

Abstract
Our previous study on retinal light exposure suggests the involvement of zinc (Zn2+) toxicity in the death of RPE and photoreceptors (LD) which could be attenuated by pyruvate and nicotinamide, perhaps through restoration of NAD+ levels. In the present study, we examined Zn2+ toxicity, and the effects of NAD+ restoration in primary retinal cultures. We then reduced Zn2+ levels in rodents by reducing Zn2+ levels in the diet, or by genetics and measured LD. Sprague Dawley albino rats were fed 2, or 61 mg Zn2+/kg of diet for 3 weeks, and exposed to 18 kLux of white light for 4h. We light exposed (70 kLux of white light for 50h) Zn2+ transporter 3 knockout (ZnT3-KO, no synaptic Zn2+), or RPE65 knockout mice (RPE65-KO, lack rhodopsin cycling), or C57/BI6/J controls and determined light damage and Zn2+ staining. Retinal Zn2+ staining was examined at 1h and 4h after light exposure. Retinas were examined after 7d by optical coherence tomography and histology. After LD, rats fed the reduced Zn2+ diet showed less photoreceptor Zn2+ staining and degeneration compared to a normal Zn2+ diet. Similarly, ZnT3-KO and RPE65-KO mice showed less Zn2+ staining, NAD+ loss, and RPE or photoreceptor death than C57/BI6/J control mice. Dietary or ZnT3-dependent Zn2+ stores, and intracellular Zn2+ release from rhodopsin recycling are suggested to be involved in light-induced retinal degeneration. These results implicate novel rhodopsin-mediated mechanisms and therapeutic targets for LD. Our companion manuscript demonstrates that pharmacologic, circadian, or genetic manipulations which maintain NAD+ levels reduce LD.
Keywords: rat, mouse, rhodopsin, RPE65, ZnT3, photoreceptors
1. Introduction
Light-induced retinal damage can occur after acute or chronic sun exposure and surgery (Fuller et al., 1978; Zigman et al., 1979; Kuhn et al., 1991; Thanos et al., 2001; Codenotti et al., 2002; Jain et al., 2009; Vojnikovic et al., 2009). Light also accelerates disease progression and neurodegeneration in many retinal diseases in which light or oxidative stress (OS) are implicated (RP, glaucoma, macular degeneration) (Wang et al., 1997; Bicknell et al., 2002; Organisciak et al., 2003; Ranchon et al., 2003; Richards et al., 2006; Vaughan et al., 2006; Yang et al., 2007; Rodriguez and Fliesler, 2009) (Reviewed in (Organisciak and Vaughan, 2010)). Intense light damage is preferentially confined to the photoreceptors of the outer nuclear layer (ONL) in the superior central retina of rats and mice (Gordon et al., 2002; Cortina et al., 2003). This damage involves apoptotic rod cell death and necrotic cone cell death (reviewed in (Gordon et al., 2002; Organisciak and Vaughan, 2010)). Protection in inferior retina attributes to shorter outer segment and lower rhodopsin level than those in superior retina (Battelle and LaVail, 1978; Penn et al., 1987). Inferior retina also has a better intra-retinal circulation and neuroprotective factor synthesis in response to intense light exposure (Liu et al., 1998; Li et al., 2003). It was previously demonstrated that Zn2+ accumulation and toxicity play a role in retinal ischemia mediated cell death (Yoo et al., 2004). We have now demonstrated that photoreceptors stain for Zn2+ before dying after light exposure, and cyclic light, pyruvate, or nicotinamide attenuated LD (Sheline et al., 2010a).
The loosely bound or free Zn2+ is histochemically reactive and present physiologically in different layers of retina, and varies between dark and light. In ambient light, it notably exists in the rod inner and outer segments (RIS, ROS) of the ONL, the outer plexiform layer (OPL), and retinal pigment epithelial (RPE) cells. In the dark, however, this histochemically-reactive Zn2+ appears in photoreceptor perikarya of ONL (Wang et al., 2006). Zn2+ plays important roles in retinal functions including dark-light adaptation (reviewed in (Ugarte and Osborne, 2001)), modulating neurotransmission and regulating intracellular metabolism (Rosenstein and Chappell, 2003; Redenti et al., 2007; Chappell et al., 2008).
Zn2+ neurotoxicity is involved in many injuries and diseases including retinal ischemia (Yoo et al., 2004; Choi et al., 2006), global ischemia (Koh et al., 1996), trophic deprivation (Sheline et al., 2010b) and hypoglycemia mediated neuronal death (Suh et al., 2004; Suh et al., 2008). Excessive Zn2+ either from extracellular Zn2+ uptake through voltage gated calcium channels under depolarization, or release from intracellular Zn2+ binding proteins or organelles under oxidation is neurotoxic (Canzoniero et al., 1999; Sheline et al., 2010b). In neurons both in vitro and in vivo, excess Zn2+ triggers NAD+ loss which in turn inhibits glycolysis. Pyruvate and nicotinamide restore NAD+ levels and attenuate Zn2+ neurotoxicity in the central nervous system (Sheline et al., 2000; Lee et al., 2001; Suh et al., 2003; Cai et al., 2006; Sheline et al., 2010b). In this study we further investigated whether dietary or genetic reduction of Zn2+ levels could attenuate light-induced damage. We decreased the Zn2+ levels in the diet and genetically reduced Zn2+ levels by knocking out Zn2+ transporter 3 (ZnT3-KO, no synaptic Zn2+) or RPE65 (RPE65-KO, no rhodopsin recycling). We propose a model in which ZnR or reduced Zn2+ release attenuates retinal Zn2+ toxicity and LD.
2. Research Design And Methods
2.1. Primary retinal culture
Primary retinal cultures were generated from retinas (16 retinas/plate) of P1 mouse pups. Retinas were isolated and mechanically dissociated into single cells by trituration with fire-polished Pasteur pupettes. Then triturated retinas were plated in DMEM, 10% FBS, 1% glutamine, 0.1% P/S, 25mM KCL solution. Retinal cultures were grown in 5% CO2, 95% humidity at 37°C. Toxic exposures were initia ted after 10 days of culturing. Cells were exposed to Zn2+ in the presence of pyruvate, nicotinamide or NAD+. Cell viability was determined after 24h by adding in PI (5 μg/ml) for 30 min at 37°C and fluorescence measured (ex 530/em 645).
2.2. Live-cell imaging
FluoZin3 AM is a sensitive and selective fluorescent probe used to measure Zn2+ in living cells. FluoZin3 AM has extremely high affinity to Zn2+ (KD=15nM) that is unperturbed by Ca2+ concentrations (Haugland, 2005). FluoZin3 AM increases in fluorescence more than 50 fold upon saturation with Zn2+ (Gee et al., 2002). PRC were washed and exposed to 0 or 150 μM Zn2+ in serum free medium for 4h (Figure 1). FluoZin3 AM (5 μM; Invitrogen/Life Technologies, Carlsbad, CA) was then loaded into washed cells for 30 min at 37°C, washed again and photomicrographs o f identical duration were taken. The specificity of increased [Zn2+]I was demonstrated using a zinc chelator, TPEN. TPEN is cell permeable and selectively chelates intracellular heavy metal ions such as Zn2+, Cu2+ and Fe2+ (Haugland, 2005). It has extremely high affinity to Zn2+ (KD=10–15.6, but no affinity for Ca2+, or Mg2+, and has been used as a Zn2+ specific chelator (Canzoniero et al., 2003). Adding TPEN to Zn2+-, or H2O2- treated cell cultures completely blocked the increased signal detected by FluoZin3 (data not shown).
