

Abstract
Pancreatic cysts are a heterogeneous group of lesions, which can be benign or malignant. Due to improved imaging techniques, physicians are more often confronted with pancreatic cysts. Little is known about the origin of pancreatic cysts in general. Von Hippel-Lindau (VHL) disease is an atypical ciliopathy and inherited tumor syndrome, caused by a mutation in the VHL tumor suppressor gene encoding the VHL protein (pVHL). VHL patients are prone to develop cysts and neuroendocrine tumors in the pancreas in addition to several other benign and malignant neoplasms. Remarkably, pancreatic cysts occur in approximately 70% of VHL patients, making it the only hereditary tumor syndrome with such a discernible expression of pancreatic cysts. Cellular loss of pVHL due to biallelic mutation can model pancreatic cystogenesis in other organisms, suggesting a causal relationship. Here, we give a comprehensive overview of various pVHL functions, focusing on those that can potentially explain pancreatic cyst development in VHL disease. Based on preclinical studies, cilia loss in ductal cells is probably an important early event in pancreatic cyst development.
Keywords: Cilia, Cytoskeleton, Pancreatic cysts or serous cystadenomas, VHL tumor suppressor protein, von Hippel-Lindau disease
Review
Introduction
Pancreatic cysts are frequent, with a prevalence of 2.4 to 13.5% in patients without known pancreatic disease. Due to increased use of cross-sectional imaging techniques, physicians are more frequently confronted with pancreatic cysts [1]. Various types of pancreatic cysts can occur, which can be benign or have malignant potential. An expectative policy is accepted for benign cysts and surgery is indicated for malignant lesions. Currently, accurate diagnostics are not available to identify malignant cysts [1]. Despite the need for mechanistic insight, little is known about the origin and pathophysiology of pancreatic cysts in general.
Von Hippel-Lindau (VHL) disease (MIM #193300) is a rare hereditary tumor syndrome that results from a germline mutation in the VHL gene. The reported incidence is 1 per 36,000 live births and a >90% penetrance is present by the age of 65 years [2]. VHL disease can lead to the development of hemangioblastomas of the central nervous system, retinal angiomas, endolymphatic sac tumors, epididymis or broad ligament cystadenomas, renal cysts and renal cell carcinomas (RCCs), pheochromocytomas, pancreatic cysts and pancreatic neuroendocrine tumors (pNETs) [3] (Figure 1). Currently, RCC and hemangioblastomas are the main causes of death [4,5]. VHL patients undergo screening for early detection of manifestations [6]. Understanding the role of the VHL gene in the oxygen-sensing pathway in the tumor micro-environment of RCC has led to major pharmaceutical successes through targeted therapies for many cancer types, such as humanized antibodies targeting vascular endothelial growth factor (VEGF), mTOR- and VEGF receptor tyrosine kinase inhibitors [7]. As a result, first-line treatment of metastasized RCCs has entirely changed in the last decade.
Figure 1.

VHL disease can affect various organs. On the left, the organs in which cysts as well as solid lesions occur, have been listed and on the right are the locations in which only hypervascular solid lesions occur. (Constructed using Servier Medical art).
pNETs are present in 10 to 17% of VHL patients [8,9] and pancreatic cysts occur in about 70% [10,11]. Because of this high prevalence, it is worthwhile examining the early cellular events that result in pancreatic cysts in VHL disease, reflecting insight into pancreatic cystic disorders in general. In this review, we conduct a complete overview of pVHL functions to explain cellular events involved in cyst development in the context of VHL. Based on knock-out mouse models, we discuss the consequences of Vhlh loss in the pancreas and the origin of pancreatic cysts.
Pancreatic involvement in VHL disease
VHL pancreatic cysts include simple cysts and serous cystadenomas. In addition to these cysts pNETs occur in VHL patients, which can have malignant potential [10]. One autopsy series of 50 VHL patients showed a prevalence of 72% for pancreatic cysts [11]. In the largest clinical study describing pancreatic involvement, 158 VHL patients underwent abdominal computed tomography scan at least once. Pancreatic involvement was observed in 77% of patients: 71% had simple cysts, 15% had serous cystadenomas and 10% had pNETs, which coincided with pancreatic cysts in 11 cases (69%) [10]. In VHL patients, a broad heterogeneity is present regarding pancreatic cyst involvement: isolated cystadenomas and small cysts occur, whereas in some patients cystic growth replaces almost the entire pancreas (Figure 2) [10,12-16].
Figure 2.
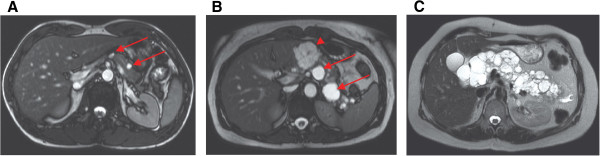
Axial MRI images of pancreatic involvement in three VHL patients. None of these patients had pancreas-related symptoms or exocrine/endocrine insufficiency. (A) Simple cysts (arrows) with size <1 cm in a 32 year old man; (B) A 4 cm sized serous microcystic cystadenoma (arrowhead) is present next to multiple simple cysts (arrows) in a 39 year old woman; (C) Shows replacement of almost the entire pancreas by multiple cysts in a 47 year old woman.
Data is limited about clinical consequences of VHL pancreatic cysts. One study [10] and numerous case-reports [12,13,17-23] have recorded clinical problems, of which compression of the biliary tract was most frequently reported (Table 1). Intervention was indicated in only 3% of VHL patients [10]. No evidence exists for an association between endocrine or exocrine pancreatic insufficiency and cyst involvement. Moreover, no cases have been described of malignant pancreatic cysts in VHL disease. Nineteen cases with a pancreatic serous cystadenoma mixed with a pNET were reported [10,14-16,24,25], but no relationship exists between presence of pancreatic cysts and pNETs. Conclusively, pancreatic cysts in VHL disease are not associated with malignancy and sporadically cause problems [26].
Table 1.
