

Abstract
Nonhealing diabetic foot ulcers (DFUs) are a common and costly complication of diabetes. Microbial burden, or “bioburden,” is believed to underlie delayed healing, although little is known of those clinical factors that may influence microbial load, diversity, and/or pathogenicity. We profiled the microbiomes of neuropathic nonischemic DFUs without clinical evidence of infection in 52 individuals using high-throughput sequencing of the bacterial 16S ribosomal RNA gene. Comparatively, wound cultures, the standard diagnostic in the clinic, vastly underrepresent microbial load, microbial diversity, and the presence of potential pathogens. DFU microbiomes were heterogeneous, even in our tightly restricted study population, but partitioned into three clusters distinguished primarily by dominant bacteria and diversity. Ulcer depth was associated with ulcer cluster, positively correlated with abundance of anaerobic bacteria, and negatively correlated with abundance of Staphylococcus. Ulcer duration was positively correlated with bacterial diversity, species richness, and relative abundance of Proteobacteria, but was negatively correlated with relative abundance of Staphylococcus. Finally, poor glycemic control was associated with ulcer cluster, with poorest median glycemic control concentrating to Staphylococcus-rich and Streptococcus-rich ulcer clusters. Analyses of microbial community membership and structure may provide the most useful metrics in prospective studies to delineate problematic bioburden from benign colonization that can then be used to drive clinical treatment.
Fifteen percent of those with diabetes will develop at least one diabetic foot ulcer (DFU) during their lifetime (1). Effective treatment strategies for DFUs are still lacking. Systemic and topical antimicrobial treatments are commonly used, even though minimal evidence supports their efficacy in DFU treatment (2). Clinical signs and symptoms of infection cannot be reliably used in chronic wounds to direct antibiotic treatment (3), and the fine line between benign colonization and problematic bioburden from which to direct antibiotic treatment remains unclear. Targeting microbial populations to promote healing and deter infection-related complications might be a novel treatment option.
To establish the role of bacteria in impaired healing and infection-related complication, it is necessary to define the full array of microorganisms colonizing the DFU. Clinical practice and most studies of DFU bioburden have relied on cultivation-dependent methods, which are biased toward those microorganisms that thrive in isolation under laboratory conditions. Importantly, cultivation-based methods tend to overlook slow-growing fastidious bacteria such as anaerobes, a subset of bacteria thought to be particularly damaging to the wound environment (4). Genomic approaches to analyze microbial communities, such as sequencing of the small subunit 16S ribosomal RNA (rRNA) gene, are increasingly accessible and provide much greater resolution by eliminating biases associated with culturing bacteria (5–7). The 16S rRNA gene is present in the genomes of all prokaryotes and contains sequences that allow identification and classification of the organism. The gene also contains highly conserved regions that allow broad-range amplification by PCR (8,9). Three dimensions of bioburden may be important in the nonhealing DFU: microbial load; microbial diversity; and/or pathogenicity (10). Microbial load is the total quantity of microbes present. Microbial diversity is the number of different bacterial taxa present. Potential pathogens of the DFU are believed to include Staphylococcus, Streptococcus, Proteobacteria (a phylum of Gram-negative bacteria), and anaerobic bacteria (4). Genomic methods to evaluate bioburden allow these dimensions to be considered together as the microbial community and structure within a wound. This ability represents a significant step forward in our quest to understand the role that the microbiota play in DFU outcomes and complications.
Previous studies of the chronic wound microbiome using 16S rRNA gene sequencing used study designs and methods that combined and analyzed heterogeneous types of wounds, including ischemic, neuropathic, and mixed-type DFUs (7,11,12). Pathophysiologically distinct DFUs likely lead to a different host/wound environment, ultimately confounding identification of microbial populations associated with DFUs and estimations of DFU microbial diversity. Furthermore, previous studies have not related the DFU microbiota to clinical factors found to be associated with nonhealing. In the absence of useful culture data and the inability to access molecular techniques, clinical factors may be useful for identifying individuals at risk for problematic bioburden because the wound environment may support or deter particular microbiota (7), which then may lead to poor ulcer outcomes. Potential patient and/or ulcer factors found to be associated with failure to heal include wound surface area, ulcer grade, arterial perfusion, necrosis, and wound duration (13–16). Although glycemic control was found to be associated with wound healing in persons with diabetes (17), it has not been found to be associated with healing of DFUs (18). Clinical factors need to be systematically examined to determine which may serve as clinically relevant biomarkers of problematic bioburden.