Fig. 1. Zn2+ exposure causes accumulation, toxicity, and NAD+ loss in PRC cultures.
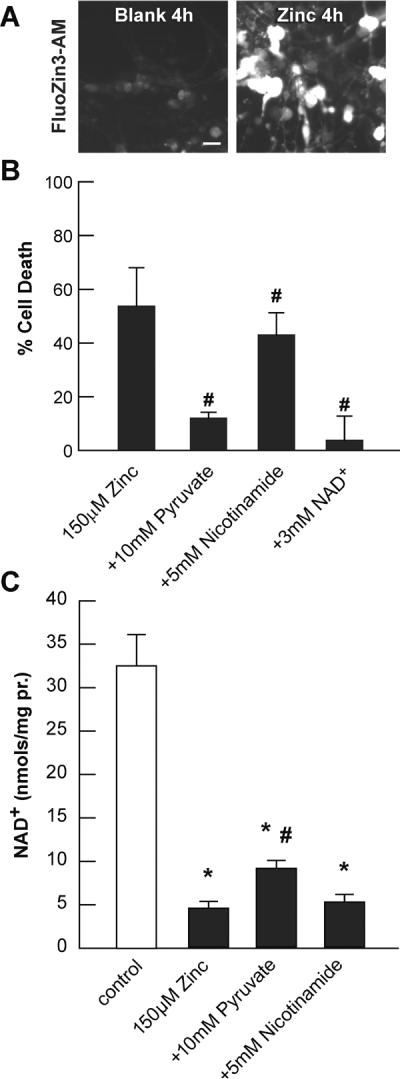
A. PRC cultures were post-loaded with 5 μM FluoZin3-AM and exposed to 150 μM Zn2+ as indicated. Photomicrographs were taken at equivalent exposures. B. Cell death was measured in sister cultures after exposure for 24 h as indicated using propidium iodide staining. C. NAD+ levels were measured in sister cultures after exposure for 3 h as indicated using enzyme-cycling reactions. * indicates difference from control, and # indicates difference from Zn2+ exposure at P < 0.05 by one-way ANOVA and a Student t-test, n>8.
2.3. Zn2+ restricted and normal diets in the rat light damage model
Four wk old Sprague-Dawley albino rats (Charles River, Wilmington, MA) weighing 70g were fed a 2 mg/kg purified Zn2+ diet, and 61mg/kg purified Zn2+ diet for 3 wk as previously reported (Sheline, J. Nutrition, accepted Takeda et al., 2003; Suh et al., 2009). Zn2+ deficient diet contains ~1 mg/kg diet, which was supplemented with 1, or 60 mg Zn2+/kg diet Zn2+ equivalent ZnSO4 in 18 mega-Ohm purified drinking water(= 2 or 61 ppm). The animals were maintained in plastic cages with plastic water bottles to minimize Zn2+ contamination. Rats fed normal standard diet which contains 61 mg/kg Zn2+ were supplemented with 18 mega-Ohm purified drinking water (normal Zn2+ diet). The Zn2+ restricted diet was from Harlan Teklad (TD.85419, Madison WI), and the normal diet (contains 61 mg/kg Zn2+) was also from Harlan Teklad (2019). The Zn2+ deficient diet is composed of: 200 g/kg of egg white solids, 634 g/kg of dextrose, 100 g/kg of corn oil, 30 g/kg of cellulose, 10 g/kg of vitamin mix, 0.004 g/kg of biotin, 25.7 g/kg of Zn2+ deficient mineral mix, 0.02 g/kg of chromium potassium dodecahydrate, and 0.02 g/kg of ethoxyquin. The 2019 normal diet is a standard extruded diet with 19% protein which is derived from ground wheat, ground corn, corn gluten meal, wheat middlings, and soybean oil with vitamins and minerals added. Each experiment is internally controlled as only Zn2+ deficient diet is used in varying the dietary Zn2+ (Figure 2), and only normal diet is used when the genotype is being varied (Figures 3–4). Animals consumed similar rates of diet and water with 1g food for 1ml water, and no developmental problems were observed.
Fig. 2. Light induced Zn2+ staining and retinal damage were attenuated by ZnR.
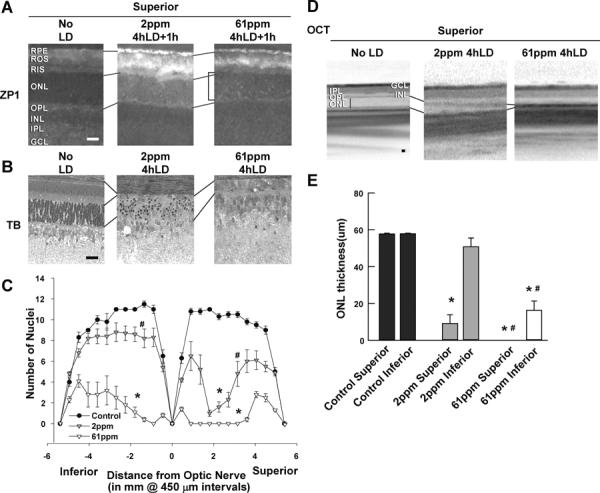
LD was performed on rats fed the diets indicated for 3 wks prior and retinas were analyzed for Zn2+ staining (A) 1h after sham exposure (No LD), or 1 h after 4h of LD. Zn2+ accumulation (white regions) was assessed by ZP1 staining (5 μM for 2 min) in fresh frozen, dried cryostat sections. Representative photomicrographs (n = 4) were taken of the mid superior regions of the retina at 0.2 second exposure. Notice the large increase in the number and intensity of Zn2+ stained cells in superior ONL (brackets) of 61 ppm animals. This is reduced in the 2 ppm animals. B. Retinas of male littermates mice were analyzed 7 days after LD by plastic sectioning cutting from eyes along a superior to inferior meridian encompassing the optic nerve and stained with 0.1% Toluidine Blue. Photomicrographs were taken of the mid superior regions of the retina. C. ONL nuclei counted at increasing distance from the optic nerve was averaged and plotted as a function of distance from the optic nerve for each of the experimental conditions above (n=6). D. Representative ONL images of central superior were taken from OCT; vertical lines show the thickness of the ONL layer. E. The mean thickness of OCT measurement in central superior and inferior hemispheres of the retina in microns is presented ± SEM (n=6). Lines align the RIS or OPL of different panels. Retinal layers are labeled, and the horizontal bars represent 25 microns. * indicates a significant difference from no LD, and # indicates a significant difference from light damage at P < 0.05 by one-way ANOVA and a Student t-test.
Fig. 3. Light induced Zn2+ accumulation and retinal damage were attenuated by ZnT3 KO.

