

Abstract
Background
Oxidative stress has been implicated as a possible mechanism for adverse health effects associated with traffic emissions. We examined the association of an estimate of traffic emissions with blood biomarkers of antioxidant capacity (glutathione, glutathione peroxidase, trolox-equivalent antioxidant capacity) and oxidative damage (thiobarbituric acid-reactive substances (TBARS)) among 1,810 healthy women, randomly selected from Erie and Niagara Counties in Western New York.
Methods
A geographic traffic emission and meteorological dispersion model was used to estimate annual polycyclic aromatic hydrocarbon (PAH) exposure from traffic emissions for each woman based on her residence at the time of study. Associations of traffic-related PAH exposure with measures of oxidative stress and antioxidant capacity were examined in multiple regression analyses with adjustment for potential confounders.
Results
Higher traffic-related PAH exposure was associated with decreased glutathione and increased glutathione peroxidase. Stronger associations between traffic-related PAH exposure and levels of glutathione and glutathione peroxidase were suggested among nonsmoking women without secondhand smoke exposure, especially among premenopausal nonsmoking women. Associations were also stronger for measurements made in warmer months.
Conclusions
These findings suggest that PAHs or other components of traffic emissions may impact anti-oxidative capacity among healthy women, particularly premenopausal non-smokers without secondhand smoke exposure.
Keywords: Traffic emissions, Polycyclic aromatic hydrocarbon, Oxidative stress
Introduction
Research in recent decades consistently indicates that outdoor air pollution adversely affects health and that air pollution stemming from transportation is an important contributor (World Health Organization, 2005). There is evidence that exposure to traffic emissions increases risk of asthma and other respiratory diseases as well as of cardiovascular diseases, lung cancer, breast cancer and childhood leukemia (Amigou et al., 2011; Crouse et al., 2010; Ito et al., 2010; Liao et al., 2011; Nie et al., 2007; Patel et al., 2010).
The underlying mechanisms behind traffic emission- related health effects are not well understood; it has been suggested that oxidative stress might be one possible mechanism (Laumbach and Kipen, 2010). Oxidative stress, defined as an imbalance between free radical production and protective radical scavenging antioxidants, can lead to damage of biologic macromolecules and dysregulation of normal metabolism and physiology (Terada, 2006). There is evidence that oxidative stress is important in the etiology of cardiovascular diseases, lung diseases and cancer (Olinski et al., 2002; Spector, 2000).
Traffic emissions contain a multitude of air contaminants with pro-oxidant properties. These include heavy metals, volatile organic compounds and polycyclic aromatic hydrocarbons (PAHs). PAHs, a class of organic compounds containing only carbon and hydrogen, and comprised of two or more fused aromatic rings (IARC, 1998; IARC, 2010) have been reported to increase production of free radicals and to possess carcinogenic, mutagenic and endocrine disrupting properties (Brunekreef et al., 2009; Kim and Lee, 1997; Kiruthiga et al., 2010). Motor vehicle exhaust is a major source of PAH exposure in urban areas (Zhang et al., 2006).
Several human studies have been conducted to examine PAH exposure and oxidative stress biomarkers in occupations with high PAH exposure and among children (Bae et al., 2010; Jeng et al., 2010; Rossner et al., 2008b; Singh et al., 2008; Wei et al., 2010; Wu et al., 2003). However, few studies have been conducted to specifically examine the health effects of traffic emissions on oxidative stress status among the general population.
In the present study, we assessed cross-sectional associations of an estimate of PAH exposure from traffic emissions with blood biomarkers of antioxidant capacity (glutathione, glutathione peroxidase, trolox-equivalent antioxidant capacity) and oxidative damage to lipids (thiobarbituric acid-reactive substances (TBARS)) among 1,810 healthy women, randomly selected from Erie and Niagara Counties in Western New York. We hypothesized that higher PAH exposure from traffic emissions would adversely affect antioxidant capacity and increase oxidative stress. Since tobacco smoke is another major source of PAHs, we further hypothesized that the associations of traffic-related PAH exposure with oxidation biomarkers might be more easily detected among never-smokers without secondhand smoke exposure. In addition, we hypothesized that pre- and post-menopausal women might differ in their responses to PAH exposure because of the estrogen-mimicking properties of some PAHs (Clemons et al., 1998).
Methods
Study population
We randomly selected 2,115 healthy women from the general population of Erie and Niagara counties in Western New York between 1996 and 2001. These women were originally selected as controls for a series of case-control studies including the Western New York Exposures and Breast Cancer study, described in detail elsewhere (Bonner et al., 2005; Nie et al., 2007). Briefly, eligible control participants were English-speaking, aged 35–79 years, who were current residents of Erie or Niagara counties, with no previous history of malignancy other than non-melanoma skin cancer. Women under age 65 years were selected from driver’s license rolls; those aged 65 years or older were selected from the rolls of the Health Care Financing Administration. In the original case-control study, the response rate for controls was 63% among those determined to be eligible. All participants provided written informed consent, and the study protocol was approved by the Institutional Review Board of the University at Buffalo.
For these analyses, we excluded individuals who had either missing measurement of blood oxidation biomarkers (n=287) or were missing the estimate of PAHs from traffic emissions (n=30), resulting in data from 1,810 women for the current analyses. The main reasons for missing blood biomarkers were lack of a blood sample or that the quantity of the sample collected was insufficient for assay. Missing traffic emissions estimates were the result either of lack of information about the participant’s residence at the time of study enrollment or inability to geocode the address. In comparisons of those excluded from these analyses to those included, body mass index and smoking status were not different. However excluded women tended to be nonwhite (16.38% versus 8.73%), older in age (mean age 62.72 versus 57.08 years) and less educated (12.75 versus 13.39 years of education) compared to those included.
