

Abstract
Iterative polyketide synthases (PKS) are large multifunctional enzymes that resemble eukaryotic fatty acid synthases, but can make highly functionalized secondary metabolites using complex and unresolved programming rules. During biosynthesis of the kinase inhibitor hypothemycin by Hypomyces subiculosus, a highly-reducing iterative PKS, Hpm8, cooperates with a non-reducing iterative PKS, Hpm3, to construct the advanced intermediate dehydrozearalenol (DHZ). The identity of putative intermediates in the formation of the highly reduced hexaketide portion of DHZ could be confirmed by incorporation of 13C-labeled N-acetylcysteamine (SNAC) thioesters using the purified enzymes. The results show that Hpm8 can accept SNAC thioesters of intermediates ready for transfer from its acyl carrier protein (ACP) domain to its ketosynthase (KS) domain and assemble them into DHZ in cooperation with Hpm3. Addition of certain structurally modified analogs of intermediates to Hpm8 and Hpm3 can produce DHZ derivatives.
Polyketides display a wide spectrum of biological activities that make them an important resource for drug discovery.1–4 Iterative PKSs, which often occur in fungi, have only a single copy of each domain (e.g. keto reduction, dehydration and enoyl reduction) that can be utilized repeatedly for the multiple cycles of chain elongation and tailoring of functionality (Figure 1).2,5–7 The control of product functionality by an iterative PKS depends on the structure of the growing chain covalently attached as a thioester to the enzyme as well as the exact protein sequence. Although understanding of the biosynthesis of polyketides is advancing rapidly, knowledge of the detailed programming by iterative PKS is still limited.8,9 A key requirement for understanding the mechanisms of PKS enzymes is the determination of the structures of intermediates that remain enzyme-bound during numerous successive steps of elongation and modification. Chemical termination at intermediate steps in conjunction with mass spectrometry has been effective for some modular bacterial PKS systems.10–12 In fungal systems, direct FT-ICR-MS has been used to examine potential intermediates loaded on the PKS.13,14 However, mass spectrometry has limitations for elucidation of stereochemistry of intermediates and analysis of possible isomers. An alternative approach pioneered by Cane, Hutchinson and coworkers to probe the mechanism of modular bacterial PKS systems relies on the use of N-acetylcysteamine (SNAC) thioesters of partially assembled precursors.15–17 Using PKS domain inactivation followed by addition of putative intermediate SNAC thioesters also allows bacterial polyketides to be produced in cell-free systems.18–19 However, it is challenging to apply this advanced precursor feeding approach to the iterative PKS systems since domain inactivation will abolish the production of polyketides. In addition, this approach often fails with whole or disrupted fungal cells due to catabolism of the precursors except in some isolated cases.20–22 Therefore, developing new methods to probe fungal PKS function using partially assembled intermediates can aid the elucidation of these complex machineries.
Figure 1.

Biosynthesis of hypothemycin. Hpm8 assembles a hexaketide intermediate, which is then transferred to Hpm3. Hpm3 extends the hexaketide intermediate to DHZ. (Red intermediates were synthesized as 13C labeled SNAC thioesters).
Hypothemycin biosynthesis has been studied in considerable detail. 23–25 Two iterative PKS proteins, Hpm8 and Hpm3 build the polyketide backbone of hypothemycin. Hpm8, a highly reducing (HR) PKS, first assembles a reduced hexaketide intermediate. With the assistance of a SAT domain (starter unit acyl-carrier protein transacylase), this newly formed hexaketide is transferred to a non-reducing (NR) PKS, Hpm3, where it is further extended to a nonaketide. The nonaketide then undergoes regioselective cyclization, and macrolactonization to afford (6′S,10′S)-7′,8′-dehydrozearalenol (DHZ).24 Subsequent post-PKS modifications of DHZ by other enzymes afford hypothemycin. Although the general functions of the two PKSs have been assigned, it remains unresolved how Hpm8 controls tailoring of the intermediates en route to its hexaketide product using its reductive domains (KR, DH and ER) in a permutative fashion. To probe the programing rules of HRPKSs, confirmation of the structures of enzyme-bound intermediates and the way they interact with HRPKSs would be desirable. Here we report efforts on confirming such intermediate structures by in vitro incorporation of partly assembled precursors into Hpm8.
