

Abstract
The catalytic effects of perdeuterating the pyridoxal phosphate dependent enzyme alanine racemase from Geobacillus stearothermophilus are reported. The heavy, perdeuterated form is ~5.5% greater in mass than the protiated form, causing kinetic isotope effects (KIEs) of ~1.3 on kcat and kcat/KM for both L- and D-alanine. These values increase when Cα-deuterated alanine is used as substrate. The heavy enzyme KIEs of ~3 on kcat/KM with deuterated substrates are greater than the product of the individual heavy enzyme and primary substrate KIEs. This breakdown of the rule of the geometric mean is likely due to coupled motion between the protein and the proton transfer reaction coordinate in the rate-limiting step. These data implicate a direct role for protein vibrational motions in barrier crossing for proton transfer steps in alanine racemase.
The role of protein motions in enzyme catalysis has been under debate over the last decade or more.1–14 Certainly, loop and domain motions on the micro- to millisecond time-scale are important to closing off active sites from bulk solvent, providing catalytically productive environments. The controversial aspect is the role of protein motions in overcoming barriers to chemical transformations in closed active sites. Once the productive enzyme-substrate complex is formed, energetic barriers from 12 to 18 kcal/mol must be traversed to form the enzyme-product complex.
The catalytic effects of isotopically substituted (i.e., light vs. heavy) enzymes can, in principle, address the involvement of protein motions in barrier crossing because of the theoretically addressable and minimal nature of the catalyst alteration.15 Heavy enzyme kinetic isotope effects (KIEs) were first measured over 40 years ago on E. coli alkaline phosphatase under conditions where hydrolysis of the phosphoenzyme intermediate is rate-limiting.16,17 The KM values for the protiated and deuterated enzymes are identical and show the same temperature dependence. The kcat values, on the other hand, are greater for the protiated enzyme by a factor of 1.8, yet the temperature dependence of kcat is the same for both. These results were not originally interpreted in terms of protein motions and catalysis.
More recently, Schramm and coworkers have measured the catalytic effects of isotopically substituted enzymes for the explicit purpose of probing the participation of protein vibrational motions in chemical barrier crossing.18,19 Their work on purine nucleoside phosphorylase shows that the rate of nucleoside phosphorolysis is decreased 20–27% by the 10% increase in mass of the heavy enzyme.18 Substrate KIEs for inosine were unaltered by the increased mass of the heavy enzyme, as were the steady-state kinetic parameters due to rate-limiting product release. Their work on HIV protease under conditions where chemistry is rate-limiting shows heavy enzyme KIEs of 1.2 and 1.9 on kcat and kcat/KM, respectively, due to an approximately 12% increase in enzyme mass.19 These results imply that protein motions facilitate chemical barrier crossing in enzyme active sites.
The experiments of Schramm and coworkers employ enzymes in which the transition states probed involve motions of primarily heavy atoms, although simple hydrogen transfers between heteroatoms are involved. This may be the reason that the substrate KIEs for purine nucleoside phosphorylase are essentially identical for light and heavy enzymes even though protein motions facilitate barrier crossing. We hypothesized that enzyme catalyzed reactions in which proton transfer is the central, rate-limiting step may show a coupling between enzyme vibrational motions and the motion of the hydrogen in the transition state, leading to nonequivalent substrate KIEs with the isotopic enzymes.
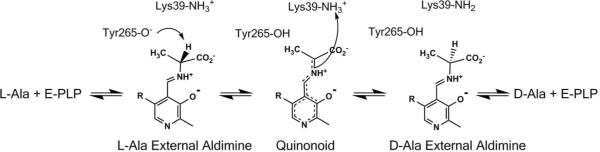
We chose alanine racemase (AR) to test this hypothesis. AR catalyzes the reversible interconversion of L- and D-alanine using pyridoxal 5'-phosphate as a coenzyme. The stepwise proton transfer mechanism (delineated using multiple KIEs)20 is shown in Scheme 1. Previously, we reported free energy profiles for AR at pH 6.9 and 8.9, as well as an isotopic free energy profile.21–23 At high pH, the two proton transfer transition states in the stepwise mechanism are jointly rate-limiting, and substrate and solvent KIEs are observed in both directions (see Supporting Information for details).21
Scheme 1.