A. LD was performed on ZnT3 mice in a C57/Bl6 RPE65Met/Met background and retinas were analyzed 4h after 50h of LD. Zn2+ accumulation (white regions) was assessed by ZP1 staining. Representative photomicrographs (n = 4) were taken of the mid superior regions of the retina at 0.3 second exposure. Notice the decrease in number and intensity of Zn2+ staining in superior retina after LD exposure in ZnT3 KO retina. B. Male littermates retinas were analyzed 7 days after LD by plastic sectioning cutting from eyes along a superior to inferior meridian encompassing the optic nerve and stained with 0.1% Toluidine Blue. Photomicrographs were taken of the mid superior regions of the retina, and the dotted lines show the alignment of the retinal layers between the panels. C. ONL nuclei counted at increasing distance from the optic nerve were averaged and plotted as a function of distance from the optic nerve for each of the experimental conditions above (n=6). * indicates a significant difference from control, and # indicates a significant difference from light damage at P < 0.05 by one-way ANOVA and a Student t-test. Layers are as marked. Bar represents 25 microns.
Fig. 4. Light induced Zn2+ accumulation was attenuated in retina of RPE65 KO mice.

LD was performed on RPE65 KO mice in a C57/Bl6/J RPE65Met/Met background and retinas were analyzed 4h after 50h of LD. Zn2+ accumulation (white regions) was assessed by ZP1 staining. Representative photomicrographs (n = 4) were taken of the mid superior regions of the retina at 0.3 second exposure. Notice the decrease in number and intensity of Zn2+ staining in superior retina after LD exposure in RPE65 KO retina (bracket). Layers are as marked, and lines show alignment. Bar represents 25 microns.
2.4. Rat light damage model
Rats were dark adapted for 60h before light exposure. Pupils were dilated with 1% tropicamide ophthalmic solution USP in room light and exposed to bright cool white fluorescent light from 8× 20W-circular fluorescent bulbs (18,000 Lux) for 4h at <25C (starting at 8:30am) (Gordon et al., 2002). The animal chamber was ventilated and gently rotated during the light exposure to ensure temperature maintenance and that the animals were awake with their eyes open. This was followed by recovery in the dark for 24h. After this, they were returned to cyclic, dim (30 Lux) overhead fluorescent light environment for six days. All studies were conducted within guidelines established by the Institutional Animal Care and Use Committee (Louisiana State University Health Sciences Center, New Orleans), and were in accordance with the PHS Guide for the Care and Use of Laboratory Animals, USDA Regulations, and the AVMA Panel on Euthanasia guidelines.
2.5. Genetically manipulated mice
ZnT3-KO, and RPE65-KO (originally prepared in a hybrid 129/Sv background) were backcrossed into C57/BI6/J for at least 13 generations. We used wild-type C57/BI6/J as the controls. The ZnT3-KO mice were kindly supplied by Dr. Palmiter (University of Washington). The RPE65-KO mice were from Jackson laboratories (Bar Harbor, ME) and contained the rd12 spontaneous mutant which was bred to C57/Bl6/J.
2.6. Mouse light damage model
C57/Bl6/J, ZnT3-KO, and RPE65-KO mice were all in the C57/Bl6/J background and were used at 7 weeks of age. They were acclimated for 5 days to a cyclic, dim overhead fluorescent light (30 Lux) followed by a 70h dark adaption. Pupils were dilated with 1% tropicamide ophthalmic solution USP under red light illumination and then exposed to white fluorescent light from 8× 17W fluorescent bulbs using a mirrored reflector (70 kLux) at <25C for 50 h (starting at 7:30pm). The animal chamber was ventilated and gently rotated during the light exposure as above. Diet gel (Clear H2O; Portland, ME) was given during the light exposure to make sure the mice were fed and hydrated. After light exposure, mice were maintained totally in the dark for 7d until retinas were examined. This paradigm was necessary to induce substantial mid-superior photoreceptor loss in these light resistant mouse strains (Bai et. al., 2012, co-submission).
2.7. Optical Coherence Tomography (OCT)
OCT is an optical signal acquisition and processing method providing extremely high-quality, micrometer-resolution, three-dimensional images from within optical scattering media (Spectralis, Heidelberg Engineering; Heidelberg, Germany) (Knott et al., 2011). On the seventh day after light damage, rats and mice were anesthetized with ketamine and xylazine and OCT was performed to measure the thickness of the mid superior outer nuclear layer (ONL). A real time eye tracker was used to couple confocal Scanning Laser Ophthalmoscopy (cSLO) and spectral domain-OCT (SD-OCT) scanners to position and stabilize the OCT scan on the retina. Scaling X was 3.24–3.31 μm/pixel; scaling Z was 3.87 μm/pixel. The built-in scale bar was used when performing OCT analysis. The thickness of the ONL was measured from the bottom edge of the outer plexiform layer to the top edge of the RIS at 3 points of the mid superior region and averaged.
2.8. Retinal Histology
After OCT, rats and mice were sacrificed by CO2 asphyxiation. Eyes were fixed in 2% formaldehyde/2% glutaraldehyde, and cut in half along a superior-to-inferior meridian through the center of the optic nerve. After a one-hour fixation period in 1% osmium tetroxide and sequential dehydration in ethanol, eyes were embedded in plastic resin (Electron Microscopy Systems; Hatfield, PA). Retinal sections of 1.5 microns were cut, mounted on glass slides and stained with 0.1% toluidine blue for 2 mins. The number of ONL nuclei was counted on sections from 8–12 different retinas at increasing distances from the optic nerve on the superior-to-inferior meridian. Pictures were taken in the area of mid-superior hemispheres.
2.9. Retinal Zn2+ staining
Eyes of Sprague Dawley rats, ZnT3-KO, RPE65-KO mice and C57/BI6/J control mice were collected 0–12h after light exposure under red light illumination. Zn2+ staining started to appear from 0–4h after LD, and was reduced by 14–24h. The peak varies from albino rats to pigmented mice (1–12 hr, data not shown). Fresh frozen cryostat sections (10 microns) were prepared, dried, and stained with 5μM ZinPyr-1 (ZP1, TefLabs, Galveston, TX) for 2 min, washed with PBS, and photomicrographs were taken immediately using the exposure times indicated (ex: 480nm; em: 530nm). ZP1 has high specificity for releasable zinc in secretory granules (KD=0.7nM). It is stably-lipophilic, and thus penetrates subcellular organelles such as the outer segments. It increases many fold in fluorescence when binding to Zn2+ (or other transition metals), but does not fluoresce in response to Mg2+, or Ca2+ (Burdette et al., 2001; Chang et al., 2004; Giblin et al., 2006). There was no autofluorescence at this wavelength either basally or after light damage, and the ZP1 staining was prevented by pretreatment of the section with the Zn2+ specific chelator, N,N,N'N'-tetrakis(−)[2-pyridylmethyl]-ethylenediamine (TPEN) (data not shown).