Based on the accepted mechanism for polyketide biosynthesis,1 we propose that there are fourteen ACP-tethered intermediates (Figure 1 and supporting information) en route to the hexaketide that is eventually transferred to Hpm3.24 Among these fourteen compounds, four are classified as “ready” precursors because they have the correct functionality to proceed to next round of chain elongation by Claisen condensation. The other ten putative intermediates are “unready” precursors because they must first be modified by KR, DH, or ER domains prior to chain extension.
Our concept was to chemically synthesize all the “ready” precursors (2, 4, 6, 8) and several “unready” precursors (1, 3, 5, 7) as 13C labeled SNAC thioesters (Table 1), and determine if these compounds can be linked to Hpm8 to give corresponding enzyme-bound intermediates that can be elaborated to the correct hexaketide and ultimately to 13C labeled DHZ. We selected “unready” precursors 1, 3, 5 and 7 because they cover the scope of oxidation states possible on the β-position of the intermediates. The stereochemistry of the β-hydroxyl of “unready” precursor 3 is based on the stereoselectivity of the KR domain of Hpm8.25
Table 1.
Summary of incorporation of partially assembled precursor analogues.
| Intermediate Analoguea,b | 13C Enrichment | Relative RAL yieldc | |
|---|---|---|---|
| 0 | none | none observed | 1.0 |
| 1 |
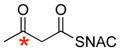
|
41% | 0.4 |
| 2 |
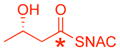
|
37% | 1.0 |
| 3 |
|
15% | 0.6 |
| 4 |
|
78% | 1.0 |
| 5 |
|
19% | 0.4 |
| 6 |
|
27% | 2.0 |
| 7 |
|
8% | 0.9 |
| 8 |
|
39% | 2.6 |
* indicates the location of 13C label;
ready precursor structures are shown in red;
resorcylic acid lactone (RAL) relative yield includes DHZ, and for unnatural precursor analogs, also the amount of corresponding DHZ analogs;
Compounds 9–12 have incorrect stereochemistry or functionality for conversion to DHZ and are therefore (except for 12) transformed to corresponding DHZ analogs;
production is the amount of DHZ analog formed compared to the total RAL production.
The first substrate tested was the ”ready” triketide 4, which was labeled with 13C at the carbonyl that is proposed to become C-6′ in DHZ (Figure 2a, syntheses in supporting information). To avoid a large background of unlabeled DHZ that could be formed by initial self-loading of unlabeled malonyl-CoA,26 we designed a “pre-loading” assay. This consisted of: 1) incubation of Hpm8 (10 μM) with a large excess of labeled substrate (e.g. 4) (0.2 mM) for 15 min (pre-load); 2) addition of 10 μM Hpm3, 4 mM NADPH, 0.4 mM malonyl-CoA, incubated for 1 h; 3) subsequent addition of 0.4 mM malonyl-CoA at 1 h intervals (9 times); addition of 0.2 mM labeled substrate every 2 h (3 times). Analysis of the reaction mixture by LC-MS after incorporation of 4 generated a compound identical to DHZ in terms of LC retention time and UV absorbance. This compound showed a major peak in the mass spectrum at 318 ([M – H]-) (Figure 2b), indicating it consists primarily of a singly 13C labeled DHZ. The incorporation ratio based on this mass spectrum27 was calculated to be 78%.
Figure 2.

(a) Incorporation of the triketide 4 into DHZ. (b) Mass spectrum of DHZ from the triketide 4 assay (left) and unlabeled DHZ standard (right). (c) 13C NMR spectra of DHZ from the triketide 4 incorporation (upper) and unlabeled DHZ standard (lower).