Complications caused by pancreatic cysts in VHL disease
| Reference [No.] | No. of patients | Symptoms | Diagnosis | Therapeutic intervention |
|---|---|---|---|---|
| [17] |
1 |
Jaundice |
Bile duct obstruction |
Surgery |
| [19] |
1 |
Jaundice |
Bile duct obstruction |
Biliary stent |
| [20] |
2 |
Jaundice |
Bile duct obstruction |
Surgery |
| [12] |
2 |
Jaundice |
Bile duct obstruction |
Surgery |
| [22] |
1 |
Abdominal pain, jaundice and fever |
Bile duct obstruction |
Surgery |
| [23] |
1 |
Pruritis |
Bile duct obstruction |
Surgery |
| [10] |
2 |
Abdominal pain |
Pressure/ obstruction |
1 Drainage |
| |
|
|
|
1 Surgery |
| [10] |
1 |
Not reported |
Necrotizing pancreatitis |
Expectative |
| [21] |
1 |
Vomiting |
Pressure on stomach and duodenum |
Surgery |
| [13] |
1 |
Abdominal swelling |
Hemosuccus pancreaticus |
Surgery |
| [18] | 1 | Abdominal swelling | Duodenal compression | Surgery |
VHL disease classification
The clearest genotype-phenotype correlation is exemplified by type 2 VHL, typically characterized by a VHL missense mutation and presence of pheochromocytomas [27,28]. Type 1 VHL is more frequently characterized by a VHL truncating mutation and absence or rare occurrence of pheochromocytomas. Type 2 VHL alleles can be further subdivided based on absence or presence of RCC; called VHL type 2A and type 2B, respectively [28,29]. A pheochromocytoma-only subtype has also been described: VHL type 2C [30]. Pancreatic involvement occurs in both VHL type 1 and type 2B, although it is unclear whether it occurs in the rare VHL types 2A and 2C [31].
The VHL gene
The VHL gene was identified in 1993 [32] and is a tumor suppressor gene; somatic inactivation of the wild-type allele or loss of heterozygosity (LOH) of the VHL gene is often observed prior to development of VHL-associated lesions [33,34]. Consisting of three exons, the human VHL gene is located on chromosome 3 (3p26-p25), encoding a 213-amino acid pVHL (30 kDa VHLp30) and a 160-amino acid shorter form (19 kDa VHLp19) [35,36]. The role of VHL in the oxygen-sensing pathway is its best-characterized function: cellular normoxic conditions enable the pVHL E3 ubiquitin ligase complex to target the α-subunit of hypoxia inducible factor (HIF) for proteosomal degradation. During hypoxia, pVHL is not able to bind HIF-α, resulting in accumulation of un-ubiquitinated HIF-α, which then translocates to the nucleus. This stimulates the transcription of various genes, including VEGF[37].
Since pVHL is still capable of functioning within the E3 ubiquitin ligase complex in VHL type 2C [38], other pVHL functions must be present to induce tumorigenesis. Indeed, VHL also regulates the assembly of the extracellular matrix (ECM) [39-45], and recent studies have shown that pVHL regulates the microtubule cytoskeleton, particularly plus-end stability [46-54].
Sporadic pancreatic serous cystadenomas
Data suggest that VHL loss through LOH of chromosome 3p might be a common mechanism initiating cystogenesis in sporadic pancreatic cystadenomas [55,56]. Recently, whole-exome sequencing was performed in various sporadic pancreatic cysts [57]. Seven out of eight serous cystadenomas lost chromosome 3p alleles, and in four, VHL gene mutations were found. This further supports that VHL loss initiates cystogenesis in pancreatic cystadenomas. Given that loss of the VHL locus was the only recurrent lesion identified, these data indicate that VHL loss alone could be sufficient for this development. Interestingly, in this same study, intraductal papillary mucinous neoplasms, mucinous cystic neoplasms, and solid pseudopapillary neoplasms did not show alterations in 3p alleles. In these lesions other genes encoding E3 ubiquitin ligases were involved, pointing to protein turnover as an underlying mechanistic theme [57].
pVHL regulates cellular architecture
Most in vitro studies exploring the influence of pVHL on the cytoskeleton have been performed in RCC cell lines. To the best of our knowledge, no comparable studies have been performed with pancreatic cell lines. Since somatic LOH of wild-type VHL allele has been verified in pancreatic cysts in VHL [58], we reviewed the existing literature based on RCC cell line studies to gain insight into the effect of pVHL on pancreatic cell regulation and cyst development. Figure 3 represents a schematic overview of pVHL functions which might also explain pancreatic cyst development in VHL disease.
Figure 3.

Schematic overview of functions of pVHL.
pVHL and the extracellular matrix
The ECM consists of proteins including collagen, fibronectin and laminin. Fibronectin plays a major role in the spread and migration of cells by binding them to the ECM. Integrins are cell surface receptors that mediate cell-cell and cell-ECM attachment [59]. pVHL promotes cell adhesion to the ECM [40]. VHL inactivation in RCC cells, mouse embryos and mouse embryo fibroblasts impair the ability to form a fibronectin assembly [39]. Fibrillar adhesions are essential to form a fibronectin assembly. Despite sufficient fibronectin, VHL−/− RCC cells fail to construct β1-integrin fibrillar adhesions due to deficient integrin regulation [41]. In RCC cell lines with wild-type VHL, collagen IV interacts with pVHL [42]. More specifically, pVHL binds the collagen IVα2 chain, part of the triple helix collagen IV; whereas in RCC cells with mutant pVHL this interaction fails [43]. This results in loss of collagen network in vitro and collagen remodeling in vivo (Figure 4) [42]. Collagen IV associates with fibronectin, suggesting that the previously observed interaction between pVHL and fibronectin is indirect [43].
Figure 4.