2.10. NAD+ measurements
The effect that Zn2+ had on NAD+ levels in PRC cultures was determined at 3hrs of Zn2+ exposure. For pigmented RPE65Met450 mice, it took 50hrs to light damage the retinas. The peak of Zn2+ was between 0 and 4 h post-LD. Therefore, the effect that LD had on NAD+ levels in ZnT3- KO, and cytNMNAT1 Tg retinal tissues was determined at 12hrs which was prior to the onset of cell death. Curved forceps were gently inserted into back of the eye, by cutting the lens in front and simultaneously pulling upwards on the back of the eye, retinas were rapidly isolated. Retinas or PRC cultures were then immediately lysed in NaOH/EDTA lysis buffer. Aliquots were acidified and heated to destroy NADH. These lysates were then neutralized and stored at −80°C for up to 1 year. The lysates generated were used for enzymatic cycling determinations of NAD+ using the malate dehydrogenase/ alcohol dehydrogenase cycling pair (Lin et al., 2001; Cai et al., 2006). The malate produced was then quantitatively converted to NADH, and measured fluorimetrically (excitation at 365nm, emission monitored at 460nm). The dynamic range of this cycling assay can be varied (10−15 to 10−9 moles) by the amount of cycling enzymes used and the duration of the cycling time. The results obtained are compared to a calibration curve, and normalized to protein content.
2.11. Data analysis and statistics
The numbers in each experiment were performed with an n=6–12 from 2–3 experiments. Means ± SEM were plotted and analyzed for significance using a one-way ANOVA followed by a Student t-test with significance achieved by P< 0.05.
2.12. Reagents
All materials were purchased from Sigma Chemical Co. (Saint Louis, MO) unless otherwise stated.
3. Results
3.1. Zn2+ induces an increase in [Zn2+]i, toxicity, and NAD+ loss in PRC
PRC were exposed to Zn2+ in the presence or absence of pyruvate, nicotinamide, or NAD+ and [Zn2+]i (A), toxicity (B), and NAD+ levels (C) were determined (Figure 1). [Zn2+]i was increased by 150 μM Zn2+ in PRC; toxicity was induced which was significantly attenuated by pyruvate, nicotinamide and NAD+. Only pyruvate and NAD+ (not shown) significantly restored NAD+ levels in PRC cultures.
3.2. Light induced Zn2+ staining was attenuated by ZnR
We have previously used this diet paradigm to depress Zn2+ levels in the brain and pancreas and have shown that it is reversible (Sheline et. al., 2012 J. Nutrition, accepted and Suh et al., 2009; Sheline et al., 2010b). LD was produced in rats fed the diets indicated for 3 wks prior. Retinas were analyzed for Zn2+ staining 1h after LD. There was an increase in Zn2+ staining in the cell bodies of photoreceptors after light exposure, which was attenuated by ZnR. These are the regions that are specifically sensitive to light exposure. The staining of the ROS is variable, and zinc deficiency was shown to induce ER stress in brain and paradoxically increase Zn2+ staining (Stoltenberg et al., 2007). The number of photoreceptors which stained for Zn2+ was significantly higher in the 61 mg/kg (ppm) diet compared to no LD controls, and was significantly reduced by ZnR (Figure 2A and Table 1).
Table 1.
LD induced Zn2+ staining of photoreceptors which was attenuated by ZnT3 and RPE65 knockout, and 2 ppm Zn2+ dieta.
| Condition | Number of Zn2+ Stained Photoreceptors/Field |
|---|---|
| No LD Control +1h | 0.2 ± 0.1 |
| 2ppm Zn2+ Diet-4hLD + 1h | 25.3 ± 2.7*# |
| 61ppm Zn2+ Diet-4hLD +1h | 58.1 ± 2.0# |
| B6-basal (No LD) | 0.3 ± 0.1 |
| B6-50hLD +0h | 85.0 ± 3.6# |
| B6-50hLD +4h | 58.3 ± 2.1# |
| ZnT3-KO no LD | 0.3 ± 0.2 |
| ZnT3-KO-50hLD +4h | 10.4 ± 2.6*# |
| RPE65-KO no LD | 1.5 ± 0.3 |
| RPE65-KO 50hLD +4h | 1.2 ± 0.5* |
sections from the indicated animals were stained for Zn2+ using ZP1, and stained cells in the mid superior photoreceptor layer fields were counted (n=12).
indicates a significant difference from no LD control at P < 0.05, n=12.
indicates a significant difference from LD control at P < 0.05, n=12.
3.3. Light induced retinal damage was attenuated by ZnR
The number of photoreceptors remaining was determined in male littermates 7 days after LD. ZnR attenuated damage to both superior and inferior retina, as shown by OCT and plastic sectioning (Figures 2B–E). Rats fed with 61 mg/kg purified Zn2+ diet showed complete loss in mid-superior retinas and severe loss in inferior retinas, whereas 2 mg/kg purified Zn2+ diet showed a less severe and smaller range of damage. Feeding 2 mg/kg Zn2+ for 2 wks followed by 1 wk of 61 mg/kg Zn2+ restored sensitivity to LD (data not shown). The numbers of photoreceptor nuclei at increasing distances superior or inferior to the optic nerve were plotted as a spider graph which is shown in figure 2C. Quantitations of mid retinal ONL thickness, measured as the distance between the RIS and the OPL were made by OCT (Figures 2D & 2E). In this study, 61 mg/kg purified Zn2+ diet rats were set as the controls for 2 mg/kg purified Zn2+ diet rats. We found that although rats fed a 61 mg/kg purified Zn2+ diet had similar photoreceptor Zn2+ staining as rats fed the normal Zn2+ diet (contains 61 mg/kg Zn2+), they showed more severe damage than the normal Zn2+ diet rats (Figure 2, and (Sheline et al., 2010a)). This may be because the normal diet contains vitamin A, carotenoids (lutein and zeaxanthin) whereas the 2 mg/kg purifed Zn2+ diet does not. Those supplements serve as important precursors of all-trans-retinol for vision generation and antioxidants for irradiant protection.
3.4. Light induced Zn2+ accumulation and retinal damage were attenuated, and NAD+ levels were not significantly reduced in ZnT3 KO mice
LD was performed on retinas from ZnT3 mice in a C57/Bl6/J RPE65Met/Met background and analyzed 4h (Zn2+, Figure 3A), or 7 days (death, Figure 3B–C) after 50h of LD. ZnT3 KO mice showed little photoreceptor Zn2+ staining and damage compared to B6 control mice (quantified in Table 1). Photoreceptors were counted and plotted in figure 3C. NAD+ levels in ZnT3-KO mice after LD were 17.9 ± 1.1 nmols/mg of protein which was not significantly different than ZnT3-KO no LD retinas, or C57/Bl6 no LD control mice (19.3 ± 0.7, and 22.8 ± 0.8, respectively, n=6). NAD+ levels were significantly reduced in C57/Bl6 mice after LD (Bai et. al., 2012, co-submission).
3.5. LD induced Zn2+ accumulation was reduced in retina of RPE65 KO mice
RPE65 knockout mice can not reisomerize all trans-retinol resulting in a block in rhodopsin recycling. RPE65 KO mice are resistant to LD at 6–10 weeks of age, and develop spontaneous retinal degeneration starting at 10 weeks of age (Redmond et al., 1998; Grimm et al., 2000). LD was performed on 7 week old RPE65 KO mice and Zn2+ staining was analyzed 4h after 50h of LD. We showed a lack of Zn2+ staining in photoreceptors after 50h LD in 7 week old RPE65 KO mice compared to C57/Bl6/J control mice (Figure 4, and Table 1).