To further confirm the location of the 13C enrichment, we isolated approximately 20 μg of 13C labeled DHZ (based on UV absorbance). The 13C NMR spectrum (Figure 2c) showed only a single resonance at 72.8 ppm, corresponding to the C-6′ position of DHZ. This demonstrates that the C-6′ position of DHZ is 13C enriched, which is consistent with the specific incorporation of the triketide portion of 4 and its subsequent conversion into the final product. These data illustrate that the KS domain of Hpm8 can be primed by trans-thioesterification with the “ready” precursor 4, which can then be elongated to the hexaketide moiety that is then transformed by the NRPKS Hpm3 into DHZ. This represents the first example of incorporation of a labeled advanced intermediate by a purified iterative HRPKS.
We next examined the incorporation of putative intermediates shown in Table 1: diketides 1 and 2, triketide 3, tetraketides 5 and 6, and pentaketides 7 and 8. The “ready” precursors 2, 6, and 8 were incorporated efficiently, with 13C enrichments of 37%, 27%, and 39%, respectively, based on mass spectra (Table 1). The relative amounts of the resorcylic acid lactone (RAL) produced compared to no precursor addition range from 1.0 to 2.6, based on UV absorbance. Interestingly, the more advanced the precursor, the more DHZ was produced, possibly because advanced precursors require fewer additional reactions to reach the final product. Alternatively, the higher yields may also be due to a higher rate of priming (transthioesterification) of more advanced precursors onto the cysteine thiol of the KS domain.
In contrast, with exception of the diketide 1, which may also mimic malonyl-CoA, conversions of the “unready” precursors 1, 3, 5, and 7 gave relatively lower incorporation ratios (41%, 15%, 19%, and 8%, respectively) into DHZ and poorer relative production of RAL (0.4, 0.6, 0.4 and 0.9, respectively). For example, the lack of α,β-dehydration in 3 compared to 4, or the unrealized enoyl reduction of 7 compared to 8, both resulted in greater than five-fold decrease in the incorporation level of the labeled precursor. Therefore the Hpm8 machinery clearly prefers precursor analogs that are structurally comparable to the correctly tailored intermediates ready for the next round of chain extension. The differences in incorporation efficiencies between the “ready” and the “unready” precursor feeding experiments provides insights into the mechanisms of the Hpm8 multi-domain enzyme. For the “ready” precursors, the subsequent enzymatic steps can be immediately initiated following priming (trans-thioesterification) of the KS domain.28 The carbon chains of ready precursors are correctly functionalized, so they may easily prime on KS domains and serve as the starting material for next round chain elongation. For incorporation of “unready” precursors, several alternative mechanisms can be considered. One possibility is that the “un-ready” precursors load directly onto the ACP by trans-esterification and are then further converted into the “ready” forms in the normal fashion. However, as free ACPs are normally loaded with malonyl-CoA by the MAT domain, competing for the vacant ACP thiol is likely to be very inefficient. An alternative path would involve loading on the active site thiol of the KS domain with subsequent trans-esterification to the ACP domain followed by modification into “ready” precursors. This “KS domain to ACP domain” transfer on incompletely tailored fragments does not occur naturally, so this process may be very slow or impossible. The most likely pathway may be direct action of reducing/tailoring domains on the “unready” SNAC esters to transform them into “ready” SNAC derivatives. In our previous studies,25 we found “unready” SNAC esters such as 1 and 5 could be reduced by Hpm8 to “ready” precursors 2 and 6, respectively. The KR domain can recognize the SNAC moiety, and this recognition can correctly direct the acyl chain to the active site for reduction. The analogous concept may apply for the direct transformation of 7 to 8 by the ER domain or of 3 to 4 by the DH domain. The DH29 domain has been shown to be inefficient at utilizing SNAC precursors, and this may account for the significantly lower incorporation of 3.
We also observed the “unready” β-keto analogs 1 and 5 produced much less DHZ. This may be because they inhibit the natural chain extension. With the unlabeled DHZ production inhibited, the incorporation ratio (13C enrichment) of 1 and 5 would also be relatively high, as observed.