Overview of direct and indirect pVHL-regulated cell processes in an epithelial cell. The cilium (1) consists a microtubule-based axoneme (2), and a mother (3) and a daughter centriole (4). In the cell, pVHL is located in the cilium (1), the cytoplasm (5), the endoplasmic reticulum (6) and microtubules (7). The endoplasmic reticulum is the cellular compartment where trihelical collagen IV (8) is produced. pVHL binds collagen IVα2 in the endoplasmic reticulum. When pVHL-collagen IVα2 binding is perturbed, defects in the collagen network results. Integrins (9) facilitate cell-ECM (10) adhesion. Upon loss of pVHL function, misregulation of β1-integrin disturbs the fibronectin matrix assembly. The epithelial cell polarity complex PAR3-PAR6-aPKC is located at the apical membrane (11) and LGL2 at the basolateral membrane (12). Moreover, pVHL and PAR3-PAR6-aPKC are necessary for the formation of adherens junctions (13) and tight junctions (14). PAR3-PAR6-aPKC and pVHL play a role in regulation of the cilium, each capable of binding kinesin-2 (15). Functional loss of pVHL destabilizes cell polarity, partially attributable to abnormal adherens junctions [with consequent effects on the actin belt (16)] and unstable tight junctions. The relevant literature supports a scenario whereby microtubule instability as a result of pVHL dysfunction might affect PAR3-PAR6-aPKC localization, subsequently destabilizing cell polarity and cilia maintenance.
In a xenograft model using RCC cell lines, VHL-ECM, VHL-HIF or both pathways were inactivated. VHL−/− 786–0 cells expressing HIF-2α failed to assemble an ECM. VHL+/+ cells retrovirally infected to produce proteasome-resistant HIF-2α, were still able to assemble an ECM, indicating that it is independent of VHL-HIF regulation. Alternatively in VHL type 2C cells, mutant pVHL regulates HIF normally, but interferes with ECM assembly [45]. Xenograft tumors from VHL type 2C cells were hypervascular and invasive, similar to tumors originating from VHL−/− 786–0 cells. In contrast, xenografts derived from VHL+/+ cells engineered to stabilize HIF-2α resulted in tumors with lower microvessel density and invasiveness, despite higher VEGF expression [45]. Thus, upregulation of VEGF alone is not sufficient for hypervascularization of tumors. Kurban et al. suggested that a strong collagen IV network, dependent on pVHL, naturally suppresses tumorigenesis [45].
pVHL and cell polarity
Epithelial cells have asymmetric specification of membrane domains. Asymmetry and polarization are regulated by partitioning defective proteins (PAR) and atypical protein kinase C (aPKC). The PAR3-PAR6-aPKC complex is essential for establishing the apical membrane domain and junction structures of epithelial cells [60]. In addition, this complex is involved in formation of the apical lumen in three-dimensional cultures [61].
The pVHL ubiquitin ligase complex targets the active form of aPKC for degradation, analogous to HIF-α [62]. VHL mutant cells fail to form intercellular junctions, resulting in lost polarity [44]. This may be due to deregulation of active aPKC. Moreover, pVHL associates with the PAR3-PAR6-aPKC complex [50]. In a follicular epithelial model in Drosophila, loss of wild-type vhl resulted in epithelial disorganization, microtubule destabilization and subsequent aPKC mislocalization [63] (Figure 4). Duchi et al. concluded that loss of VHL in vivo can destabilize strict planar cell polarity control, resulting in architectural changes permissive to cyst development [63].
pVHL and microtubule dynamics
Microtubules are polymerized filaments composed of α- and β-tubulin monomers. Microtubules continuously shrink at their “minus-ends” and grow at their “plus-ends” [64]. pVHL promotes microtubule stabilization by reducing tubulin turnover [46,47,54]; it binds microtubules through kinesin-2 [48]. In vitro inhibition of tubulin GTP-ase activity by pVHL at microtubule plus-ends contributes to this stability, which is compromised by VHL patient-associated mutant alleles [54]. Furthermore, cellular inactivation of pVHL results in spindle misorientation, spindle checkpoint weakening and chromosomal instability attributed to microtubule instability [53]. Interestingly, pVHL directs growth of microtubule orientation towards the outer plasma membrane [50].
pVHL and cilia
Microtubules form the backbone of cilia, which project from the apical cell surface. Cilia sense outside the cell and are involved in signaling pathways. Intraflagellar transport of ciliary components is required for ciliary functions, which is powered by kinesin-2. The heterotrimeric motor kinesin-2 comprises motor subunits of kinesin superfamily protein 3 (KIF3A, KIF3B) and kinesin-associated protein 3 (KAP3) [65]. pVHL binds KIF3A and KAP3 of kinesin-2 [48], and in the cell, mobility of pVHL is at least partially regulated by kinesin-2 [49].
In renal tissue from VHL patients, cilia are lost in cysts, while cilia are still present in normal tissue [51]. Accordingly, RCC cell lines with or without reconstitution of wild-type VHL, show that pVHL contributes to ciliary maintenance and stability [51,52]. Cilia loss in kidney tubules due to pVHL dysfunction likely results from disoriented microtubule growth and decreased microtubule stability [50], and is associated with renal cyst development (Figure 4) [51]. It has not been confirmed that pancreatic cysts in VHL patients are the consequence of cilia loss. However, in a pancreatic-specific Kif3a knock-out mouse model, cyst development was attributed to cilia loss (see “Cilia loss in pancreatic cells in vivo”) [66]. Therefore, it is likely that pancreatic cysts in VHL are a result of similar consequences.
Histopathology of the pancreas in VHL
Embryonic epithelial cells that express the transcription factor pancreatic and duodenal homeobox 1 (Pdx1) give rise to pancreatic tissue, consisting of exocrine (acinar and duct) and endocrine (islet) cells [67]. Centro-acinar cells are duct cells, which connect acini with intralobular ducts. Recently centro-acinar cells were isolated based on specific expression of aldehyde dehydrogenase 1. In vitro, centro-acinar cell suspensions were able to proliferate with a capacity to differentiate into both exocrine and endocrine cells [68]. Centro-acinar cells might therefore act as facultative progenitor cells in the mature pancreas for acinar, duct and islet cells (Figure 5).
Figure 5.

Pancreatic progenitor or Pdx-cells can differentiate into endocrine or exocrine cells, including acinar, ductal and centro-acinar cells. Strong evidence supports the assertion that centro-acinar cells, similar to Pdx-cells, can differentiate into both endocrine and exocrine cells (Constructed using Servier Medical art).