4. Discussion
We previously showed that intense light can induce early Zn2+ accumulation in rats, specifically in severely damaged superior retinal layers, including RPE cells, ROS, OPL and especially ONL. This early (before cell loss), preferential Zn2+ accumulation suggested a role for Zn2+ toxicity in light-induced damage (Sheline et al., 2010a). In the present study, we find that: 1) Zn2+ causes toxicity in primary retinal cultures dependent on Zn2+ accumulation and loss of NAD+ levels which is attenuated by pyruvate, nicotinamide, and NAD+. 2) a reduced Zn2+ diet (2 mg/kg) attenuated photoreceptor degeneration after LD in albino rats compared to normal Zn2+ diet (61 mg/kg). The number of photoreceptors staining for Zn2+ was reduced in the rats receiving 2 mg/kg diets 1h after LD. 3) ZnT3-KO mice showed less Zn2+ staining and death in photoreceptor and RPE cells than those of C57/BI6/J control mice after light exposure. 4) NAD+ levels after the LD were not significantly reduced in ZnT3-KO mice. 5) RPE65-KO mice are resistant to light damage, and showed no RPE or photoreceptor Zn2+ staining after light exposure compared to C57/Bl6/J Zn2+ staining.
Zn2+ is involved in retinal health and the importance of Zn2+ is being demonstrated in an increasing number of disease conditions in both animals and humans. Excess Zn2+ accumulation has been shown to be toxic in retinal ischemia (Yoo et al., 2004; Choi et al., 2006), global ischemia (Koh et al., 1996), trophic deprivation (Sheline et al., 2010b) and hypoglycemia mediated neuronal death (Suh et al., 2004; Suh et al., 2008). Acute Zn2+ chelation or Zn2+ therapeutics were efficacious (Lee et al., 2001; Choi et al., 2006; Sheline et al., 2010b). Also, in Alzheimer disease (AD), Zn2+ increases the aggregation of beta-amyloid protein (Lee et al., 1999; Lynch et al., 2000; Lee et al., 2002). Knocking out Zn2+ transporter protein (ZnT3) was found to significantly decrease beta-amyloid plaque formation (Lee et al., 2002). The metal chelating agents desferrioxamine and clioquinol have been suggested as potential therapeutics for AD (Finefrock et al., 2003).
In the present study, we further examined the role of Zn2+ in light induced retinal damage. Previous studies showed that retinal cell-line cultures were susceptible to Zn2+, and oxygen radicals causing Zn2+ accumulation, NAD+ loss, glycolytic inhibition and death that was attenuated by pyruvate, nicotinamide, and NAD+ (Sheline et al., 2010a) (accompanying NAD+ paper). We now demonstrate that PRC cultures (Figure 1) are similarly affected by Zn2+, though nicotinamide is less effective at restoring NAD+. Retinal neurons require high metabolic energy (ATP) from glycolysis to survive (Winkler, 1981), and GAPDH has been shown to be enriched on retinal outer segment membranes (Hsu and Molday, 1990). We went on to examine the affects of a reduced Zn2+ diet in vivo. This diet can significantly decrease the total Zn2+ content in the brain, and switching back to a Zn2+ adequate diet can restore Zn2+ levels (Takeda et al., 2003; Suh et al., 2009). The Zn2+ transporters involved in organismal Zn2+ homeostasis have been studied, and their expression profiles in different cell types, cellular localization, and response to dietary Zn2+ have been studied. Vision deficits have not been reported due to this dietary restriction (reviewed in Lichten and Cousins, 2009). Here we showed a reduced Zn2+ diet (2 mg/kg) attenuated photoreceptor Zn2+ staining and death after light exposure in albino rats compared to a diet containing normal levels of Zn2+ (61 mg/kg). Feeding 2 mg/kg Zn2+ for 2 wks followed by 1 wk of 61 mg/kg Zn2+ restores sensitivity to LD (data not shown), suggesting specificity of these effects for Zn2+ over effects this diet could have on uptake of other metals. Our studies were performed on an acute dietary reduction of only 3 wks to minimize the potential complications such as defects in rhodopsin synthesis which requires a longer term Zn2+ deficiency (Dorea and Olson, 1986).
In addition to dietary manipulations to alter Zn2+ levels, we also tested strains of mice that have genetic manipulations which reduce retinal Zn2+ levels. Genetic strains that increase retinal Zn2+ levels are not available. Unlike rats, susceptibility of light damage in mouse is determined genetically by the expression levels of a retinal pigment epithelial protein RPE65 (Danciger et al., 2000; Wenzel et al., 2001; Iseli et al., 2002). RPE65 is an essential enzyme for the visual cycle, catalyzing the conversion of all-transretinyl esters to 11-cis retinal for rhodopsin regeneration. Substitution of leucine by methionine at position 450 of the RPE65 protein (RPE65Met450) reduced RPE65 expression, and decreased the rate of rhodopsin regeneration after bleaching. This caused a lower susceptibility towards light exposure compared to RPE65Leu450, demonstrating that rapid visual cycling increases LD (Wenzel et al., 2001). Similarly, RPE65 KO mice are resistant to LD if examined prior to spontaneous retinal degeneration which starts at 10 weeks of age (Redmond et al., 1998; Grimm et al., 2000). The C57/BI6/J control and ZnT3-KO mice in this study are all pigmented RPE65Met450 mice. To achieve light damage in these resistant mice required modifications to the LD protocol (Bai et. al., 2012, co-submission).
Zn2+ is present physiologically in different layers of the retina. The expression and localization of Zn2+ transporters suggest that Zn2+ transporter 7, (ZnT7, slc30a7), and ZnT3 (slc30a3), as well as ZIP transporters (slc39a1–16) (Wang et al., 2006; Leung et al., 2008) could be important sources for physiologic or patho-physiologic Zn2+ in the eye (reviewed in Ugarte and Osborne, 2001). ZnT3 is expressed in specific layers in the retina. In inner and outer plexiform layers, it is associated with synaptic interactions. In photoreceptor inner segments and outer limiting membrane, it co-localizes with the Zn2+ staining pattern after light adaptation. ZnT3 has also been found to be expressed in Müller cells which transverse throughout all of the retinal layers and may be involved in Zn2+ homeostasis (Redenti and Chappell, 2004, 2007). ZnT3 KO mice have a 20% reduction in total Zn2+ in brain regions where histochemically reactive Zn2+ is usually detected, and were not reported to have vision deficits (Cole et al., 1999). ZnT3 KO mice have reduced neuronal injury after LD, rescuing more than half of the photoreceptors in the superior retina (Figure 3). Injury in ZnT3 KO mice is also reduced after global ischemia and hypoglycemia (Sheline and Wei, unpublished observation, and (Suh et al., 2008)). We also showed in this study that NAD+ levels in ZnT3-KO mice were not significantly reduced after LD, as expected due to the lack of Zn2+ toxicity. This suggests that ZnT3-dependent Zn2+ is released and accumulates in photoreceptors contributing to their loss. This Zn2+ appears in perikaya of photoreceptors especially after a dark/depolarized period. This results in NAD+ loss causing potentiation of metabolic inhibition and cell loss.