Data collection
Information on demographics, reproductive history, smoking and secondhand smoke exposure, diet, alcohol consumption, non-steroidal anti-inflammatory drugs use, and residential history was collected through in-person interviews. Women were classified as current smokers if they had smoked more than 100 cigarettes in their lifetime and reported smoking at the time of interview. They were classified as former smokers if they had smoked more than 100 cigarettes in their lifetime but were not smokers at the time of interview. The most recent information on alcohol drinking, non-steroidal anti-inflammatory drugs use and vitamin supplement use collected in this study was for the period 12–24 months prior to the interview. Recent alcohol nondrinkers were defined as those who had had less than one drink per month during the 12 to 24 months prior to interview. Those who had never had at least 12 drinks of alcohol in any 12-month period were classified as lifetime nondrinkers. Recent non-steroidal anti-inflammatory drugs use, including aspirin and ibuprofen use, 12–24 months prior to the interview was categorized as non-users (0 days/month), infrequent users (≤14 days/month), and regular users (>14 days/month). Current height and weight at the time of interview were measured by trained interviewers according to a standardized protocol. Body mass index was calculated by dividing weight (in kilograms) by height (in meters) squared. Daily vitamin C and vitamin E intakes 12 to 24 months prior to interview were calculated by combining dietary and supplemental use. Dietary intakes were assessed through a modified version of the Health Habits and History food frequency questionnaire. Dietary vitamin C and vitamin E intakes were calculated from the food frequency questionnaire using the DietSys (version 3.7) nutrient analysis software utilizing food composition data from the United States Department of Agriculture (Block et al., 1986). Vitamin C and vitamin E supplement intakes were queried in the questionnaire. Those who reported no secondhand smoke exposure at home or at work in the past ten years were defined as being without secondhand smoke exposure. For assessment of occupational exposure, women were dichotomized based on their likelihood of being exposed to PAHs in the working environment. Included in the high exposure category were women who had ever had worked as coke oven workers, auto mechanics, professional drivers, or cooks. Season at time of interview was classified as follows: spring (March to May), summer (June to August), autumn (September to November), and winter (December to next February). In addition to this classification of season, we also divided the year into two periods: a warm season (April to October) and cold season (November to March), based on usual temperatures in the study area and the likelihood of opening windows in the residence.
Traffic-related PAH exposure assessment
Residential addresses of the participants at the time of interview were geocoded using ArcView 3.2 (ESRI, Inc., Redlands, CA), with GDT/Dynamap 2000 (GDT, Inc., Lebanon, NH) as the reference theme. ZP4 (Semaphore Co., Aptos, CA) software was used to correct and update the zip code for each address before the geocoding process. A validation study showed very good positional accuracy of the geocoded addresses (Bonner et al., 2003).
A geographic traffic emissions and meteorological dispersion model was applied to estimate PAH exposure from traffic emissions in the calendar year of blood collection based on each participant’s residence. In this model, benzo[a]pyrene, the most commonly used marker of PAHs exposure, was used as a surrogate for total PAH exposure from traffic emissions. The geographic traffic exposure model, originally developed for the Long Island Breast Cancer Study Project, was modified for the Western New York region with region-specific meteorological and traffic data (Nie et al., 2007). Meteorological data were obtained from the National Climatic Data Center and traffic count data were obtained from the Greater Buffalo-Niagara Regional Transportation Council (Appendix Table A-1). Briefly, annual average traffic density, distance from road, wind speed, direction and other meteorological conditions, as well as tailpipe emission including cruise emissions and excess emissions at intersections and during engine warm-up were modeled to estimate traffic exposures for each residence of the study participants. Cold start emissions were not explicitly included in the final model, because we had previously found them to contribute only negligibly to a regression model for soil PAH data, the preferred environmental dataset (Beyea et al., 2006). We did include intersection data in the model. Cold start emissions, highest at intersections, would contribute to this term. The model produced relative rather than absolute estimates of PAH exposure, because the former are less sensitive to uncertainties in model parameters (Nie et al., 2007).
This estimate of exposure to traffic emissions was validated and calibrated in a subset of the Long Island Breast Cancer Study participants (Beyea et al., 2006; Beyea et al., 2005). The model was shown to predict successfully measured PAH, including soil benzo[a]pyrene concentrations, PAH-DNA adducts in study participants’ blood and carbon monoxide level at a U.S. Environmental Protection Agency monitoring station (Beyea et al., 2006; Beyea et al., 2005). To examine if the model were valid in our study region and also to further calibrate the model parameters, we performed an additional validation study using measured historical data on benzo[a]pyrene in air and carbon monoxide concentrations from the Erie and Niagara areas. The correlation coefficient between historical measured and predicted benzo[a]pyrene was 0.54 and 0.43 for the Pearson and Spearman correlations, respectively (Appendix Figure A-1). The model, adapted to predict carbon monoxide concentrations at four U.S. Environmental Protection Agency monitoring stations, was also able to reproduce the patterns in hourly carbon monoxide concentrations reasonably well (Appendix Figures A-2 to A-5). Annual average values predicted for the four stations followed the trend in measurement between stations (Appendix Figure A-6).
Laboratory analysis of oxidation biomarkers
Fasting blood samples were collected in the morning of the interview and were processed within 30 minutes for glutathione measurement and within two hours for the other oxidative stress biomarkers. Samples were stored at −80°C until assayed (Trevisan et al., 2001).
Total erythrocyte glutathione (mg/dl of packed red blood cells) was measured in EDTA whole blood using the method of Browne and Armstrong (Browne and Armstrong, 1998). Plasma glutathione peroxidase activity (IU/liter) was measured using a Cobas Mira automated chemistry analyzer (Pippenger et al., 1998). Trolox-equivalent antioxidant capacity was measured using EDTA plasma and is expressed as a percent inhibition of the radical-generating reaction relative to the vitamin E analogue trolox (Miller et al., 1993). TBARS (nmol/ml of malondialdehyde equivalents) were measured in EDTA plasma (Armstrong and Browne, 1994).
Several quality control procedures were used. External standards and “in-house” control specimens were included in all assays on every run. Recovery experiments were performed on all assays by standard addition methodology for assessment of the ability of each particular method to accurately quantify the analyte present in the sample. Intra-assay reproducibility was calculated from 20 determinations in the same run. Long-term inter-assay reproducibility and control ranges were generated by running five samples per day over a period of 20 days. The coefficients of variation of the mean values for the intra-assay coefficients were as follows: glutathione 3.3 percent, glutathione peroxidase 4.4 percent, trolox-equivalent antioxidant capacity 8.5 percent and TBARS 7.6 percent; for the inter-assay coefficients, they were glutathione 4.0 percent, glutathione peroxidase 8.6 percent, trolox-equivalent antioxidant capacity 10.0 percent and TBARS 9.2 percent.