Histology of pancreatic cysts and pNETs
Pancreatic cysts and pNETs in VHL disease have distinct features. pNETs have a solid, trabecular and/or glandular architecture with stromal collagen bands and neurosecretory dense core granules [69], which are absent in VHL pancreatic cysts [58]. Immunohistochemical staining for chromogranin A, S-100, synaptophysin and neuron-specific enolase showed positive expression in VHL pNETs [69]. Pancreatic cysts stained negative for chromogranin A and S-100 [58]. The histology of 119 pancreatic cysts was examined in detail from nine VHL patients [58]. All demonstrated a mixture of clear and/or amphophilic glycogen-rich epithelial cells, endothelial cells and smooth muscle cells. Cysts contained and were surrounded by fibrous tissue. In both pNETs and pancreatic cysts, LOH of VHL wild-type allele was confirmed [58,69].
The pancreas in VHL mouse models
Constitutional inactivation of Vhlh results in embryonic lethality at 10.5 to 12.5 days of gestation, due to placental vasculogenesis defects [70]. To investigate the development of VHL-associated pancreatic manifestations, conditional mouse models have been generated using Cre/LoxP technology [71]. In other mouse models Vhlh, Hif-1α or both, were conditionally inactivated in pancreatic β-cells in order to investigate the role of pVHL in glucose metabolism [72-75].
In the first study [71], the Vhlh gene was inactivated in pancreatic progenitor cells by driving Cre recombinase with the Pdx-1 promoter. Postnatal death was observed in the Pdx1-Cre;Vhlh f/f mice (n = 22), of which 18 were dead within five days. Histological analyses by pathologists blinded for genotypes showed no abnormalities. However, five individual Pdx1-Cre;Vhlh f/f mice survived longer. At 6 to 7 months of age, no pancreatic abnormalities were found in two Pdx1-Cre;Vhlh f/f mice. The remaining three were sacrificed at 16 to 18 months of age. At this time point, pancreatic tissue was replaced by fat deposition and pancreatic cysts, and microcystic adenomas were present. The epithelial lining and endothelial cells of the microcystic adenomas expressed cytokeratin MAK6 and CD31, respectively, comparable to findings of pancreatic cysts in VHL patients [58]. In Pdx1-Cre;Vhlh f/f mice, all pancreatic islets were characterized by complex and dilated hypervascularity. Some islets had a small, abnormally shaped appearance and others were hyperplastic [71]. In another study Pdx1-Cre;Vhlh f/f mice were born in the normal Mendelian frequencies. At 12 months of age no cysts or tumors were seen, but a slightly increased pancreatic vascularization was present, compared to control mice [73].
In other knock-out mouse models the Vhlh gene was inactivated by targeting endocrine pancreatic cells for Cre recombinase [71]. In mice with conditional Vhlh inactivation of endocrine α-cells or β-cells, no pancreatic abnormalities were observed (n = 16). Deletion of Vhlh alleles in α- or β-cells was confirmed by polymerase chain reaction analysis [71]. However, others did find increased vascularization in islets in conditional Vhlh knock-out mice in β-cells [73-75]. Additionally, disrupted islet morphology with α-cells scattered throughout the islets were found in Vhlh knock-out mice in β-cells [75]. In all these models, no pNETs were observed. Table 2 shows an overview of knock-out mouse models, serving as a VHL pancreatic model.
Table 2.
VHL (related) pancreatic knock-out mouse models
| Reference [No.] | Knock-out gene(s) | Target cells | Age (months) | Pancreatic manifestations |
|---|---|---|---|---|
| [71] |
Vhlh |
Progenitor |
6-8 |
None |
| [73] |
Vhlh |
Progenitor |
12 |
Increased pancreatic vascularization |
| [71] |
Vhlh |
Progenitor |
16-18 |
Cysts, microcystic adenomas, fat depositions, abnormal shaped and hyper-vascular islet cells |
| [71] |
Vhlh |
Endocrine α |
10-23 |
None |
| [71] |
Vhlh |
Endocrine β |
15 |
None |
| [73] |
Vhlh |
Endocrine β |
12 |
Increased vascularization in islets |
| [72] |
Vhlh |
Endocrine β |
6.5 |
None |
| [72] |
Vhlh; Hif-1α |
Endocrine β |
6.5 |
None |
| [72] |
Hif-1α |
Endocrine β |
6.5 |
None |
| [74] |
Vhlh |
Endocrine β |
N/A |
Increased vascularization in islets |
| [75] |
Vhlh |
Endocrine β |
2 |
Increased vascularization and α-cells scattered throughout the islets |
| [66] |
Kif3a |
Progenitor |
2 |
Compromised acinar tissue |
| [66] | Kif3a | Progenitor | 12 | Fibrosis, ductal dilation, cysts |
Origin of pancreatic lesions in VHL
Whether pNETs originate from exocrine or endocrine cells remains unknown [76]. In pancreatic tissue of 13 VHL patients who underwent surgery because of pNETs, microadenomas were found ranging from 1 to 25 per patient. Expression of cyclin D1, carbonic anhydrase 9 and HIF-1α suggests that these microadenomas occur due to LOH of VHL in clonal lesions, which might be an early stage of pNETs. Most microadenomas were located close to acinar cells, but were also found close to duct or islet cells [76].
In contrast, evidence exists that pancreatic serous cystadenomas originate from duct cells. In these cysts and duct cells, co-expression of cytokeratin patterns is present, as determined immunohistochemically [77,78]. Moreover, of 38 serous cysts including two VHL-related, 70% stained positive for mucin 6 [79], which is also expressed in duct and essentially centro-acinar cells [80]. This suggests a ductal/centro-acinar origin for pancreatic serous cysts [79].