Rhodopsin regeneration is positively correlated to severity of light damage (Grimm et al., 2000). Zn2+ has been shown to bind to rhodopsin at a high affinity site comprised of Glu122 (at the end of transmembrane region 3, TM 3) and His211 (at the end of transmembrane region 5, TM 5) to stabilize the inactive rhodopsin structure in the dark (Shuster et al., 1992; Stojanovic et al., 2004). Additional Zn2+ ions (>1) binding at low affinity sites His100 and His195 are required for destabilizing the structure of Rho in RPE cells where [Zn2+]i is high, potentially allowing its degradation (Gleim et al., 2009). Light induces changes in rhodopsin structure that moves TM 3 and TM 5 away from each other, increasing the tetrahedral binding distance for Zn2+ which is inconsistent with continued Zn2+ binding. We therefore predict rhodopsin could serve as a pool for Zn2+ release after injuries. A single episode of complete photobleaching of rhodopsin does not induce retinal degeneration, but block of rhodopsin recycling does prevent retinal degeneration. It is the repeated photobleaching and rhodopsin recycling that is required to induce retinal degeneration induced by light damage. Others have also shown that the visual cycle must be allowed to cycle in order to induce damage. Therefore, we postulate that it is the repeated cycling that is required to induce zinc accumulation in photoreceptors which contributes to their death (Figures 3 and 4). After extensive bleaching, the initial rate of rhodopsin regeneration in control rats and zinc-deficient rats was the same, and no vision deficits were noted. The only difference was that the extent of rhodopsin regeneration in control rats kept in the dark for 2 hrs was higher than that in zinc-deficient rats. The explanation is that the rhodopsin concentration is lower in ROS in zinc-deficient rats (Dorea and Olson, 1986). Therefore, manipulating Zn2+ levels may not affect rhodopsin's capability to absorb photons or to regenerate. Zn2+ restriction may reduce the rhodopsin pool available to store zinc, which in turn reduces zinc release and cell death.
RPE65 protein is required for rhodopsin regeneration. RPE65-KO (no RPE65 protein) prevents the production of 11-cis-retinal and rhodopsin regeneration (Grimm et al., 2000). Therefore, in RPE65-KO mice less cycling occurs resulting in less Zn2+ release and staining, and less LD. Our findings that light exposure does not induce an increase in photoreceptor Zn2+ staining in RPE65-KO mice compared to wildtype mice supports this possibility, as shown in Figure 4 and Table 1. We postulate that ZnT3-, rhodopsin-, and oxidation-dependent release of zinc each contribute to the zinc accumulation, and light-induced damage.
These studies suggest: 1) Zn2+ toxicity in PRC may be mediated by reduced NAD+ levels, consistent with previous studies in other neuronal culture, 2) reduced dietary Zn2+ reduces [Zn2+]i leading to reduced light damage, 3) rhodospin recycling and release of bound Zn2+ is a possible source of excessive [Zn2+]i, as is ZnT3-dependent stores. Zn2+ toxicity is partially mediated by reducing NAD+ levels in cortical and retinal neuronal cell death. We therefore have examined the efficacy of nicotinamide mononucleotide adenyl-transferase-1 (NMNAT1, an NAD+ synthetic enzyme) overexpression on reducing LD and increasing NAD+ levels in a companion manuscript (Bai et. al., 2012, co-submission).
Zn2+ accrual and NAD+ loss induce PRC toxicity which is reduced by P, N, and NAD+.
A Zn2+ reduced diet reduced photoreceptor Zn2+ staining and degeneration after LD.
ZnT3-KO mice have less Zn2+ staining and photoreceptor and RPE cell death after LD.
NAD+ levels after the LD were not significantly reduced in ZnT3-KO mice.
RPE65-KO mice are resistant to LD, and have no photoreceptor Zn2+ staining after LD.
Acknowledgements
This work was supported by NIH NIDDK grant #073446 to C.T.S., RPB and Lions Eye grants to the department, and by departmental funds. We would like to thank Dr. Nicolas Bazan and his lab for their help, and Dr. Minghao Jin for the RPE65 mice, and helpful discussions.
Abbreviations
- (TPEN)
d-N,N,N'N'-tetrakis(-)[2-pyridylmethyl]-ethylenediamine
- (GCL)
ganglion cell layer
- (LD)
light damage
- (INL)
inner nuclear layer
- (ONL)
outer nuclear layer
- (IPL)
inner plexiform layer
- (OPL)
outer plexiform layer
- (ROS)
rod outer segments
- ([Zn2+]i)
intracellular zinc concentration
- (OS)
oxidative stress
- (ppm)
part per million = mg/kg
- (ZnR)
reduced zinc diet
- (ZnE)
supplemented zinc diet
- (RP)
retinitis pigmentosa
- (ZnT3)
Zinc transporter 3
- (ZP1)
ZinPyr-1
Footnotes
Aspects of this paper were presented at the 2011 annual meeting of the Association for Research in Vision and Ophthalmology in Ft. Lauderdale, FL; and at the 2010 and 2011 Society for Neuroscience annual meetings in San Diego, CA, and Washington DC.
Author Contributions C.T.S. is the guarantor of this manuscript and had primary responsibility for research design and conduct, writing, and final content. S.B. designed and performed research, collected data, and revised and reviewed the final manuscript. Y.Z. and C.R.S. performed research, collected data, and reviewed the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure The authors have no disclosures or conflicts to be reported.