To further probe the substrate tolerance of the KS domain of Hpm8, the unnatural precursors 9, 10, 11 and 12 were chemically synthesized for use in the assay to potentially produce DHZ analogues. Compounds 9 and 10 have an epimeric distal alcohol, whereas 11 is lacking the conjugated double bond. As the functions of the tailoring domains of PKS enzymes are processive and limited to the two carbons adjacent to the thioester carbonyl,1–7 such “incorrect” functionality cannot be modified to give the natural DHZ product. Hpm8 is able to accept 9 or 10 to form 10″-epi-DHZ, but with much lower efficiency (relative ratios of epi-DHZ to DHZ formed directly from malonate are 25:75, Table 1). This is consistent with our in vivo observation that Hpm8 showed discrimination toward the stereochemistry of that hydroxyl group.25 Compound 11, which represents an over-reduced precursor, was similarly incorporated by this system to generate β-zearalenol (relative ratio of β-zearalenol to DHZ is 3 to 7). Compound 12 is a diastereomer of the “unready” precursor 3. Our previous work25 showed that the KR domain from Hpm8 displays strict stereospecificity toward β-keto intermediates that is dependent on chain length. We selected 12, which is not an expected intermediate, to test whether the DH domain is able to eliminate a triketide alcohol of unnatural stereochemistry. The results showed that very little if any 12 is incorporated into DHZ. This demonstrates that the (S) hydroxyl group is not a good substrate in the DH active site, which is consistent with other PKS and FAS systems.30 The overall lowered yield of RAL suggests that the system is inhibited by this unnatural precursor analog.
In summary, a series of 13C labeled intermediate SNAC thioesters have been chemically synthesized and incorporated into the DHZ using purified Hpm8 and Hpm3. Our results show an interesting pattern of the incorporation of partially assembled precursor analogs at the HRPKS stage: 1) “ready” precursors are easily recognized and taken up by Hpm8; 2) “unready” precursors are incorporated less effectively by Hpm8, but some incorporation is still observed, albeit with lower yields of DHZ; 3) unnatural precursor analogs can be incorporated, but the efficiency is dependent on the nature of the structural changes. Our findings not only further support the processive nature of polyketide biosynthesis, but also provide guidelines for precursor-directed biosynthesis to generate novel polyketides with improved biological profiles.
Supplementary Material
Acknowledgments
We are grateful to Mark Miskolzie for assistance with NMR experiments. We thank the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Research Chair in Bioorganic and Medicinal Chemistry and the US National Institutes of Health (1R01GM085128 and 1DP1GM106413) for financial support.
Footnotes
Synthetic procedures for labeled precursors and experimental details for incorporations by Hpm8 and Hpm3. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Cox RJ. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 3.Li JH, Vederas JC. Science. 2009;325:165–169. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 4.Zabala AO, Cacho RA, Tang YJ. Ind Microbiol Biotechnol. 2012;39:227–241. doi: 10.1007/s10295-011-1044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Science. 1999;284:1368–1372. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]; (b) Hutchinson CR, Kennedy J, Park C, Auclair K, Vederas JC. In: Handbook of Industrial Mycology. An Z, editor. Chapter 17. Marcel Dekker Inc; New York: 2005. pp. 479–492. [Google Scholar]