Cilia loss in pancreatic cells in vivo
A study with conditionally inactivated Kif3a in pancreatic tissue in mice suggests that pancreatic cysts originate from duct cells, as a result of cilia loss [66]. Pdx1-Creearly;Kif3a f/f mice were sacrificed at different time points: at two days postnatal, loss of acini was observed and enhancement of lumen between acinar cells and interstitial cells in acini; at 15 days progressive lumen between acini and duct dilation were found. These pathologies all progressed with age and led to acinar tissue being replaced by adipose tissue, severe fibroses, fluid-filled cysts and extensive ductal dilation (age 6 to 12 months). Cilia were lacking in all pancreatic cells. To further identify the cells of origin, conditional knock-out mice were developed for Kif3a inactivation in islet cells only, as well as in islet and acinar cells. In mice lacking Kif3a in islet cells, no morphological abnormalities were observed. Cilia were only present in duct and reduced in islet cells, suggesting that pancreatic cysts originate from ductal cells [66].
Conclusions
We argue that VHL disease can serve as an excellent model to improve the understanding of pathophysiology of pancreatic cysts in general. In vitro studies support a role for pVHL in microtubule stabilization and subsequent cilia maintenance. Loss of cilia is directly related to renal cyst development in VHL and in other renal cystic syndromes [51]. Other cell aspects are also involved; pVHL influences assembly of the extracellular matrix as well as the cytoskeleton, including cell polarity.
In Pdx1-Cre;Vhlh f/f mice with pancreatic-specific loss of Vhlh, pancreatic cysts were observed after 16 to 18 months [71]. Conditional Kif3a knock-out in pancreatic duct cells in mice also resulted in cysts, but changes were already observed after 2 days [66]. Assuming that cysts mainly result from cilia loss, these data suggest that loss of additional alleles might be necessary for Vhlh-driven pancreatic cyst development. In contrast, exome sequencing of sporadic human pancreatic serous cysts only found chromosome 3p loss/VHL mutations as recurrent genetic lesions, suggesting that VHL loss is sufficient for pancreatic cyst development. The high prevalence of pancreatic cysts in patients with VHL disease supports this notion. Differences in findings might be attributed to differences in species, since in general mice seem to be relatively resistant to VHL loss when compared to humans [81], indicating that in humans VHL loss alone might be sufficient for pancreatic serous cyst development.
Monogenetic diseases like VHL provide insights which can be translated to pancreatic cysts in general. Better understanding and identification of genes regarding pancreatic cyst development will probably provide new directions for diagnostics, follow-up and treatment options. Future studies should focus more on VHL and also other E3 ubiquitin ligase genes, which appear to be involved in pancreatic cysts.
Abbreviations
aPKC: Atypical protein kinase C; ECM: Extracellular matrix; HIF: Hypoxia inducible factor; KAP: Kinesin-associated protein; LOH: Loss of heterozygosity; PAR: Partitioning defective proteins; Pdx1: Pancreatic and duodenal homeobox 1; pNET: Pancreatic neuroendocrine tumor; pVHL: von Hippel-Lindau protein; RCC: Renal cell carcinoma; VEGF: Vascular endothelial growth factor; VHL: von Hippel-Lindau.
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
SA: planning/conducting the review, collecting and analysis/interpretation of literature, drafting the manuscript. EV: analysis/interpretation of literature, critical review and revision of the manuscript. HD: critical review and revision of the manuscript. AB: critical review and revision of the manuscript. AW: critical review and revision of the manuscript. RG: analysis/interpretation of literature, critical review and revision of the manuscript. TL: analysis/interpretation of literature, critical review and revision of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Sophie J van Asselt, Email: s.j.van.asselt@umcg.nl.
Elisabeth GE de Vries, Email: e.g.e.de.vries@umcg.nl.
Hendrik M van Dullemen, Email: h.m.dullemen@umcg.nl.
Adrienne H Brouwers, Email: a.h.brouwers@umcg.nl.
Annemiek ME Walenkamp, Email: a.walenkamp@umcg.nl.
Rachel H Giles, Email: r.giles@umcutrecht.nl.
Thera P Links, Email: t.p.links@umcg.nl.
Acknowledgments
Supported by Grant 2008–4188 from the Dutch Cancer Society and a grant from the VHL Family Alliance. RHG acknowledges support from EU FP7/2009 241955 “SYSCILIA”. The work was independent of the funding. We would like to thank Esther M van Straten for assistance of illustration of Figure 4.
References
- de Jong K, Bruno MJ, Fockens P. Epidemiology, diagnosis, and management of cystic lesions of the pancreas. Gastroenterol Res Pract. 2012;2012:147465. doi: 10.1155/2012/147465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, Sampson J, Williams A, Ferguson-Smith MA, Morton N. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443–447. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, Ferguson-Smith MA. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151–1163. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Eggert HR, Scheremet R, Schumacher M, Mohadjer M, Wakhloo AK, Volk B, Hettmannsperger U, Riegler P, Schollmeyer P. Central nervous system lesions in von Hippel-Lindau syndrome. J Neurol Neurosurg Psychiatry. 1992;55:898–901. doi: 10.1136/jnnp.55.10.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The VHL Handbook. 2012. http://www.vhl.org/patients-caregivers/resources/vhl-handbooks/
- Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruine AP. VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010;221:125–138. doi: 10.1002/path.2689. [DOI] [PubMed] [Google Scholar]
- Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, Seidel G, Shutack Y, Yuldasheva N, Eugeni M, Bartlett DL, Glenn GM, Middelton L, Linehan WM, Libutti SK. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs) Surgery. 2007;142:814–818. doi: 10.1016/j.surg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlic Z, Ploeckinger U, Cascon A, Hoffmann MM, von Duecker L, Winter A, Kammel G, Bacher J, Sullivan M, Isermann B, Fischer L, Raffel A, Knoefel WT, Schott M, Baumann T, Schaefer O, Keck T, Baum RP, Milos I, Muresan M, Peczkowska M, Januszewicz A, Cupisti K, Tonjes A, Fasshauer M, Langrehr J, von Wussow P, Agaimy A, Schlimok G, Lamberts R. VHL-ICT Consortium, German NET Registry et al. Systematic comparison of sporadic and syndromic pancreatic islet cell tumors. Endocr Relat Cancer. 2010;17:875–883. doi: 10.1677/ERC-10-0037. [DOI] [PubMed] [Google Scholar]
- Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas JM, Chauveau D, Balian A, Beigelman C, O’Toole D, Bernades P, Ruszniewski P, Richard S. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087–1095. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- Horton WA, Wong V, Eldridge R. Von Hippel-Lindau disease: clinical and pathological manifestations in nine families with 50 affected members. Arch Intern Med. 1976;136:769–777. doi: 10.1001/archinte.1976.03630070017007. [DOI] [PubMed] [Google Scholar]
- Cheng TY, Su CH, Shyr YM, Lui WY. Management of pancreatic lesions in von Hippel-Lindau disease. World J Surg. 1997;21:307–312. doi: 10.1007/s002689900234. [DOI] [PubMed] [Google Scholar]
- Kanno A, Satoh K, Hamada S, Hirota M, Masamune A, Motoi F, Egawa S, Unno M, Ishida K, Kimura K, Shuin T, Shimosegawa T. Serous cystic neoplasms of the whole pancreas in a patient with von hippel-lindau disease. Intern Med. 2011;50:1293–1298. doi: 10.2169/internalmedicine.50.4946. [DOI] [PubMed] [Google Scholar]
- Hough DM, Stephens DH, Johnson CD, Binkovitz LA. Pancreatic lesions in von Hippel-Lindau disease: prevalence, clinical significance, and CT findings. AJR Am J Roentgenol. 1994;162:1091–1094. doi: 10.2214/ajr.162.5.8165988. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay B, Sahdev A, Monson JP, Besser GM, Reznek RH, Chew SL. Pancreatic lesions in von Hippel-Lindau disease. Clin Endocrinol (Oxf) 2002;57:603–608. doi: 10.1046/j.1365-2265.2002.01637.x. [DOI] [PubMed] [Google Scholar]
- Delman KA, Shapiro SE, Jonasch EW, Lee JE, Curley SA, Evans DB, Perrier ND. Abdominal visceral lesions in von Hippel-Lindau disease: incidence and clinical behavior of pancreatic and adrenal lesions at a single center. World J Surg. 2006;30:665–669. doi: 10.1007/s00268-005-0359-4. [DOI] [PubMed] [Google Scholar]
- Beerman MH, Fromkes JJ, Carey LC, Thomas FB. Pancreatic cystadenoma in von Hippel-Lindau disease: an unusual cause of pancreatic and common bile duct obstruction. J Clin Gastroenterol. 1982;4:537–540. doi: 10.1097/00004836-198212000-00011. [DOI] [PubMed] [Google Scholar]
- Jackaman FR. Polycystic pancreas: Lindau’s disease. J R Coll Surg Edinb. 1984;29:121–122. [PubMed] [Google Scholar]
- Deboever G, Dewulf P, Maertens J. Common bile duct obstruction due to pancreatic involvement in the von Hippel-Lindau syndrome. Am J Gastroenterol. 1992;87:1866–1868. [PubMed] [Google Scholar]
- Issar SK, Kumar N, Sachdeva AK, Jain P, Puri SK. von Hippel-Lindau syndrome presenting as obstructive jaundice with involvement of pancreas in two siblings. Trop Gastroenterol. 1996;17:30–32. [PubMed] [Google Scholar]
- Kunzli BM, Shrikhande SV, Buchler MW, Friess H. Pancreatic lesions in von Hippel-Lindau syndrome: report of a case. Surg Today. 2004;34:626–629. doi: 10.1007/s00595-004-2769-6. [DOI] [PubMed] [Google Scholar]
- Boujaoude J, Samaha E, Honein K, Noun R, Abboud B, Ghorra C, Sayegh R. A benign cause of obstructive jaundice with von Hippel-Lindau disease. A case report and review of the literature. JOP. 2007;8:790–794. [PubMed] [Google Scholar]
- Gupta R, Chettri D, Sharma A, Duseja A, Dhiman RK, Chawla YK, Kalra N, Gupta A, Behera A. Pancreatic cysts causing biliary obstruction in von Hippel-Lindau syndrome. JOP. 2008;9:313–316. [PubMed] [Google Scholar]
- Baek SY, Kang BC, Choi HY, Lee SW. Pancreatic serous cystadenoma associated with islet cell tumour. Br J Radiol. 2000;73:83–86. doi: 10.1259/bjr.73.865.10721327. [DOI] [PubMed] [Google Scholar]
- Agarwal N, Kumar S, Dass J, Arora VK, Rathi V. Diffuse pancreatic serous cystadenoma associated with neuroendocrine carcinoma: a case report and review of literature. JOP. 2009;10:55–58. [PubMed] [Google Scholar]
- Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature. J Gastrointest Surg. 2012;16:1422–1428. doi: 10.1007/s11605-012-1847-0. [DOI] [PubMed] [Google Scholar]
- Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, Lerman MI, Zbar B, Affara NA, Ferguson-Smith MA. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994;3:1303–1308. doi: 10.1093/hmg/3.8.1303. [DOI] [PubMed] [Google Scholar]
- Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995;5:66–75. doi: 10.1002/humu.1380050109. [DOI] [PubMed] [Google Scholar]
- Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richard S, Lips CH, Lerman M. Germline mutations in the von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8:348–357. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ritter MM, Frilling A, Crossey PA, Hoppner W, Maher ER, Mulligan L, Ponder BA, Engelhardt D. Isolated familial pheochromocytoma as a variant of von Hippel-Lindau disease. J Clin Endocrinol Metab. 1996;81:1035–1037. doi: 10.1210/jc.81.3.1035. [DOI] [PubMed] [Google Scholar]
- Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, van Brussel A, Voest EE, Giles RH. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31:521–537. doi: 10.1002/humu.21219. [DOI] [PubMed] [Google Scholar]
- Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- Maher ER, Yates JR, Ferguson-Smith MA. Statistical analysis of the two stage mutation model in von Hippel-Lindau disease, and in sporadic cerebellar haemangioblastoma and renal cell carcinoma. J Med Genet. 1990;27:311–314. doi: 10.1136/jmg.27.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossey PA, Foster K, Richards FM, Phipps ME, Latif F, Tory K, Jones MH, Bentley E, Kumar R, Lerman MI. Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Hum Genet. 1994;93:53–58. doi: 10.1007/BF00218913. [DOI] [PubMed] [Google Scholar]
- Schoenfeld A, Davidowitz EJ, Burk RD. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci U S A. 1998;95:8817–8822. doi: 10.1073/pnas.95.15.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos O, Ohh M, Kaelin WG Jr. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A. 1998;95:11661–11666. doi: 10.1073/pnas.95.20.11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase VH. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006;69:1302–1307. doi: 10.1038/sj.ki.5000221. [DOI] [PubMed] [Google Scholar]
- Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10:1029–1038. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG Jr, Iliopoulos O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–968. doi: 10.1016/S1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- Kamada M, Suzuki K, Kato Y, Okuda H, Shuin T. von Hippel-Lindau protein promotes the assembly of actin and vinculin and inhibits cell motility. Cancer Res. 2001;61:4184–4189. [PubMed] [Google Scholar]
- Esteban-Barragan MA, Avila P, Alvarez-Tejado M, Gutierrez MD, Garcia-Pardo A, Sanchez-Madrid F, Landazuri MO. Role of the von Hippel-Lindau tumor suppressor gene in the formation of beta1-integrin fibrillar adhesions. Cancer Res. 2002;62:2929–2936. [PubMed] [Google Scholar]
- Grosfeld A, Stolze IP, Cockman ME, Pugh CW, Edelmann M, Kessler B, Bullock AN, Ratcliffe PJ, Masson N. Interaction of hydroxylated collagen IV with the von Hippel-Lindau tumor suppressor. J Biol Chem. 2007;282:13264–13269. doi: 10.1074/jbc.M611648200. [DOI] [PubMed] [Google Scholar]
- Kurban G, Duplan E, Ramlal N, Hudon V, Sado Y, Ninomiya Y, Pause A. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene. 2008;27:1004–1012. doi: 10.1038/sj.onc.1210709. [DOI] [PubMed] [Google Scholar]
- Calzada MJ, Esteban MA, Feijoo-Cuaresma M, Castellanos MC, Naranjo-Suarez S, Temes E, Mendez F, Yanez-Mo M, Ohh M, Landazuri MO. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006;66:1553–1560. doi: 10.1158/0008-5472.CAN-05-3236. [DOI] [PubMed] [Google Scholar]
- Kurban G, Hudon V, Duplan E, Ohh M, Pause A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006;66:1313–1319. doi: 10.1158/0008-5472.CAN-05-2560. [DOI] [PubMed] [Google Scholar]
- Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5:64–70. doi: 10.1038/ncb899. [DOI] [PubMed] [Google Scholar]
- Lolkema MP, Mehra N, Jorna AS, van Beest M, Giles RH, Voest EE. The von Hippel-Lindau tumor suppressor protein influences microtubule dynamics at the cell periphery. Exp Cell Res. 2004;301:139–146. doi: 10.1016/j.yexcr.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Lolkema MP, Mans DA, Snijckers CM, van Noort M, van Beest M, Voest EE, Giles RH. The von Hippel-Lindau tumour suppressor interacts with microtubules through kinesin-2. FEBS Lett. 2007;581:4571–4576. doi: 10.1016/j.febslet.2007.08.050. [DOI] [PubMed] [Google Scholar]
- Mans DA, Lolkema MP, van Beest M, Daenen LG, Voest EE, Giles RH. Mobility of the von Hippel-Lindau tumour suppressor protein is regulated by kinesin-2. Exp Cell Res. 2008;314:1229–1236. doi: 10.1016/j.yexcr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Schermer B, Ghenoiu C, Bartram M, Muller RU, Kotsis F, Hohne M, Kuhn W, Rapka M, Nitschke R, Zentgraf H, Fliegauf M, Omran H, Walz G, Benzing T. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol. 2006;175:547–554. doi: 10.1083/jcb.200605092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban MA, Harten SK, Tran MG, Maxwell PH. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J Am Soc Nephrol. 2006;17:1801–1806. doi: 10.1681/ASN.2006020181. [DOI] [PubMed] [Google Scholar]
- Lutz MS, Burk RD. Primary cilium formation requires von Hippel-Lindau gene function in renal-derived cells. Cancer Res. 2006;66:6903–6907. doi: 10.1158/0008-5472.CAN-06-0501. [DOI] [PubMed] [Google Scholar]
- Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, Hergovich A, Moch H, Meraldi P, Krek W. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol. 2009;11:994–1001. doi: 10.1038/ncb1912. [DOI] [PubMed] [Google Scholar]
- Thoma CR, Matov A, Gutbrodt KL, Hoerner CR, Smole Z, Krek W, Danuser G. Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J Cell Biol. 2010;190:991–1003. doi: 10.1083/jcb.201006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortmeyer AO, Lubensky IA, Fogt F, Linehan WM, Khettry U, Zhuang Z. Allelic deletion and mutation of the von Hippel-Lindau (VHL) tumor suppressor gene in pancreatic microcystic adenomas. Am J Pathol. 1997;151:951–956. [PMC free article] [PubMed] [Google Scholar]