References
- Bai S, Sheline CT. NAD+ maintenance attenuates light induced photoreceptor degeneration. Exp. Eye. Res. 2012 doi: 10.1016/j.exer.2012.12.007. in press, http://dx.doi.org/10.1016/j.exer.2012.12.007. [DOI] [PMC free article] [PubMed]
- Battelle BA, LaVail MM. Rhodopsin content and rod outer segment length in albino rat eyes: modification by dark adaptation. Exp Eye Res. 1978;26:487–497. doi: 10.1016/0014-4835(78)90134-3. [DOI] [PubMed] [Google Scholar]
- Bicknell IR, Darrow R, Barsalou L, Fliesler SJ, Organisciak DT. Alterations in retinal rod outer segment fatty acids and light-damage susceptibility in P23H rats. Mol Vis. 2002;8:333–340. [PubMed] [Google Scholar]
- Burdette SC, Walkup GK, Spingler B, Tsien RY, Lippard SJ. Fluorescent sensors for Zn(2+) based on a fluorescein platform: synthesis, properties and intracellular distribution. J Am Chem Soc. 2001;123:7831–7841. doi: 10.1021/ja010059l. [DOI] [PubMed] [Google Scholar]
- Cai AL, Zipfel GJ, Sheline CT. Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway. Eur J Neurosci. 2006;24:2169–2176. doi: 10.1111/j.1460-9568.2006.05110.x. [DOI] [PubMed] [Google Scholar]
- Canzoniero LM, Turetsky DM, Choi DW. Measurement of intracellular free zinc concentrations accompanying zinc- induced neuronal death. J Neurosci. 1999;19:RC31. doi: 10.1523/JNEUROSCI.19-19-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzoniero LM, Manzerra P, Sheline CT, Choi DW. Membrane-permeant chelators can attenuate Zn2+-induced cortical neuronal death. Neuropharmacology. 2003;45:420–428. doi: 10.1016/s0028-3908(03)00171-0. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Nolan EM, Jaworski J, Burdette SC, Sheng M, Lippard SJ. Bright fluorescent chemosensor platforms for imaging endogenous pools of neuronal zinc. Chem Biol. 2004;11:203–210. doi: 10.1016/j.chembiol.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Chappell RL, Anastassov I, Lugo P, Ripps H. Zinc-mediated feedback at the synaptic terminals of vertebrate photoreceptors. Exp Eye Res. 2008;87:394–397. doi: 10.1016/j.exer.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Kim KA, Yoon YJ, Fujikado T, Joo CK. Inhibition of cyclooxygenase-2 expression by zinc-chelator in retinal ischemia. Vision Res. 2006;46:2721–2727. doi: 10.1016/j.visres.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Codenotti M, Patelli F, Brancato R. OCT findings in patients with retinopathy after watching a solar eclipse. Ophthalmologica. 2002;216:463–466. doi: 10.1159/000067540. [DOI] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A. 1999;96:1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortina MS, Gordon WC, Lukiw WJ, Bazan NG. Light-induced photoreceptor damage triggers DNA repair: differential fate of rods and cones. Adv Exp Med Biol. 2003;533:229–240. doi: 10.1007/978-1-4615-0067-4_29. [DOI] [PubMed] [Google Scholar]
- Danciger M, Matthes MT, Yasamura D, Akhmedov NB, Rickabaugh T, Gentleman S, Redmond TM, La Vail MM, Farber DB. Mamm Genome. United States: 2000. A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors; pp. 422–427. [DOI] [PubMed] [Google Scholar]
- Dorea JG, Olson JA. The rate of rhodopsin regeneration in the bleached eyes of zinc-deficient rats in the dark. J Nutr. 1986;116:121–127. doi: 10.1093/jn/116.1.121. [DOI] [PubMed] [Google Scholar]
- Finefrock AE, Bush AI, Doraiswamy PM. J Am Geriatr Soc. United States: 2003. Current status of metals as therapeutic targets in Alzheimer's disease; pp. 1143–1148. [DOI] [PubMed] [Google Scholar]
- Fuller D, Machemer R, Knighton RW. Retinal damage produced by intraocular fiber optic light. Am J Ophthalmol. 1978;85:519–537. doi: 10.1016/s0002-9394(14)75250-x. [DOI] [PubMed] [Google Scholar]
- Gee KR, Zhou ZL, Ton-That D, Sensi SL, Weiss JH. Measuring zinc in living cells. A new generation of sensitive and selective fluorescent probes. Cell Calcium. 2002;31:245–251. doi: 10.1016/S0143-4160(02)00053-2. [DOI] [PubMed] [Google Scholar]
- Giblin LJ, Chang CJ, Bentley AF, Frederickson C, Lippard SJ, Frederickson CJ. Zinc-secreting Paneth Cells Studied by ZP Fluorescence. J Histochem Cytochem. 2006;54:311–316. doi: 10.1369/jhc.5A6724.2005. [DOI] [PubMed] [Google Scholar]
- Gleim S, Stojanovic A, Arehart E, Byington D, Hwa J. Conserved rhodopsin intradiscal structural motifs mediate stabilization: effects of zinc. Biochemistry. 2009;48:1793–1800. doi: 10.1021/bi800968w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WC, Casey DM, Lukiw WJ, Bazan NG. DNA damage and repair in light-induced photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2002;43:3511–3521. [PubMed] [Google Scholar]
- Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat Genet. 2000;25:63–66. doi: 10.1038/75614. [DOI] [PubMed] [Google Scholar]
- Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. Tenth Edition. Molecular Probes; Eugene, OR: 2005. [Google Scholar]
- Hsu SC, Molday RS. Glyceraldehyde-3-phosphate dehydrogenase is a major protein associated with the plasma membrane of retinal photoreceptor outer segments. J Biol Chem. 1990;265:13308–13313. [PubMed] [Google Scholar]
- Iseli HP, Wenzel A, Hafezi F, CE RE, Grimm C. Exp Eye Res. England: 2002. Light damage susceptibility and RPE65 in rats; pp. 407–413. [PubMed] [Google Scholar]
- Jain A, Desai RU, Charalel RA, Quiram P, Yannuzzi L, Sarraf D. Solar retinopathy: comparison of optical coherence tomography (OCT) and fluorescein angiography (FA) Retina. 2009;29:1340–1345. doi: 10.1097/IAE.0b013e3181b0da88. [DOI] [PubMed] [Google Scholar]
- Knott EJ, Sheets KG, Zhou Y, Gordon WC, Bazan NG. Exp Eye Res. 2010 Elsevier Ltd.; England: 2011. Spatial correlation of mouse photoreceptor-RPE thickness between SD-OCT and histology; pp. 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Kuhn F, Morris R, Massey M. Photic retinal injury from endoillumination during vitrectomy. Am J Ophthalmol. 1991;111:42–46. doi: 10.1016/s0002-9394(14)76894-1. [DOI] [PubMed] [Google Scholar]
- Lee JY, Mook-Jung I, Koh JY. Histochemically reactive zinc in plaques of the Swedish mutant beta-amyloid precursor protein transgenic mice. J Neurosci. 1999;19:RC10. doi: 10.1523/JNEUROSCI.19-11-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim YH, Koh JY. Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci. 2001;21:RC171. doi: 10.1523/JNEUROSCI.21-20-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci U S A. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KW, Liu M, Xu X, Seiler MJ, Barnstable CJ, Tombran-Tink J. Expression of ZnT and ZIP zinc transporters in the human RPE and their regulation by neurotrophic factors. Invest Ophthalmol Vis Sci. 2008;49:1221–1231. doi: 10.1167/iovs.07-0781. [DOI] [PubMed] [Google Scholar]
- Li F, Cao W, Anderson RE. Alleviation of constant-light-induced photoreceptor degeneration by adaptation of adult albino rat to bright cyclic light. Invest Ophthalmol Vis Sci. 2003;44:4968–4975. doi: 10.1167/iovs.03-0140. [DOI] [PubMed] [Google Scholar]
- Lichten LA, Cousins RJ. Mammalian zinc transporters: nutritional and physiologic regulation. Annu Rev Nutr. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. [DOI] [PubMed] [Google Scholar]