Heavy, perdeuterated AR (DAR) was expressed in minimal medium in D2O using deuterated glycerol as the carbon source (see Supporting Information). Light AR (HAR) was similarly expressed using protium in place of deuterium. The enzymes were purified by identical procedures to near homogeneity. The measured increase in mass of 5.5% is identical to the theoretical value for perdeuterated enzyme with all exchangeable deuterons equilibrated with the H2O solvent.
Michaelis-Menten saturation curves for HAR and DAR with protiated L- and D-alanine are presented in Figure 1. Heavy enzyme KIEs are detected on both kcat and kcat/KM (Table 1; reported errors are based on standard errors from nonlinear regression presented in Table S1, and standard error propagation techniques24). They are similar in magnitude (~30%) to those observed by Schramm and coworkers on purine nucleoside phosphorylase and HIV protease.18,19 Figure 2 shows saturation curves for HAR and DAR with Cα-deuterated L-and D-alanine, which effect primary substrate KIEs on kcat and kcat/KM.20,23 The heavy enzyme KIEs are significantly larger with deuterated than with protiated alanines. The same kinetic data (Figures 1 and 2, Table S1) can be alternatively presented as substrate KIEs (Table 2).
Figure 1.
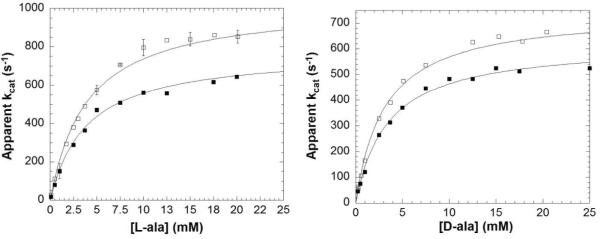
Michaelis-Menten kinetics for HAR (□) and DAR (■) with L-alanine (left) and D-alanine (right) (pH 8.9, 25 °C).
Table1.
Heavy enzyme KIEs for alanine and [2-2H]-alanine (pH 8.9, 25 °C).
| Substrate |
|
|
||
|---|---|---|---|---|
| L-ala | 1.32 ± 0.04 | 1.3 ± 0.1 | ||
| D-ala | 1.21 ± 0.03 | 1.3 ± 0.1 | ||
| [2-2H]-L-ala | 1.67 ± 0.05 | 3.2 ± 0.4 | ||
| [2-2H]-D-ala | 1.40 ± 0.04 | 2.9 ± 0.4 |
Figure 2.
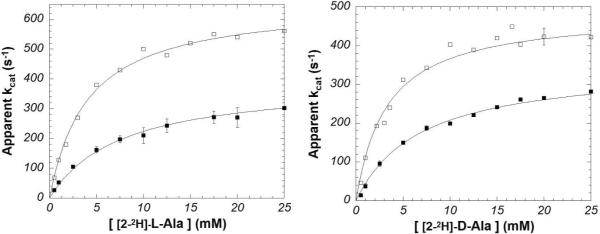
Michaelis-Menten kinetics for HAR (□) and DAR (■) with [2-2H]-L-alanine (left) and [2-2H]-D-alanine (right) (pH 8.9, 25 °C).
Table 2.
Primary substrate KIEs for HAR and DAR (pH 8.9, 25 °C).
| Substrate | Enzyme |
|
|
||
|---|---|---|---|---|---|
| L-ala | HAR | 1.56 ± 0.04 | 1.6 ± 0.2 | ||
| DAR | 1.98 ± 0.07 | 3.8 ± 0.4 | |||
| D-ala | HAR | 1.53 ± 0.04 | 1.6 ± 0.2 | ||
| DAR | 1.77 ± 0.04 | 3.5 ± 0.3 |
The larger increase in primary substrate KIEs on kcat/KM versus kcat when comparing HAR and DAR raises concern regarding a potential change in substrate affinity. This was addressed by measuring the competitive inhibition constant for 2-methylalanine, which binds to AR to form an unreactive external aldimine intermediate. Under conditions identical to those used in KIE measurements, the KI for HAR is 24 ± 1 mM while that for DAR is 27 ± 2 mM (Table S2). These values are identical within error and support the conclusion that the changes in KM are due to effects other than simply lower substrate affinity for DAR. The increase in heavy enzyme KM for deuterated substrate likely originates in the substrate binding isotope effect of ~1.26 on external aldimine formation with HAR,21 and its enhancement by enzyme deuteration. The binding isotope effect is due to hyperconjugation of the Cα-H bond with the electrophilic π system of the coenzyme, and has been independently observed with another PLP dependent enzyme, aspartate aminotransferase.25
Considering the kinetic results either as heavy enzyme KIEs (Table 1) or as substrate KIEs (Table 2) leads to the same critical conclusion: isotope effects due to either enzyme or substrate deuteration are not independent of each other. This violates the rule of the geometric mean, a principle of isotope effect theory, which holds that isotope effects are independent of each other.26,27 The parsimonius explanation for the violation of the rule of the geometric mean observed here is that the different isotopic species (enzyme and substrate) are coupled in a rate-limiting event. In the present case, the enzyme is vibrationally coupled to the motion of the proton undergoing transfer from Cα of the external aldimine intermediate to an enzymic base in the rate-limiting step. Another interpretation of these data, which we consider less viable, is discussed in the Supporting Information.