Statistical methods
To describe basic characteristics of the study participants, we calculated means and standard deviations for continuous variables and frequencies and percentages for categorical variables. We calculated medians and inter-quartile ranges for all the biomarkers.
The association of PAH exposure with oxidation biomarkers was estimated with a multiple regression model. To avoid an assumption of a linear association, traffic-related PAH exposure was categorized into quartiles and treated as dummy variables in the regression models. The mean levels of oxidative biomarkers among women in the second, third, and fourth quartiles of PAH exposure categories were compared to the level of biomarker among women in the first PAH quartile and the differences were presented as regression coefficients (95% confidence intervals). Oxidation biomarkers were normally distributed except for the TBARS measurement, which was slightly right skewed. Logarithmic transformed and non-transformed TBARS measures produced similar results; we present here results using non-transformed TBARS. Potential confounders examined for inclusion in the regression model included age, education, body mass index, menopausal status, smoking status, secondhand smoke exposure, alcohol consumption, recent non-steroidal anti-inflammatory drugs use, daily vitamin C intake, daily vitamin E intake, season and occupation. The potential covariates were selected on the basis of factors in the literature that potentially influence biomarkers of oxidative stress, factors affecting other PAH exposures and those affecting hormones. Education was used as a proxy for socioeconomic status, which might affect residential traffic exposure levels and dietary characteristics. A reduced model was examined, removing covariates that altered the partial regression coefficients of the highest quartiles by 10% or less. To test potential effect modification by smoking and menopausal status, interaction terms with PAH exposure were also included in the regression models, and stratified analyses were also examined.
Quadratic spline regression was used to examine the associations between traffic-related PAH exposure and levels of oxidative stress biomarkers. Knots were selected based on the categorical analyses. The end categories were restricted to be linear segments to prevent instability (Greenland, 1995).
Results
Selected characteristics of the study participants are presented in Table 1. The mean age of the women was 57 years. The majority of the women were white. Because this sample was originally selected as controls for a breast cancer case-control study, the women were disproportionately post-menopausal. Eight hundred and seventy nine of the women were never-smokers and 676 of them were never-smokers without secondhand smoke exposure in their home or workplace (data not shown). Distributions of anti-oxidative and oxidative damage biomarkers are also shown in Table 1.
Table 1.
Descriptive characteristics of study participants, Erie and Niagara Counties
| Variables | Distribution |
|---|---|
| Mean ± Standard deviation | |
| Age (years) | 57.08±11.65 |
| Body mass index (kg/m2) | 28.16±6.28 |
| Number (%) | |
| Race | |
| White | 1652 (91.27) |
| Others | 158 (8.73) |
| Education | |
| <12 years | 164 (9.06) |
| 12–16 years | 1437 (79.39) |
| ≥16 years | 209 (11.55) |
| Menopausal status | |
| Pre-menopausal | 551 (30.44) |
| Post-menopausal | 1259 (69.56) |
| Smoking status | |
| Never smoker | 879 (48.64) |
| Former smoker | 646 (35.75) |
| Current smoker | 282 (15.61) |
| Secondhand smoke exposure | |
| No | 1286 (75.12) |
| Yes | 426 (24.88) |
| Alcohol drinking status | |
| Lifetime nondrinker | 260 (14.50) |
| Not-current drinker | 634 (35.34) |
| Current drinker | 900 (50.16) |
| Recent aspirin use | |
| Non-user | 984 (54.79) |
| Infrequent user | 582 (32.41) |
| Regular user | 230 (12.81) |
| Recent ibuprofen use | |
| Non-user | 788 (44.00) |
| Infrequent user | 848 (47.35) |
| Regular user | 155 (8.65) |
| Median (Inter-quartile range) | |
| Glutathione (mg/dl packed red blood cells) | 55.98 (47.30, 64.83) |
| Glutathione peroxidase (IU/liter) | 613 (550, 677) |
| Trolox-equivalent antioxidant capacity (%) | 71.2 (68.0, 75.2) |
| TBARS (nmol/ml) | 1.26 (1. 08, 1.50) |
| Daily vitamin C intake (mg) | 169.48 (104.98, 430.99) |
| Daily vitamin E intake (IU) | 33.94 (8.49, 258.17) |
Abbreviation: TBARS, thiobarbituric acid-reactive substances
Results for multiple regression analyses among all study participants are presented in Table 2. Higher traffic-related PAH exposure was associated with decreased glutathione and increased glutathione peroxidase. Mean blood glutathione among women in the third and fourth quartile of PAH exposure were 2.12 (95% confidence interval: 0.09, 4.15) and 3.31 (95% confidence interval: 1.26, 5.36) mg/dl packed red blood cells lower than for women in the first quartile. Mean blood glutathione peroxidase among women in the third and fourth quartile of PAH exposure were 12.49 (95% confidence interval: 0.18, 24.81) and 16.69 (95% confidence interval: 4.23, 29.15) IU/liter higher than women in the first quartile. Similar patterns were observed in the quadratic spline regression analyses (Figure 1). No associations were found between PAH exposure with trolox-equivalent antioxidant capacity or with TBARS.
Table 2.
Differences in mean blood oxidative stress biomarker concentrations (95% confidence intervals), for PAH exposure categories, among all participants (n=1,810)
| PAH exposure categories | Glutathione (mg/dl packed red blood cells) | Glutathione peroxidase (IU/liter) | Trolox-equivalent antioxidant capacity (%) | TBARS (nmol/ml) |
|---|---|---|---|---|
| 2nd vs. 1st quartile | −0.43 (−2.45, 1.58) | 8.51 (−3.76, 20.77) | 0.09 (−0.63, 0.81) | 0.02 (−0.04, 0.07) |
| 3rd vs. 1st quartile | −2.12 (−4.15, −0.09) | 12.49 (0.18, 24.81) | −0.65 (−1.38, 0.07) | −0.01 (−0.06, 0.04) |
| 4th vs. 1st quartile | −3.31 (−5.36, −1.26) | 16.69 (4.23, 29.15) | −0.60 (−1.34, 0.13) | 0.03 (−0.03, 0.08) |
Abbreviations: PAH, polycyclic aromatic hydrocarbon; TBARS, thiobarbituric acid-reactive substances
Adjusted for age, education, body mass index, menopausal status, smoking status, secondhand smoke exposure, season, dietary vitamin C intake, and dietary vitamin E intake.