- Lin SS, Manchester JK, Gordon JI. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem. 2001;276:36000–36007. doi: 10.1074/jbc.M103509200. [DOI] [PubMed] [Google Scholar]
- Liu C, Peng M, Laties AM, Wen R. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci. 1998;18:1337–1344. doi: 10.1523/JNEUROSCI.18-04-01337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch T, Cherny RA, Bush AI. Exp Gerontol. England: 2000. Oxidative processes in Alzheimer's disease: the role of abeta-metal interactions; pp. 445–451. [DOI] [PubMed] [Google Scholar]
- Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010;29:113–134. doi: 10.1016/j.preteyeres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci. 2003;44:486–492. doi: 10.1167/iovs.02-0708. [DOI] [PubMed] [Google Scholar]
- Penn JS, Naash MI, Anderson RE. Effect of light history on retinal antioxidants and light damage susceptibility in the rat. Exp Eye Res. 1987;44:779–788. doi: 10.1016/s0014-4835(87)80041-6. [DOI] [PubMed] [Google Scholar]
- Ranchon I, LaVail MM, Kotake Y, Anderson RE. Free radical trap phenyl-N-tertbutylnitrone protects against light damage but does not rescue P23H and S334ter rhodopsin transgenic rats from inherited retinal degeneration. J Neurosci. 2003;23:6050–6057. doi: 10.1523/JNEUROSCI.23-14-06050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redenti S, Chappell RL. Localization of zinc transporter-3 (ZnT-3) in mouse retina. Vision Res. 2004;44:3317–3321. doi: 10.1016/j.visres.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Redenti S, Chappell RL. Muller cell zinc transporter-3 labeling suggests a role in outer retina zinc homeostasis. Mol Med. 2007;13:376–379. doi: 10.2119/2007-00041.Redenti. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redenti S, Ripps H, Chappell RL. Zinc release at the synaptic terminals of rod photoreceptors. Exp Eye Res. 2007;85:580–584. doi: 10.1016/j.exer.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- Richards MJ, Nagel BA, Fliesler SJ. Lipid hydroperoxide formation in the retina: correlation with retinal degeneration and light damage in a rat model of Smith-Lemli-Opitz syndrome. Exp Eye Res. 2006;82:538–541. doi: 10.1016/j.exer.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez IR, Fliesler SJ. Photodamage generates 7-keto- and 7-hydroxycholesterol in the rat retina via a free radical-mediated mechanism. Photochem Photobiol. 2009;85:1116–1125. doi: 10.1111/j.1751-1097.2009.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein FJ, Chappell RL. Endogenous zinc as a retinal neuromodulator: evidence from the skate (Raja erinacea) Neurosci Lett. 2003;345:81–84. doi: 10.1016/s0304-3940(03)00472-5. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J Neurosci. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline CT, Zhou Y, Bai S. Light-induced photoreceptor and RPE degeneration involve zinc toxicity and are attenuated by pyruvate, nicotinamide, or cyclic light. Mol Vis. 2010a;16:2639–2652. [PMC free article] [PubMed] [Google Scholar]
- Sheline CT, Cai AL, Zhu J, Shi C. Serum or target deprivation-induced neuronal death causes oxidative neuronal accumulation of Zn2+ and loss of NAD+ Eur J Neurosci. 2010b;32:894–904. doi: 10.1111/j.1460-9568.2010.07372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster TA, Nagy AK, Conly DC, Farber DB. Direct zinc binding to purified rhodopsin and disc membranes. Biochem J. 1992;282(Pt 1):123–128. doi: 10.1042/bj2820123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovic A, Stitham J, Hwa J. Critical role of transmembrane segment zinc binding in the structure and function of rhodopsin. J Biol Chem. 2004;279:35932–35941. doi: 10.1074/jbc.M403821200. [DOI] [PubMed] [Google Scholar]
- Stoltenberg M, Bush AI, Bach G, Smidt K, Larsen A, Rungby J, Lund S, Doering P, Danscher G. Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience. 2007;150:357–369. doi: 10.1016/j.neuroscience.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Suh SW, Garnier P, Aoyama K, Chen Y, Swanson RA. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol Dis. 2004;16:538–545. doi: 10.1016/j.nbd.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E, Liu J, Swanson RA. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci. 2003;23:10681–10690. doi: 10.1523/JNEUROSCI.23-33-10681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SW, Hamby AM, Gum ET, Shin BS, Won SJ, Sheline CT, Chan PH, Swanson RA. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. J Cereb Blood Flow Metab. 2008;28:1697–1706. doi: 10.1038/jcbfm.2008.61. [DOI] [PubMed] [Google Scholar]
- Suh SW, Won SJ, Hamby AM, Yoo BH, Fan Y, Sheline CT, Tamano H, Takeda A, Liu J. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J Cereb Blood Flow Metab. 2009;29:1579–1588. doi: 10.1038/jcbfm.2009.80. [DOI] [PubMed] [Google Scholar]
- Takeda A, Hirate M, Tamano H, Nisibaba D, Oku N. J Neurochem. England: 2003. Susceptibility to kainate-induced seizures under dietary zinc deficiency; pp. 1575–1580. [DOI] [PubMed] [Google Scholar]
- Thanos S, Heiduschka P, Romann I. Exposure to a solar eclipse causes neuronal death in the retina. Graefes Arch Clin Exp Ophthalmol. 2001;239:794–800. doi: 10.1007/s004170100362. [DOI] [PubMed] [Google Scholar]
- Ugarte M, Osborne NN. Zinc in the retina. Prog Neurobiol. 2001;64:219–249. doi: 10.1016/s0301-0082(00)00057-5. [DOI] [PubMed] [Google Scholar]
- Vaughan DK, Peachey NS, Richards MJ, Buchan B, Fliesler SJ. Light-induced exacerbation of retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Exp Eye Res. 2006;82:496–504. doi: 10.1016/j.exer.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojnikovic B, Micovic V, Coklo M, Vojnikovic D. Sun exposure and visual field damage among children on the Adriatic Island Rab--possible initial risk factor in development of age-related macular degeneration. Coll Antropol. 2009;33:747–749. [PubMed] [Google Scholar]
- Wang M, Lam TT, Tso MO, Naash MI. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci. 1997;14:55–62. doi: 10.1017/s0952523800008750. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang ZY, Gao HL, Danscher G, Huang L. Localization of ZnT7 and zinc ions in mouse retina--immunohistochemistry and selenium autometallography. Brain Res Bull. 2006;71:91–96. doi: 10.1016/j.brainresbull.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. J Neurosci. United States: 2001. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration; pp. 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler BS. Glycolytic and oxidative metabolism in relation to retinal function. J Gen Physiol. 1981;77:667–692. doi: 10.1085/jgp.77.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LP, Wu LM, Guo XJ, Tso MO. Activation of endoplasmic reticulum stress in degenerating photoreceptors of the rd1 mouse. Invest Ophthalmol Vis Sci. 2007;48:5191–5198. doi: 10.1167/iovs.07-0512. [DOI] [PubMed] [Google Scholar]
- Yoo MH, Lee JY, Lee SE, Koh JY, Yoon YH. Protection by pyruvate of rat retinal cells against zinc toxicity in vitro, and pressure-induced ischemia in vivo. Invest Ophthalmol Vis Sci. 2004;45:1523–1530. doi: 10.1167/iovs.03-1315. [DOI] [PubMed] [Google Scholar]
- Zigman S, Datiles M, Torczynski E. Sunlight and human cataracts. Invest Ophthalmol Vis Sci. 1979;18:462–467. [PubMed] [Google Scholar]