The present results are in accord with the concepts developed by Schwartz and coworkers.1,28–34 They advocate the idea that protein motions on the femto- to picosecond time scale can be directly involved in barrier crossing, through transient formation of high energy active site structures highly favorable to reaction (i.e., that allow bond making/breaking to occur with a low energetic barrier). Slower protein motions are also important to formation of these transient active site structures via larger amplitude fluctuations (i.e., conformational changes) on longer time scales, but the higher frequency motions can potentially be more strongly coupled vibrationally to the reaction coordinate.
This picture is satisfying on many levels, and does not demean the important role of electrostatics. The unimolecular enzyme-substrate complex is formed from multiple relatively strong interactions between the two molecules. Hydrogen bonding, hydrophobic, and electrostatic interactions would prevent the active site bound substrate from becoming vibrationally hot (as required for barrier crossing if protein motions are not involved) relative to the protein structure by facilitating rapid vibrational energy redistribution. The Schwartz ideas avoid this conundrum since much of the activation energy is distributed into protein vibrations. These coordinately act to provide a relatively high energy active site structure that allows bond making/breaking to occur with a low barrier (i.e., the substrate does not have to become vibrationally hot in the midst of a relatively cold protein). This is analogous to the picture painted by Hynes and coworkers for SN2 reactions in water, where a substantial portion of the activation energy goes into pre-organization of the polar solvent shell to a high energy structure that allows nucleophilic substitution to occur with a low intrinsic barrier within it.35–38
Given the small KIEs and the vibrational coupling proposed here, the present results must be considered within the context of hydrogen tunneling.39,40 QM/MM studies show that proton transfers between Cα of alanine and either Lys39 or Tyr265 are very nearly symmetric, yet the intrinsic KIEs are small (1.66 ± 0.09 in the L→D direction, and 1.57 ± 0.05 in the D→L direction).20,21,23,41 Huskey has discussed the role of coupled motions and hydrogen tunneling in the breakdown of the rule of the geometric mean.26 Although the present results are not conclusive, they suggest that tunneling may occur in the AR catalyzed reaction and that protein motions may be involved in promoting proton tunneling between Cα and either Tyr265 or Lys39.
Supplementary Material
ACKNOWLEDGMENT
Funded by U.S. National Institutes of Health grant GM54779.
Footnotes
Supporting Information Experimental procedures, tables of kinetic parameters, and inhibition data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Schwartz SD, Schramm VL. Nat Chem Biol. 2009;5:551. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zhang J, Klinman JP. J Am Chem Soc. 2011;133:17134. doi: 10.1021/ja207467d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Adamczyk AJ, Cao J, Kamerlin SC, Warshel A. Proc Natl Acad Sci U S A. 2011;108:14115. doi: 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Klinman JP. Nat Chem. 2010;2:907. doi: 10.1038/nchem.886. [DOI] [PubMed] [Google Scholar]
- (5).Kamerlin SC, Sharma PK, Chu ZT, Warshel A. Proc Natl Acad Sci U S A. 2010;107:4075. doi: 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kamerlin SC, Warshel A. J Phys Org Chem. 2010;23:677. doi: 10.1002/poc.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, Hubner CG, Kern D. Nature. 2007;450:838. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- (8).Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. Nature. 2007;450:913. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- (9).Klinman JP. Philos Trans R Soc Lond B Biol Sci. 2006;361:1323. doi: 10.1098/rstb.2006.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hammes-Schiffer S, Benkovic SJ. Annu Rev Biochem. 2006;75:519. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- (11).Gao J, Ma S, Major DT, Nam K, Pu J, Truhlar DG. Chem Rev. 2006;106:3188. doi: 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. Science. 2004;303:186. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- (13).Benkovic SJ, Hammes-Schiffer S. Science. 2003;301:1196. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- (14).Bruice TC. Acc Chem Res. 2002;35:139. doi: 10.1021/ar0001665. [DOI] [PubMed] [Google Scholar]