Figure 1.
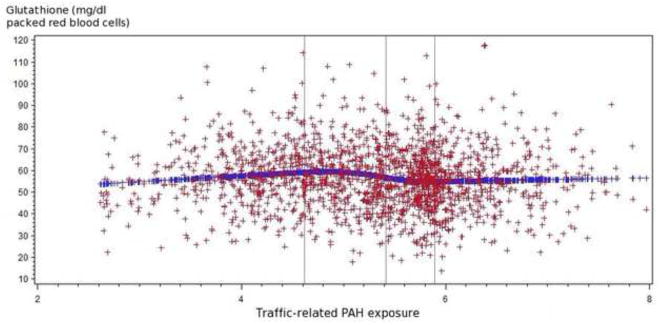
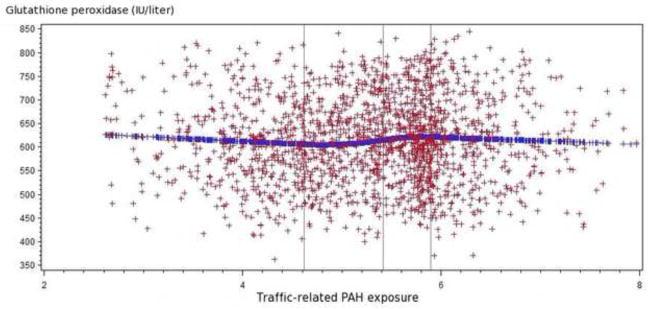
Figure 1.1 Relationship of traffic-related PAH emissions and glutathione levels (mg/dl packed red blood cells) among all participants
Figure 1.2 Relationship of traffic-related PAH emissions and glutathione peroxidase levels (IU/liter) among all participants
The spline regression indicated that higher traffic-related PAH exposure tended to be associated with increased glutathione peroxidase and decreased glutathione.
There was a multiplicative interaction for glutathione by menopausal status. For pre-menopausal women (n=551), mean glutathione concentrations were 6.18 (95% confidence interval: 2.83, 9.53) mg/dl packed red blood cells lower for women in the fourth compared to the first quartile of PAH exposure; for post-menopausal women (n=1259), mean glutathiones were only 1.99 mg/dl packed red blood cells lower for women in the fourth to the lowest quartile of PAH exposure and the confidence interval for that difference included the null (data not shown).
In analyses limited to 676 never-smokers without secondhand smoke exposure, there was some suggestion of stronger associations of traffic-related PAH exposure with glutathione and glutathione peroxidase than for all the participating women. Mean glutathione and glutathione peroxidase among women in the fourth quartile of PAH exposure were 5.98 (95% confidence interval: 2.42, 9.54) mg/dl packed red blood cells lower and 18.02 (95% confidence interval: −1.82, 37.86) IU/liter higher, respectively, than among women in the lowest quartile (Table 3). Moreover, we found multiplicative interactions between glutathione and glutathione peroxidase with menopausal status for the association with traffic-related PAH, in this subgroup of women. Among 228 pre-menopausal never-smokers without secondhand smoke exposure, higher exposure was associated with lower glutathione and higher glutathione peroxidase; while PAH exposure from traffic emissions had little impact on the glutathione and glutathione peroxidase levels among 448 post-menopausal never-smokers without secondhand smoke exposure (Table 3). We found no differences in associations in analyses stratified on use of hormone therapy in the non-smoking postmenopausal women (data not shown).
Table 3.
Differences in mean oxidative stress biomarkers (95% confidence intervals for different PAH exposure categories, among women without tobacco smoke exposure
| PAH exposure categories | Glutathione (mg/dl packed red blood cells) | Glutathione peroxidase (IU/liter) |
|---|---|---|
| All women without tobacco smoke exposure (n=676) | ||
| 2nd vs. 1st quartile | −2.52 (−5.86, 0.83) | 8.45 (−10.18, 27.08) |
| 3rd vs. 1st quartile | −2.15 (−5.58, 1.28) | 9.50 (−9.63, 28.63) |
| 4th vs. 1st quartile | −5.98 (−9.54, −2.42) | 18.02 (−1.82, 37.86) |
| Pre-menopausal women without tobacco smoke exposure (n=228) | ||
| 2nd vs. 1st quartile | −2.83 (−7.37, 1.72) | 17.30 (−12.97, 47.57) |
| 3rd vs. 1st quartile | −4.29 (−9.82, 1.23) | 31.17 (−5.63, 67.97) |
| 4th vs. 1st quartile | −11.20 (−16.28, −6.12) | 35.91 (2.09, 69.74) |
| Post-menopausal women without tobacco smoke exposure (n=448) | ||
| 2nd vs. 1st quartile | −2.06 (−6.63, 2.52) | 1.32 (−22.69, 25.33) |
| 3rd vs. 1st quartile | −0.35 (−4.74, 4.04) | −3.23 (−26.27, 19.81) |
| 4th vs. 1st quartile | −3.14 (−7.94, 1.67) | 3.52 (−21.69, 28.74) |
Abbreviation: PAH, polycyclic aromatic hydrocarbon; TBARS, thiobarbituric acid-reactive substances Adjusted for age, education, body mass index, season, dietary vitamin C intake, and dietary vitamin E intake.