Here, we report the first systematic exploratory analysis of the microbiome of a homogenous sample of neuropathic nonischemic DFUs (N = 52 subjects) by high-throughput sequencing of the 16S rRNA gene in relation to clinical factors that may influence the microbiome. Specific aims were to compare the three dimensions of DFU bioburden detected by 16S rRNA gene sequencing to those obtained with traditional quantitative cultures, to examine the microbial community structure of DFUs, and to examine clinical factors associated with DFU microbiome.
RESEARCH DESIGN AND METHODS
Design, setting, and sample.
This study used a cross-sectional design. Subjects with DFUs were assessed for microbiota colonizing the DFU using traditional cultures and 16S gene sequencing methods. Clinical factors were concurrently measured. Study protocols were approved by the University of Iowa Institutional Review Board, the National Institutes of Health Office of Human Subjects Research, and the University of Pennsylvania Institutional Review Board. Data were collected at the University of Iowa Hospitals and Clinics. A convenience sample of subjects was enrolled using the following criteria: 18 years of age or older; plantar neuropathic DFU; free of systemic antibiotics over the course of the past 2 weeks; negative for clinical signs of infection; negative for wound deterioration; and negative for osteomyelitis. Plantar neuropathic DFUs were defined as open lesions on the plantar surface of the foot exclusive of the lesser toes in persons with diabetes, insensate to 10-g monofilament on the plantar surface of the foot on one or more plantar locations, and toe–brachial indexes >0.5. Enrolled subjects signed a written informed consent.
Clinical factors.
Protocols for measuring clinical factors have been published elsewhere (19–22). Level of glycemic control was measured as hemoglobin A1c values. Wound tissue oxygen was measured using transcutaneous oxygen measures (Model TCM400; Radiometer America) on the dorsum of the ipsilateral foot. Necrotic tissue was measured using an item from the Pressure Sore Status Tool (23). Ulcer size, including surface area and depth, was measured using digital images and proprietary software, which produces surface area, depth, and volume measures (22). Duration of the study ulcer was measured as the number of weeks from the time of soft tissue loss to the baseline visit obtained through subject report and review of medical records.
Ulcer cultures.
Ulcer specimens were obtained using the Levine technique and established protocols (19). We have demonstrated this technique to have accuracy (area under the receiver-operator characteristic curve) of 0.80 when compared with culture results based on tissue specimens (19). The DFU was cleansed with nonbacteriostatic saline and an Amies with charcoal transport swab (Copan, Brescia, Italy) was rotated over a 1-cm2 area of viable non-necrotic wound tissue for 5 seconds using sufficient pressure to extract wound tissue fluid. Swabs were vortexed in 1 mL tryptic soy broth, and then diluted and plated on Columbia blood agar (Remel), eosin-methylene blue agar (Remel), CHROMagarMRSA (BD), and Brucella Agar supplemented with blood, hemin, and vitamin K (Remel). Organisms that grew on eosin-methylene blue agar plates and stained Gram-negative were further identified using Vitek Legacy (Biomerieux).
16S rRNA gene sequencing, quality control, and analysis.
DNA was isolated from the DFU samples as previously described (24). Briefly, the swab was placed in 300 μL yeast cell lysis solution (from Epicentre MasterPure Yeast DNA Purification kit) and 0.5 μL ReadyLyse lysozyme solution (Epicentre) was added before incubation for 1 h at 37°C with shaking. Samples were then processed in a TissueLyser (Qiagen) at maximum speed for 2 min, followed by 30-min incubation at 65°C with shaking; 150 μL protein precipitation reagent (Epicentre) was added and samples were spun for 10 min at maximum speed. The supernatant was removed and mixed with 500 μL isopropanol and applied to a column from the PureLink Genomic DNA Mini Kit (Invitrogen). Subsequently, the instructions for the Invitrogen PureLink kit were followed exactly, and DNA was eluted in 50 μL elution buffer (Invitrogen). The 16S rRNA gene was amplified from 2 μL of each DFU DNA sample using forward primer 27F and reverse primer 534R containing a unique error-correcting barcode. PCR using the Accumprime kit (Invitrogen) was performed in duplicate to reduce potential amplification bias attributable to the complex mixture of template. Cycling conditions were: 95°C for 2 min, followed by 30 cycles of 95°C for 20 s, 56°C for 30 s, and 72°C for 5 min. PCR products were purified with the Agencourt AMPure kit following manufacturer’s instructions (Beckman Coulter). Negative (no template and mock) controls were treated similarly and failed to produce a visible PCR product or sequencing reads; 50 ng of each PCR product was pooled, and the pool was purified using the MinElute PCR Purification kit (Qiagen) according to manufacturer’s instructions. Pyrosequencing was performed on the Roche 454 FLX Titanium Instrument at the National Institutes of Health Intramural Sequencing Center.