- (15).Phillipson PE. J Mol Biol. 1968;31:319. doi: 10.1016/0022-2836(68)90448-8. [DOI] [PubMed] [Google Scholar]
- (16).Rokop S, Gajda L, Parmerter S, Crespi HL, Katz JJ. Biochim Biophys Acta. 1969;191:707. doi: 10.1016/0005-2744(69)90365-9. [DOI] [PubMed] [Google Scholar]
- (17).Han R, Coleman JE. Biochemistry. 1995;34:4238. doi: 10.1021/bi00013a013. [DOI] [PubMed] [Google Scholar]
- (18).Silva RG, Murkin AS, Schramm VL. Proc Natl Acad Sci U S A. 2011;108:18661. doi: 10.1073/pnas.1114900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kipp DR, Silva RG, Schramm VL. J Am Chem Soc. 2011;133:19358. doi: 10.1021/ja209391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Spies MA, Toney MD. Biochemistry. 2003;42:5099. doi: 10.1021/bi0274064. [DOI] [PubMed] [Google Scholar]
- (21).Spies MA, Toney MD. J Am Chem Soc. 2007;129:10678. doi: 10.1021/ja067643k. [DOI] [PubMed] [Google Scholar]
- (22).Spies MA, Woodward JJ, Watnik MR, Toney MD. J Am Chem Soc. 2004;126:7464. doi: 10.1021/ja049579h. [DOI] [PubMed] [Google Scholar]
- (23).Sun S, Toney MD. Biochemistry. 1999;38:4058. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- (24).Bevington PR. Data reduction and error analysis for the physical sciences. McGraw-Hill; New York: 1969. [Google Scholar]
- (25).Griswold WR, Castro JN, Fisher AJ, Toney MD. J Am Chem Soc. 2012;134:8436. doi: 10.1021/ja302809e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Huskey WP. Journal of Physical Organic Chemistry. 1991;4:361. [Google Scholar]
- (27).Bigeleisen J. Journal of Chemical Physics. 1955;23:2264. [Google Scholar]
- (28).Dametto M, Antoniou D, Schwartz SD. Mol Phys. 2012;110:531. doi: 10.1080/00268976.2012.655337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Quaytman SL, Schwartz SD. J Phys Chem A. 2009;113:1892. doi: 10.1021/jp804874p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Saen-Oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Proc Natl Acad Sci U S A. 2008;105:16543. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Saen-Oon S, Schramm VL, Schwartz SD. Z Phys Chem (N F) 2008;222:1359. doi: 10.1524/zpch.2008.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Quaytman SL, Schwartz SD. Proc Natl Acad Sci U S A. 2007;104:12253. doi: 10.1073/pnas.0704304104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Basner JE, Schwartz SD. J Am Chem Soc. 2005;127:13822. doi: 10.1021/ja043320h. [DOI] [PubMed] [Google Scholar]
- (34).Antoniou D, Abolfath MR, Schwartz SD. J Chem Phys. 2004;121:6442. doi: 10.1063/1.1782813. [DOI] [PubMed] [Google Scholar]
- (35).Gertner BJ, Whitnell RM, Wilson KR, Hynes JT. Journal of the American Chemical Society. 1991;113:74. doi: 10.1021/ja00001a014. [DOI] [PubMed] [Google Scholar]
- (36).Gertner BJ, Wilson KR, Hynes JT. Journal of Chemical Physics. 1989;90:3537. [Google Scholar]
- (37).Gertner BJ, Bergsma JP, Wilson KR, Lee SY, Hynes JT. Journal of Chemical Physics. 1987;86:1377. [Google Scholar]
- (38).Bergsma JP, Gertner BJ, Wilson KR, Hynes JT. Journal of Chemical Physics. 1987;86:1356. [Google Scholar]
- (39).Nagel ZD, Klinman JP. Chem Rev. 2006;106:3095. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- (40).Nagel ZD, Klinman JP. Chem Rev. 2010;110:PR41. doi: 10.1021/cr1001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Major DT, Gao J. J Am Chem Soc. 2006;128:16345. doi: 10.1021/ja066334r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.