Associations between traffic-related PAH exposure and antioxidant biomarkers tended to be stronger for women interviewed during the warm season. For the subgroup of women whose blood was drawn during the warm season, mean blood glutathione was 4.38 (95% confidence interval: 2.11, 6.66) mg/dl packed red blood cells lower for women in the highest than in the lowest quartile of traffic-related PAH exposure. For the women with blood drawn in the cold season, mean glutathiones were only 1.33 mg/dl packed red blood cells lower for women in the fourth to the lowest quartile of PAH exposure; the confidence interval for that difference included the null. Mean glutathione peroxidase was 19.67 (95% confidence interval: 4.16, 35.18) and 9.89 (95% confidence interval: −9.79, 29.59) IU/liter higher for women in the fourth compared to the first quartile of PAH exposure in the warm and cold seasons, respectively. Similar seasonal patterns were found in the analyses limited to the nonsmokers (data not shown).
Discussion
In this study of healthy women living in Western New York, traffic-related PAH emissions at their place of residence were associated with decreased glutathione and increased glutathione peroxidase levels; these associations were strongest among premenopausal, non-smoking women, who were not exposed to secondhand smoke. Associations were also stronger for measurements made during the warmer season.
Several in vitro and in vivo studies have implicated PAH exposure in induction of oxidative stress (Bravo et al., 2010; Kim and Lee, 1997). In addition, there is evidence that occupational exposures to traffic emissions are associated with oxidative stress. Oxidative damage to DNA, protein, and lipids was observed among individuals with high exposure to traffic emissions, including bus drivers and security guards working near heavily trafficked roads (Rossner et al., 2008a; Rossner et al., 2008b; Wei et al., 2010). Several studies have shown strong correlations of blood biomarkers of PAH exposure with oxidative stress and altered antioxidant status in children (Bae et al., 2010; Singh et al., 2008). To our knowledge, ours is the first such study examining the association of traffic-related PAH exposure with biomarkers of oxidative stress in a population based study of adults.
The observed alterations of glutathione and glutathione peroxidase levels associated with traffic emissions in the present study could be an indication of oxidative stress. Glutathione is one of the key non-enzymatic antioxidants. Reduced glutathione can directly scavenge free radicals and be oxidized to form glutathione disulfide (Wu et al., 2004). Glutathione also serves as a co-factor required for glutathione peroxidase activity. Glutathione peroxidase is a major antioxidant enzyme, contributing to the reduction of peroxides and the protection of the cell against oxidative damage (Comhair and Erzurum, 2005). The observed reduction in glutathione associated with PAH exposure may result from direct depletion of glutathione via its scavenging mechanism or its consumption as a co-factor for glutathione peroxidase in response to oxidative stress (Romero et al., 1997). We observed a positive association between PAH exposure and glutathione peroxidase; this association may result from induction of the antioxidant enzyme in response to traffic emission exposure. Previous studies have suggested that synthesis of extracellular antioxidant enzymes, including glutathione peroxidase, can be induced in response to xenobiotic-induced oxidative insults (Mates and Sanchez-Jimenez, 1999).
Oxidative damage to lipids, proteins, and DNA within cells occurs when the production of free radicals exceeds the antioxidant capacity of the cell (Terada, 2006). In contrast to previous studies conducted among occupational groups with high exposure to traffic emissions, we did not find any evidence that traffic-related PAH exposure in this population sample of females was associated with oxidative damage to the lipids. The lack of association between PAH exposure and TBARS suggests that antioxidant defense was sufficient protection for the amount of exposure in this study. It may also be that the TBARS assay lacks specificity to measure any lipid peroxidation in this population sample with lower overall exposure. The assay measures malondialdehyde, a marker of lipid peroxidation. However the assay is not specific to malondialdehyde and can be affected by reactions with other related compounds. Further, malondialdehyde can also arise from mechanisms other than lipid peroxidation (Armstrong and Browne, 1994).
Exposure to tobacco smoke has been shown to produce systemic oxidative stress. Animal studies have shown a decrease in glutathione and an increase in 8-hydroxy-2′-deoxyguanosine level in blood and lung tissues several hours after smoke inhalation (Aoshiba et al., 2003). Both active smoking and secondhand smoke exposure have been shown to decrease antioxidant capacity and increase lipid peroxidation in humans (Kosecik et al., 2005; Morrison et al., 1999). To minimize the impact of tobacco smoke on these oxidation biomarkers, we performed an analysis limited to never-smokers who were not exposed to secondhand smoke. There were more pronounced associations between traffic emissions and levels of glutathione and glutathione peroxidase in this group. Among active and passive smokers, PAH exposure from tobacco smoke might be already be high, such that additional exposure from traffic emissions might not make a detectable difference.
In these analyses of non-smoking women without exposure to second hand smoke, we found stronger associations of PAH exposure with glutathione and glutathione peroxidase among pre- than among post-menopausal women. Previous studies have suggested that some of the products of fuel combustion, such as benzo[a]pyrene, are isosteric to sex steroids and have the ability to bind the estrogen receptor (Oberdorster et al., 1999). It may be that the higher blood estrogen among the pre-menopausal women prevents these products from binding to estrogen receptors, resulting in higher circulating concentrations compared to the post-menopausal women exposed to the same amount of PAHs from traffic emissions. However, we did not observe different responses among postmenopausal women who used hormone therapy compared to those who did not. Further experimental and human studies need to be conducted to better understand the underlying mechanism of the difference in response for pre- and postmenopausal women.