Sequence quality control and analysis were performed using the mothur package version 1.23 (25). Sequences were removed if they contained ambiguous bases, more than eight homopolymers, primer and/or barcode mismatches, or were <200 nt long. Low-quality sequences were removed using the criteria of average quality score of >35 over 50-nt sliding windows. Sequences were aligned to the SILVA reference set using mothur’s NAST-based aligner. Chimeras were identified and removed using the mothur implementation of UChime (26) and the chimera-free GOLD reference dataset (27). Sequences were assigned to operational taxonomic units (OTUs) using an average-neighbor clustering algorithm at a threshold of 0.03 (28). OTUs are molecular proxies for describing organisms based on their phylogenetic relationships to other organisms. Because α and β diversity metrics are sensitive to sampling effort, we standardized the number of sequences per sample by random subsampling using the subsample in mothur. OTUs were assigned to taxonomy using the mothur-implemented naïve Bayesian classifier trained on the Ribosomal Database Project taxonomy training set 4 (29). Staphylococcus OTUs were speciated using pplacer (30) and a custom curated collection of Staphylococcus reference sequences.
To measure microbial load, quantitative real-time PCR assays of the 16S rRNA gene were performed as previously described (6,31).
Data analysis.
Culture findings were compared with sequencing findings using paired t tests (two-tailed; α = 0.05). Agreement in the predominant organism identified by the two methods was examined with kappas. Clustering was performed by partitioning around medoids (PAM). PAM clusters samples by minimizing the distance between samples in a cluster. Each cluster is defined by a point designated as the center (the “medoid”). The input to PAM was a Euclidean distance matrix of normalized species-level OTU counts. Euclidean distance is the straight-line “ordinary” distance between two objects in multidimensional space. Validity of clustering and number of centroids to choose for the data were determined using the average silhouette score, for which a higher score indicates better quality and more natural clustering (32). Clusters were examined for associations with three dimensions of bioburden using Kruskal-Wallis test. Post hoc pair-wise comparisons were completed using Wilcoxon rank-sum tests (two-tailed; α = 0.05). Covariation of species within a DFU were examined with Spearman correlation coefficients (ρ), a measure of statistical dependence between two variables for which a value of 1 indicates complete positive correlation and −1 indicates complete negative correlation. Significance of ρ was assessed using false discovery rate control (q = 0.05). Associations between bioburden and clinical factors were assessed using Spearman rank correlation coefficients (ρ). Overall associations of clinical factors with microbiome community structure and membership were calculated using analysis of molecular variance in the freely available mothur software package. Kruskal-Wallis test was used to assess differences in Euclidean clusters and associations with clinical factors. Post hoc pair-wise comparisons were completed using Wilcoxon rank-sum tests.
RESULTS
Subjects and DFU microbiome measures.
Fifty-two subjects were enrolled with a mean age of 53.9 (±11.89) years. Forty-three (82.7%) were male and 48 (92.3%) were white. Forty-three (82.7%) had type 2 diabetes, whereas the remainder had type 1 diabetes. All subjects (100%) had sensory neuropathy. Forty-two (80.8%) of the DFUs were located on the plantar forefoot. The remaining DFUs were located on the plantar mid foot or hind foot. Mean toe–brachial index was 0.85 (±0.26), indicating no significant problems with arterial perfusion and that these ulcers were primarily neuropathic. The mean hemoglobin A1c was 8.5% (±2.07), the mean wound tissue oxygen was 49.3 mmHg (±9.23; n = 51), the mean ulcer surface area was 2.0 cm (±2.92), the mean ulcer duration was 34.6 weeks (±42.56), and the mean ulcer depth was 0.2 cm (±0.26). Twenty-five ulcers (48%) had no depth or volume. Forty-two (80.8%) ulcers had no necrotic tissue in the wound bed. Five (9.6%) had <25% wound bed necrotic tissue; one (1.9%) had 25–50% necrotic tissue and four (7.7%) had 75–100% necrotic tissue.
We surveyed DFU microbiomes by pyrosequencing the V1–V3 hypervariable regions of the bacterial 16S rRNA gene. We generated 300,660 high-quality sequences, with an average of 5,634 sequences per sample. A total of 13 phyla were identified, but the majority of sequences classified to Firmicutes (67%), Actinobacteria (14%), Proteobacteria (9.8%), Bacteroidetes (7.3%), and Fusobacteria (1.4%). Clustering sequences into species-level OTUs at a threshold of 97% identity revealed 867 OTUs across all samples. After normalizing the number of sequences present in each sample by random subsampling, 477 OTUs were included for further analyses. The average number of OTUs per sample was 30, with a range of 7–64. The most abundant OTU was classified as Staphylococcus and was present in 49 of 52 DFU samples, comprising 29.6% of the total sequences. Because Staphylococcus aureus is believed to be particularly pathogenic to the DFU, we performed a phylogenetic speciation analysis to distinguish among Staphylococcus species. The majority of Staphylococcus sequences (96.5%) were classified as S. aureus. Only 0.4% of the sequences were determined to be Staphylococcus epidermidis, a skin commensal. The second and third most abundant OTUs were Streptococcus (8.8% of the total sequences; present in 15 DFUs) and Lactococcus (3.9% of the total sequences; present in 38 DFUs), respectively.