- 6.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 7.Crawford JM, Townsend CA. Nature Rev Microbiol. 2010;8:879–889. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma SM, Li JW-H, Choi JW, Zhou H, Lee MKK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisch KM, Bakeer W, Yakasai AA, Song Z, Pedrick J, Wasil Z, Bailey AM, Lazarus CM, Simpson TJ, Cox RJ. J Am Chem Soc. 2011;133:16635–16641. doi: 10.1021/ja206914q. [DOI] [PubMed] [Google Scholar]
- 10.Tosin M, Spiteller D, Spencer JB. ChemBioChem. 2009;10:1714–1723. doi: 10.1002/cbic.200900093. [DOI] [PubMed] [Google Scholar]
- 11.Tosin M, Betancor L, Stephens E, Li WMA, Spencer JB, Leadlay PF. ChemBioChem. 2010;11:539–546. doi: 10.1002/cbic.200900772. [DOI] [PubMed] [Google Scholar]
- 12.Tosin M, Demydchuk Y, Parascandolo JS, Per CB, Leeper FJ, Leadlay PF. ChemComm. 2011;47:3460–3462. doi: 10.1039/c0cc05077f. [DOI] [PubMed] [Google Scholar]
- 13.Meehan MJ, Xie X, Zhao X, Xu W, Tang Y, Dorrestein PC. Biochemistry. 2011;50:287–299. doi: 10.1021/bi1014776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vagstad AL, Bumpus SB, Belecki K, Kelleher NL, Townsend CA. J Am Chem Soc. 2012;134:6865–6877. doi: 10.1021/ja3016389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Cane DE, Yang C. J Am Chem Soc. 1987;109:1255–1257. [Google Scholar]; (b) Yue S, Duncan JS, Yamamoto Y, Hutchinson CR. J Am Chem Soc. 1987;109:1253–1255. [Google Scholar]
- 16.Cane DE, Lambalot RH, Prabhakaran PC, Ott WR. J Am Chem Soc. 1993;115:522–526. [Google Scholar]
- 17.(a) Cane DE, Tan WT, Ott WR. J Am Chem Soc. 1993;115:527–535. [Google Scholar]; (b) Cane DE, Luo GL. J Am Chem Soc. 1995;117:6633–6634. [Google Scholar]
- 18.Kim CY, Alekseyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]
- 19.Yuzawa S, Kapur S, Cane DE, Khosla C. Biochemistry. 2012;51:3708–3710. doi: 10.1021/bi300399u. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Yoshizawa Y, Li Z, Reese PB, Vederas JC. J Am Chem Soc. 1990;112:3212–3213. [Google Scholar]; (b) Li Z, Martin FM, Vederas JC. J Am Chem Soc. 1992;114:1531–1533. [Google Scholar]; (c) Harrison PH, Noguchi H, Vederas JC. J Am Chem Soc. 108:3833–3834. [Google Scholar]; (d) Liu Y, Li Z, Vederas JC. Tetrahedron. 1998;54:15937–15958. [Google Scholar]
- 21.Brobst S, Townsend CA. Can J Chem. 1994;72:200–207. [Google Scholar]
- 22.Tsantrizos YS, Zhou F, Famili P, Yang X. J Org Chem. 1995;60:6922–6929. [Google Scholar]
- 23.Reeves CD, Hu ZH, Reid R, Kealey JT. Appl Environ Microbiol. 2008;74:5121–5129. doi: 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H, Qiao K, Gao Z, Meehan MJ, Li JWH, Zhao X, Dorrestein PC, Vederas JC, Tang Y. J Am Chem Soc. 2010;132:4530–4531. doi: 10.1021/ja100060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Gao Z, Qiao K, Wang J, Vederas JC, Tang Y. Nature Chem Biol. 2012;8:331–333. doi: 10.1038/nchembio.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma SM, Tang Y. FEBS J. 2007;274:2854–2864. doi: 10.1111/j.1742-4658.2007.05818.x. [DOI] [PubMed] [Google Scholar]
- 27.Biemann K. Mass Spectrometry: Organic Chemical Applications. McGraw-Hill Book Co; New York: 1962. [Google Scholar]
- 28.Tsukamoto N, Chuck JA, Luo G, Kao CM, Khosla C, Cane DE. Biochemistry. 1996;35:15244–15248. doi: 10.1021/bi961972f. [DOI] [PubMed] [Google Scholar]
- 29.Valenzano CR, You YO, Garg A, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2010;132:14697–14699. doi: 10.1021/ja107344h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwan DH, Schulz F. Molecules. 2011;16:6092–6115. doi: 10.3390/molecules16076092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.