Stronger associations were observed in the warmer season compared to the colder season in this study. Our finding was consistent with previous studies using disease incidence or mortality as endpoints. More pronounced effects for traffic related air pollutants with hospitalization for first acute myocardial infarction have been found during the warm season than during cold season (Lanki et al., 2006). Seasonal pattern was observed between ambient particulate matter exposure and mortality in the northeast US (with a peak in summer) and little seasonal variation in the southern regions of the country (Peng et al., 2005). In the study region, air conditioning is not frequently used in residences; residential windows are more likely to be open during the warmer season. In addition, people may spend more time outdoors during those months. These factors may make the modeled PAH exposures from traffic emissions in warm season a better proxy for the actual exposures. However, other factors also change by season. Meteorological conditions, including the level of ultraviolet light, also vary by season and may affect emission concentrations and mixtures differently by season (Rachel S. Russo, 2011). In our study, residential traffic emissions in the calendar year of blood collection were estimated and the exposure values were averaged over seasons. Exposure values were not adjusted by season due to the absence of seasonal data on traffic flows and uncertainty over seasonal variation in emission rates (Beyea et al., 2008). Seasonal values for atmospheric dispersion, including variations in mixing layer height and wind speed, were incorporated into the annual exposure estimates. However, seasonal variations in PAH exposure were likely to be modest (Beyea et al., 2008). Measurements of atmospheric benzo[a]pyrene in Ontario, the closest location for which data are available, are about 2-times higher in winter than in summer (Beyea et al., 2008). After accounting for the seasonal contribution from space heating, which also emits PAH selectively by season, we estimate that traffic-related PAH exposure around the yearly average at a particular location in the study region varies by a factor of approximately 1.4 (Appendix Figure A-7 showing the modest variation by season in carbon monoxide concentrations in the study area). PAH exposure from indoor heating adds additional misclassification to winter exposures, suggesting a greater bias towards the null in winter than in summer, which may explain the stronger association observed in warm season in this study.
This study has some limitations that need to be taken into account in interpretation of these findings. Residential address was used as a proxy for overall location to estimate traffic emission exposure; we were not able to account for exposures in other locations outside the residence. In one survey it was found that American adults spent about 16 hours per day at home (Leech et al., 2002); residential exposures likely account for a substantial proportion of the total. We did not have measurements of where participants were during the time that they were outside their residence; clearly missed exposures would be a source of misclassification. This error was likely non-differential with bias of effect estimates toward the null. Our model was not able to differentiate traffic emissions from trucks and light duty vehicles. A previous review of the literature suggests that emissions of high-molecular PAHs, such as benzo[a]pyrene, from diesel trucks do not differ greatly per kilometer than emissions from light vehicles (Beyea et al., 2008). However, emissions of the lighter PAHs are dominated by diesel engines, and there is the possibility of misclassification of the traffic-related lighter PAHs exposure when diesel engines cannot be measured separately. In the current study area, heavy vehicles account for 3.1–9.0 % of total vehicles on all types of roads other than interstate principal arterials, and we expect the high molecular PAHs to contribute most to oxidative stress (Jeng et al., 2010), therefore impact of this potential source of misclassification is likely limited. Further, any potential misclassification is likely non-differential, biasing results toward the null. In addition, there are no good data on emission rates of PAH by vehicle speed, however we were able to account for the most important locations of variation in speed, namely at intersections. The traffic exposure model entails the choice of a scale factor corresponding to higher emissions at intersections, where vehicles are accelerating and decelerating (Beyea et al., 2006) (Appendix Table A-1). The modest correlations between measured and predicted traffic-related PAH exposures imply potential measurement error. Due to the use relative rather than absolute estimates of PAH exposure, the estimated traffic-related PAH exposure and measured PAH have different scales, making it difficult estimate an error-corrected result. Moreover, PAH monitoring stations used in our validation study were deliberately placed relatively close to industrial emissions and thus these measurements might not be appropriate to calibrate the traffic-related PAH emissions for the region more generally. Another issue is that only twelve locations were used in the validation study and regression calibration based on such small number of observations might not provide reliable results, even if the calibration were feasible. The model that we used was developed to estimate PAH exposures. Nonetheless, traffic emissions are a complex mixture of compounds. We were not able to distinguish which of those compounds was important in the associations we observed. Further, these measures were developed to reflect conditions averaged over a calendar year; they would not necessarily correspond exactly with exposure level at the time of blood collection. A model of traffic emissions over a brief period of time before blood measures would be more informative. We did not have data on short-term traffic patterns required for such an analysis.
The biomarkers of oxidation used in this study also have limitations. In particular, as discussed above, the TBARS test does not measure malondialdehyde exclusively and malondialdehyde is also not generated exclusively by breakdown of lipid hydroperoxide (Janero, 1990). However, some experimental studies have shown that good correlations of TBARS with levels of isoprostane, a more specific lipid peroxidation marker (Gopaul et al., 1994; Nourooz-Zadeh et al., 1998).
There were also some issues with the available data for potential confounders. We assessed alcohol drinking and vitamin supplement intake 12–24 months prior to each interview, and secondhand smoke exposure was based on the information for the most recent decade. There is some possibility that the exposure status at the time of blood collection might differ from the data we used in the study; however while it is not likely that many of the participants’ status changed markedly, any change would lead to incomplete control for confounding. In addition, we did not have data to address consumption of grilled or barbecued foods, another source of PAHs. It is also possible that individuals with different socioeconomic status differed in dietary characteristics as well as residential traffic exposures. While we controlled for education as a proxy for socioeconomic status, there could be uncontrolled variation. Another limitation is the use of cross-sectional design, which precludes establishing temporality. A study of changes in these markers in response to changes in PAH exposure would be informative in understanding better these associations. Lastly, study participants in the present study consisted of women only and were predominantly white, limiting the generalizability of the study results to other population groups.
Nonetheless the study had some important strengths. In this population-based study of traffic emissions in a region with both urban and rural residences, we found evidence that traffic emissions were associated with anti-oxidative capacity of healthy women, particularly premenopausal women who were not exposed to smoke. Further, there appeared to be compensatory mechanisms to increase protection against oxidation and protect macromolecules from oxidative damages. Given the ubiquitous nature of traffic emissions, these findings are of significance. Further investigations with more specific measurements of both exposure and of oxidation would contribute to our ability to further understand the biological impact of this prevalent environmental exposure.
Supplementary Material
Highlights.
We assess the association between traffic emissions and oxidative stress biomarkers.
Higher traffic emissions were associated with decreased glutathione and elevated glutathione peroxidase.
Stronger associations were suggested among women without tobacco smoke exposure.
Associations were also stronger for measurements made in warmer months.