- Moore PS, Zamboni G, Brighenti A, Lissandrini D, Antonello D, Capelli P, Rigaud G, Falconi M, Scarpa A. Molecular characterization of pancreatic serous microcystic adenomas: evidence for a tumor suppressor gene on chromosome 10q. Am J Pathol. 2001;158:317–321. doi: 10.1016/S0002-9440(10)63971-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Jiao Y, Dal Molin M, Maitra A, de Wilde RF, Wood LD, Eshleman JR, Goggins MG, Wolfgang CL, Canto MI, Schulick RD, Edil BH, Choti MA, Adsay V, Klimstra DS, Offerhaus GJ, Klein AP, Kopelovich L, Carter H, Karchin R, Allen PJ, Schmidt CM, Naito Y, Diaz LA Jr, Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011;108:21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr VH, Vortmeyer AO, Zhuang Z, Libutti SK, Walther MM, Choyke PL, Zbar B, Linehan WM, Lubensky IA. Histopathology and molecular genetics of multiple cysts and microcystic (serous) adenomas of the pancreas in von Hippel-Lindau patients. Am J Pathol. 2000;157:1615–1621. doi: 10.1016/S0002-9440(10)64799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotran RS, Kumar V, Collins T. In: Robbins pathologic basis of disease. Cotran RS, Kumar V, Collins T, editor. WB Saunders Company, Philadelphia; 1999. Tissue repair: cellular growth, fibrosis and wound healing; pp. 89–112. [Google Scholar]
- Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda H, Saitoh K, Hirai S, Iwai K, Takaki Y, Baba M, Minato N, Ohno S, Shuin T. The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J Biol Chem. 2001;276:43611–43617. doi: 10.1074/jbc.M107880200. [DOI] [PubMed] [Google Scholar]
- Duchi S, Fagnocchi L, Cavaliere V, Hsouna A, Gargiulo G, Hsu T. Drosophila VHL tumor-suppressor gene regulates epithelial morphogenesis by promoting microtubule and aPKC stability. Development. 2010;137:1493–1503. doi: 10.1242/dev.042804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofrad MR, Kamm RD. In: Cytoskeletal mechanics: models and measurements. 1. Mofrad MR, Kamm RD, editor. Cambridge University Press, Cambridge; 2006. Introduction, with the biological basis for cell mechanics; pp. 1–17. [Google Scholar]
- D’Angelo A, Franco B. The dynamic cilium in human diseases. Pathogenetics. 2009;2:3. doi: 10.1186/1755-8417-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano DA, Sekine S, Hebrok M. Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology. 2006;131:1856–1869. doi: 10.1053/j.gastro.2006.10.050. [DOI] [PubMed] [Google Scholar]
- Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A. 2010;107:75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubensky IA, Pack S, Ault D, Vortmeyer AO, Libutti SK, Choyke PL, Walther MM, Linehan WM, Zhuang Z. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol. 1998;153:223–231. doi: 10.1016/S0002-9440(10)65563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, Emmert-Buck MR, Westphal H, Klausner RD, Linehan WM. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HC, Adem A, Ylaya K, Wilson A, He M, Lorang D, Hewitt SM, Pechhold K, Harlan DM, Lubensky IA, Schmidt LS, Linehan WM, Libutti SK. Deciphering von Hippel-Lindau (VHL/Vhl)-associated pancreatic manifestations by inactivating Vhl in specific pancreatic cell populations. PLoS One. 2009;4:e4897. doi: 10.1371/journal.pone.0004897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehetner J, Danzer C, Collins S, Eckhardt K, Gerber PA, Ballschmieter P, Galvanovskis J, Shimomura K, Ashcroft FM, Thorens B, Rorsman P, Krek W. PVHL is a regulator of glucose metabolism and insulin secretion in pancreatic beta cells. Genes Dev. 2008;22:3135–3146. doi: 10.1101/gad.496908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley J, Selman C, Shukla D, Abramov AY, Forstreuter F, Esteban MA, Claret M, Lingard SJ, Clements M, Harten SK, Asare-Anane H, Batterham RL, Herrera PL, Persaud SJ, Duchen MR, Maxwell PH, Withers DJ. Deletion of the von Hippel-Lindau gene in pancreatic beta cells impairs glucose homeostasis in mice. J Clin Invest. 2009;119:125–135. doi: 10.1172/JCI26934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri S, Cano DA, Hebrok M. A role for von Hippel-Lindau protein in pancreatic beta-cell function. Diabetes. 2009;58:433–441. doi: 10.2337/db08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D, Cai EP, Schroer SA, Wang L, Woo M. Vhl is required for normal pancreatic beta cell function and the maintenance of beta cell mass with age in mice. Lab Invest. 2011;91:527–538. doi: 10.1038/labinvest.2010.207. [DOI] [PubMed] [Google Scholar]
- Perigny M, Hammel P, Corcos O, Larochelle O, Giraud S, Richard S, Sauvanet A, Belghiti J, Ruszniewski P, Bedossa P, Couvelard A. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: characterization by VHL/HIF pathway proteins expression. Am J Surg Pathol. 2009;33:739–748. doi: 10.1097/PAS.0b013e3181967992. [DOI] [PubMed] [Google Scholar]
- Alpert LC, Truong LD, Bossart MI, Spjut HJ. Microcystic adenoma (serous cystadenoma) of the pancreas. A study of 14 cases with immunohistochemical and electron-microscopic correlation. Am J Surg Pathol. 1988;12:251–263. doi: 10.1097/00000478-198804000-00001. [DOI] [PubMed] [Google Scholar]
- Egawa N, Maillet B, Schroder S, Mukai K, Kloppel G. Serous oligocystic and ill-demarcated adenoma of the pancreas: a variant of serous cystic adenoma. Virchows Arch. 1994;424:13–17. doi: 10.1007/BF00197387. [DOI] [PubMed] [Google Scholar]
- Buisine MP, Devisme L, Degand P, Dieu MC, Gosselin B, Copin MC, Aubert JP, Porchet N. Developmental mucin gene expression in the gastroduodenal tract and accessory digestive glands. II. Duodenum and liver, gallbladder, and pancreas. J Histochem Cytochem. 2000;48:1667–1676. doi: 10.1177/002215540004801210. [DOI] [PubMed] [Google Scholar]
- Kosmahl M, Wagner J, Peters K, Sipos B, Kloppel G. Serous cystic neoplasms of the pancreas: an immunohistochemical analysis revealing alpha-inhibin, neuron-specific enolase, and MUC6 as new markers. Am J Surg Pathol. 2004;28:339–346. doi: 10.1097/00000478-200403000-00006. [DOI] [PubMed] [Google Scholar]
- Hsu T. Complex cellular functions of the von Hippel-Lindau tumor suppressor gene: insights from model organisms. Oncogene. 2012;31:2247–2257. doi: 10.1038/onc.2011.442. [DOI] [PMC free article] [PubMed] [Google Scholar]