diermayr V., Ong C. T., Cao X. (2003) GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J. 22, 1325–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang J., Yang J., Roy S. K., Tininini S., Hu J., Bromberg J. F., Poli V., Stark G. R., Kalvakolanu D. V. (2003) The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc. Natl. Acad. Sci. U.S.A. 100, 9342–9347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sepuri N. B., Yadav S., Anandatheerthavarada H. K., Avadhani N. G. (2007) Mitochondrial targeting of intact CYP2B1 and CYP2E1 and N-terminal truncated CYP1A1 proteins in Saccharomyces cerevisiae. Role of protein kinase A in the mitochondrial targeting of CYP2E1. FEBS J 274, 4615–4630 [DOI] [PubMed] [Google Scholar]
- 30. Sepuri N. B., Gorla M., King M. P. (2012) Mitochondrial lysyl-tRNA synthetase independent import of tRNA lysine into yeast mitochondria. PLoS One 7, e35321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anandatheerthavarada H. K., Sepuri N. B., Avadhani N. G. (2009) Mitochondrial targeting of cytochrome P450 proteins containing NH2-terminal chimeric signals involves an unusual TOM20/TOM22 bypass mechanism. J. Biol. Chem. 284, 17352–17363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Budas G. R., Churchill E. N., Disatnik M. H., Sun L., Mochly-Rosen D. (2010) Mitochondrial import of PKCϵ is mediated by HSP90. A role in cardioprotection from ischaemia and reperfusion injury. Cardiovasc. Res. 88, 83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kerscher O., Sepuri N. B., Jensen R. E. (2000) Tim18p is a new component of the Tim54p-Tim22p translocon in the mitochondrial inner membrane. Mol. Biol. Cell 11, 103–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang J. K., Prabu S. K., Sepuri N. B., Raza H., Anandatheerthavarada H. K., Galati D., Spear J., Avadhani N. G. (2007) Site-specific phosphorylation of cytochrome c oxidase subunits I, IVi1, and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 581, 1302–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sepuri N. B., Schülke N., Pain D. (1998) GTP hydrolysis is essential for protein import into the mitochondrial matrix, J. Biol. Chem. 273, 1420–1424 [DOI] [PubMed] [Google Scholar]
- 36. Eilers M., Oppliger W., Schatz G. (1987) Both ATP and an energized inner membrane are required to import a purified precursor protein into mitochondria. EMBO J. 6, 1073–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pfanner N., Neupert W. (1986) Transport of F1-ATPase subunit β into mitochondria depends on both a membrane potential and nucleoside triphosphates. FEBS Lett. 209, 152–156 [DOI] [PubMed] [Google Scholar]
- 38. Schleyer M., Schmidt B., Neupert W. (1982) Requirement of a membrane potential for the posttranslational transfer of proteins into mitochondria, Eur. J. Biochem. 125, 109–116 [DOI] [PubMed] [Google Scholar]
- 39. Delage L., Dietrich A., Cosset A., Maréchal-Drouard L. (2003) In vitro import of a nuclearly encoded tRNA into mitochondria of Solanum tuberosum. Mol. Cell. Biol. 23, 4000–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen J. Q., Yager J. D. (2004) Estrogen's effects on mitochondrial gene expression. Mechanisms and potential contributions to estrogen carcinogenesis. Ann. N.Y. Acad. Sci. 1028, 258–272 [DOI] [PubMed] [Google Scholar]
- 41. Mootha V. K., Bunkenborg J., Olsen J. V., Hjerrild M., Wisniewski J. R., Stahl E., Bolouri M. S., Ray H. N., Sihag S., Kamal M., Patterson N., Lander E. S., Mann M. (2003) Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115, 629–640 [DOI] [PubMed] [Google Scholar]
- 42. Gabaldón T., Huynen M. A. (2004) Shaping the mitochondrial proteome, Biochim. Biophys. Acta. 1659, 212–220 [DOI] [PubMed] [Google Scholar]
- 43. Taylor S. W., Fahy E., Zhang B., Glenn G. M., Warnock D. E., Wiley S., Murphy A. N., Gaucher S. P., Capaldi R. A., Gibson B. W., Ghosh S. S. (2003) Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281–286 [DOI] [PubMed] [Google Scholar]
- 44. Young J. C., Hoogenraad N. J., Hartl F. U. (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112, 41–50 [DOI] [PubMed] [Google Scholar]
- 45. Diekert K., Kispal G., Guiard B., Lill R. (1999) An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc. Natl. Acad. Sci. U.S.A. 96, 11752–11757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anandatheerthavarada H. K., Biswas G., Mullick J., Sepuri N. B., Otvos L., Pain D., Avadhani N. G. (1999) Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic A-dependent phosphorylation at Ser-128. EMBO J. 18, 5494–5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robin M. A., Prabu S. K., Raza H., Anandatheerthavarada H. K., Avadhani N. G. (2003) Phosphorylation enhances mitochondrial targeting of GSTA4-4 through increased affinity for binding to cytoplasmic Hsp70. J. Biol. Chem. 278, 18960–18970 [DOI] [PubMed] [Google Scholar]
- 48. Shulga N., Pastorino J. G. (2012) GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J. Cell Sci. 125, 2995–3003 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]