Acknowledgments
This work has been supported in part by U.S. Army Medical Research Grants (DAMD170010417 and DAMD179616202), NCI Grant (R21CA8713801), NIH Grant (RO1CA092040) and National Institute on Alcohol Abuse and Alcoholism Grant (P50-AA09802).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amigou A, et al. Road Traffic and Childhood Leukemia: The ESCALE Study (SFCE) Environ Health Perspect. 2011;119:566–72. doi: 10.1289/ehp.1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoshiba K, et al. Immunohistochemical evaluation of oxidative stress in murine lungs after cigarette smoke exposure. Inhal Toxicol. 2003;15:1029–38. doi: 10.1080/08958370390226431. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Browne R. The analysis of free radicals, lipid peroxides, antioxidant enzymes and compounds related to oxidative stress as applied to the clinical chemistry laboratory. Adv Exp Med Biol. 1994;366:43–58. doi: 10.1007/978-1-4615-1833-4_4. [DOI] [PubMed] [Google Scholar]
- Bae S, et al. Exposures to particulate matter and polycyclic aromatic hydrocarbons and oxidative stress in schoolchildren. Environ Health Perspect. 2010;118:579–83. doi: 10.1289/ehp.0901077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyea J, et al. Validation and calibration of a model used to reconstruct historical exposure to polycyclic aromatic hydrocarbons for use in epidemiologic studies. Environ Health Perspect. 2006;114:1053–8. doi: 10.1289/ehp.8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyea J, et al. Development of a traffic model for predicting airborne PAH exposures since 1960 on Long Island, New York. Report to the National Cancer Institute, and the National Institute of Environmental Health Sciences for work completed under USPHS Grant U01-CA/ES-66572. 2005;2011 [Google Scholar]
- Beyea J, et al. Airborne emissions from 1961 to 2004 of benzo[a]pyrene from U.S. vehicles per km of travel based on tunnel studies. Environ Sci Technol. 2008;42:7315–20. doi: 10.1021/es8000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block G, et al. A data-based approach to diet questionnaire design and testing. Am J Epidemiol. 1986;124:453–69. doi: 10.1093/oxfordjournals.aje.a114416. [DOI] [PubMed] [Google Scholar]
- Bonner MR, et al. Positional accuracy of geocoded addresses in epidemiologic research. Epidemiology. 2003;14:408–12. doi: 10.1097/01.EDE.0000073121.63254.c5. [DOI] [PubMed] [Google Scholar]
- Bonner MR, et al. Breast cancer risk and exposure in early life to polycyclic aromatic hydrocarbons using total suspended particulates as a proxy measure. Cancer Epidemiol Biomarkers Prev. 2005;14:53–60. [PubMed] [Google Scholar]
- Bravo CF, et al. Biomarker responses and disease susceptibility in juvenile rainbow trout Oncorhynchus mykiss fed a high molecular weight PAH mixture. Environ Toxicol Chem. 2010 doi: 10.1002/etc.439. [DOI] [PubMed] [Google Scholar]
- Browne RW, Armstrong D. Reduced glutathione and glutathione disulfide. Methods Mol Biol. 1998;108:347–52. doi: 10.1385/0-89603-472-0:347. [DOI] [PubMed] [Google Scholar]
- Brunekreef B, et al. Effects of long-term exposure to traffic-related air pollution on respiratory and cardiovascular mortality in the Netherlands: the NLCS-AIR study. Res Rep Health Eff Inst. 2009:5–71. discussion 73–89. [PubMed] [Google Scholar]
- Clemons JH, et al. Evidence of estrogen- and TCDD-like activities in crude and fractionated extracts of PM10 air particulate material using in vitro gene expression assays. Environmental Science & Technology. 1998;32:1853–1860. [Google Scholar]
- Comhair SA, Erzurum SC. The regulation and role of extracellular glutathione peroxidase. Antioxid Redox Signal. 2005;7:72–9. doi: 10.1089/ars.2005.7.72. [DOI] [PubMed] [Google Scholar]
- Crouse DL, et al. Postmenopausal breast cancer is associated with exposure to traffic-related air pollution in Montreal, Canada: a case-control study. Environ Health Perspect. 2010;118:1578–83. doi: 10.1289/ehp.1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopaul NK, et al. Formation of PGF2-isoprostanes during the oxidative modification of low density lipoprotein. Biochem Biophys Res Commun. 1994;200:338–43. doi: 10.1006/bbrc.1994.1453. [DOI] [PubMed] [Google Scholar]
- Greenland S. Dose-response and trend analysis in epidemiology: alternatives to categorical analysis. Epidemiology. 1995;6:356–65. doi: 10.1097/00001648-199507000-00005. [DOI] [PubMed] [Google Scholar]
- IARC; Polynuclear Aromatic Compounds, Part 1, Chemical, Environmental and Experimental Data. I. A. f. R. o. Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 1998;32 [PubMed] [Google Scholar]
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 92. World Health Organization, International agency for research on cancer; 2010. Some Non-heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures. [PMC free article] [PubMed] [Google Scholar]
- Ito K, et al. Fine Particulate Matter Constituents Associated with Cardiovascular Hospitalizations and Mortality in New York City. Environ Health Perspect. 2010 doi: 10.1289/ehp.1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med. 1990;9:515–40. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- Jeng HA, et al. Polycyclic aromatic hydrocarbon-induced oxidative stress and lipid peroxidation in relation to immunological alteration. Occup Environ Med. 2010 doi: 10.1136/oem.2010.055020. [DOI] [PubMed] [Google Scholar]
- Kim KB, Lee BM. Oxidative stress to DNA, protein, and antioxidant enzymes (superoxide dismutase and catalase) in rats treated with benzo(a)pyrene. Cancer Lett. 1997;113:205–12. doi: 10.1016/s0304-3835(97)04610-7. [DOI] [PubMed] [Google Scholar]
- Kiruthiga PV, et al. Silymarin protects PBMC against B(a)P induced toxicity by replenishing redox status and modulating glutathione metabolizing enzymes--an in vitro study. Toxicol Appl Pharmacol. 2010;247:116–28. doi: 10.1016/j.taap.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Kosecik M, et al. Increased oxidative stress in children exposed to passive smoking. Int J Cardiol. 2005;100:61–4. doi: 10.1016/j.ijcard.2004.05.069. [DOI] [PubMed] [Google Scholar]
- Lanki T, et al. Associations of traffic related air pollutants with hospitalisation for first acute myocardial infarction: the HEAPSS study. Occup Environ Med. 2006;63:844–51. doi: 10.1136/oem.2005.023911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumbach RJ, Kipen HM. Acute effects of motor vehicle traffic-related air pollution exposures on measures of oxidative stress in human airways. Ann N Y Acad Sci. 2010;1203:107–12. doi: 10.1111/j.1749-6632.2010.05604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech JA, et al. It’s about time: a comparison of Canadian and American time-activity patterns. J Expo Anal Environ Epidemiol. 2002;12:427–32. doi: 10.1038/sj.jea.7500244. [DOI] [PubMed] [Google Scholar]
- Liao CM, et al. Lung cancer risk in relation to traffic-related nano/ultrafine particle-bound PAHs exposure: A preliminary probabilistic assessment. J Hazard Mater. 2011 doi: 10.1016/j.jhazmat.2011.03.017. [DOI] [PubMed] [Google Scholar]
- Mates JM, Sanchez-Jimenez F. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci. 1999;4:D339–45. doi: 10.2741/mates. [DOI] [PubMed] [Google Scholar]
- Miller NJ, et al. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin Sci (Lond) 1993;84:407–12. doi: 10.1042/cs0840407. [DOI] [PubMed] [Google Scholar]
- Morrison D, et al. Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am J Respir Crit Care Med. 1999;159:473–9. doi: 10.1164/ajrccm.159.2.9804080. [DOI] [PubMed] [Google Scholar]
- Nie J, et al. Exposure to traffic emissions throughout life and risk of breast cancer: the Western New York Exposures and Breast Cancer (WEB) study. Cancer Causes Control. 2007;18:947–55. doi: 10.1007/s10552-007-9036-2. [DOI] [PubMed] [Google Scholar]
- Nourooz-Zadeh J, et al. F4-isoprostanes: a novel class of prostanoids formed during peroxidation of docosahexaenoic acid (DHA) Biochem Biophys Res Commun. 1998;242:338–44. doi: 10.1006/bbrc.1997.7883. [DOI] [PubMed] [Google Scholar]
- Oberdorster E, et al. Interaction of PAHs and PCBs with ecdysone-dependent gene expression and cell proliferation. Toxicol Appl Pharmacol. 1999;160:101–8. doi: 10.1006/taap.1999.8745. [DOI] [PubMed] [Google Scholar]
- Olinski R, et al. Oxidative DNA damage: assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome. Free Radic Biol Med. 2002;33:192–200. doi: 10.1016/s0891-5849(02)00878-x. [DOI] [PubMed] [Google Scholar]
- Patel MM, et al. Traffic-related particulate matter and acute respiratory symptoms among New York City area adolescents. Environ Health Perspect. 2010;118:1338–43. doi: 10.1289/ehp.0901499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng RD, et al. Seasonal analyses of air pollution and mortality in 100 US cities. Am J Epidemiol. 2005;161:585–94. doi: 10.1093/aje/kwi075. [DOI] [PubMed] [Google Scholar]
- Pippenger CE, et al. Regulatory antioxidant enzymes. Methods Mol Biol. 1998;108:299–313. doi: 10.1385/0-89603-472-0:299. [DOI] [PubMed] [Google Scholar]
- Russo Rachel S, MLW, Zhou Yong, Haase Karl B, Ambrose Jesse L, Conway Leanna, Mentis Elizabeth, Talbot Robert, Sive Barkley C. Spatial Variation, Sources and Emission Rates of Volatile Organic Compounds Over the Northeastern U.S. In: Mazzeo N, editor. Air Quality-Models and Applications. 2011. [Google Scholar]
- Romero DL, et al. Depletion of glutathione by benzo(a)pyrene metabolites, ionomycin, thapsigargin, and phorbol myristate in human peripheral blood mononuclear cells. Toxicol Appl Pharmacol. 1997;144:62–9. doi: 10.1006/taap.1997.8113. [DOI] [PubMed] [Google Scholar]
- Rossner P, Jr, et al. Seasonal variability of oxidative stress markers in city bus drivers. Part I. Oxidative damage to DNA. Mutat Res. 2008a;642:14–20. doi: 10.1016/j.mrfmmm.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Rossner P, Jr, et al. Seasonal variability of oxidative stress markers in city bus drivers. Part II. Oxidative damage to lipids and proteins. Mutat Res. 2008b;642:21–7. doi: 10.1016/j.mrfmmm.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Singh VK, et al. Blood levels of polycyclic aromatic hydrocarbons in children and their association with oxidative stress indices: an Indian perspective. Clin Biochem. 2008;41:152–61. doi: 10.1016/j.clinbiochem.2007.11.017. [DOI] [PubMed] [Google Scholar]
- Spector A. Review: Oxidative stress and disease. J Ocul Pharmacol Ther. 2000;16:193–201. doi: 10.1089/jop.2000.16.193. [DOI] [PubMed] [Google Scholar]
- Terada LS. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol. 2006;174:615–23. doi: 10.1083/jcb.200605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisan M, et al. Correlates of markers of oxidative status in the general population. Am J Epidemiol. 2001;154:348–56. doi: 10.1093/aje/154.4.348. [DOI] [PubMed] [Google Scholar]
- Wei Y, et al. Personal exposure to particulate PAHs and anthraquinone and oxidative DNA damages in humans. Chemosphere. 2010;81:1280–5. doi: 10.1016/j.chemosphere.2010.08.055. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Health Effects of Transport-related Air Pollution. World Health Organization, Regional Office for Europe; Copenhagen: 2005. [Google Scholar]
- Wu G, et al. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–92. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Wu MT, et al. Urinary excretion of 8-hydroxy-2-deoxyguanosine and 1-hydroxypyrene in coke-oven workers. Environ Mol Mutagen. 2003;42:98–105. doi: 10.1002/em.10176. [DOI] [PubMed] [Google Scholar]
- Zhang HB, et al. Distributions and concentrations of PAHs in Hong Kong soils. Environ Pollut. 2006;141:107–14. doi: 10.1016/j.envpol.2005.08.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.