Culture-based assays underestimate bioburden of DFUs.
In addition to community profiling of the 16S rRNA gene, we performed culture-based assessments of DFU bioburden. We first compared measures of microbial load using a colony-forming unit culture-based estimate and a 16S rRNA quantitative PCR-based estimate. We found that, on average, culturing underestimated bacterial load by 2.34 logs (P < 0.0001), and in some cases >6 logs (Fig. 1A). For each DFU, we also compared the number of species recovered by culture methods to the number of species-level OTUs recovered by sequencing of the 16S rRNA gene. Culture-based techniques failed to capture, on average, 26 bacterial species per DFU (P < 0.0001) (Fig. 1B).
FIG. 1.

Comparison of cultivation-based data to genomic 16S rRNA gene data for characterizing DFU bioburden. A: Prediction of bacterial load (log-transformed) by total counts of colony-forming units (CFUs; circles) compared with estimating bacterial load based on quantitative PCR (qPCR) of the 16S rRNA gene (diamonds). B: Number of different isolates recovered by culturing (circles) vs. the number of species-level OTUs by 16S rRNA gene sequencing (diamonds). Each point represents a DFU sample and the line through the points represents the median of the data.
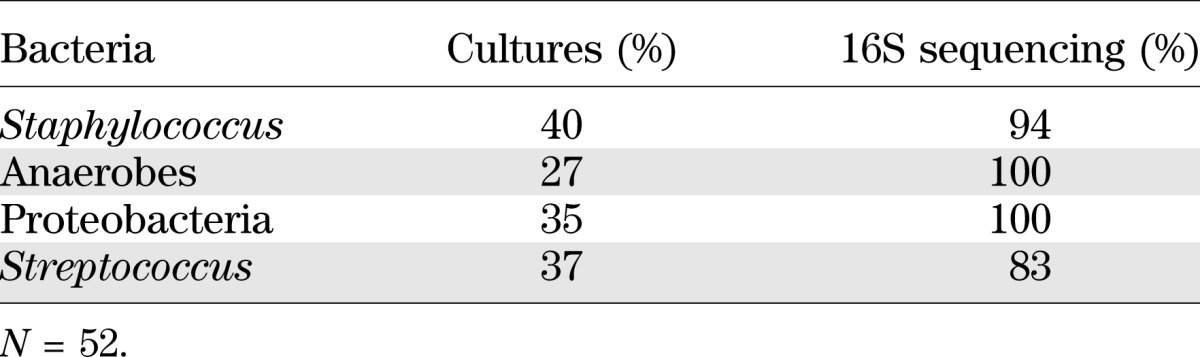
We also compared the relative abundance of organisms believed to be potential pathogens including Staphylococcus, anaerobes, Proteobacteria, and Streptococcus. Culturing overestimated the relative abundance of Staphylococcus, on average, by >15 percentage points (0.47 vs. 0.32; P = 0.0001), whereas culturing underrepresented anaerobic bacteria, on average, by 7.3 percentage points (0.11 vs. 0.18; P = 0.0063). Overall, the agreement between the two methods in identifying the predominant organism was 0.45 (κ: 95% CI = 0.30–0.61), indicating only fair agreement (33). Cultures identified Staphylococcus as the predominant organism in 24 (46%) of the DFUs as compared with 20 (39%) of the DFUs by 16S rRNA gene sequencing. Based on cultures, the number of DFUs that contained S. aureus regardless of abundance was 21 (40%), whereas 16S rRNA gene sequencing revealed that 49 (94%) of the ulcers contained S. aureus (Table 1). By culture, anaerobes were identified as the predominant organism in 6 (12%) of the DFUs, whereas sequencing identified anaerobes as the predominant organism in 12 (23%) of the DFUs. Culturing revealed that 14 (27%) of DFUs contained anaerobes as compared with 52 (100%) DFUs containing anaerobes by sequencing (Table 1). Proteobacteria and Streptococcus were cultured from 18 (35%) and 19 (37%) ulcers, respectively. This is in contrast to 16S rRNA sequencing, which revealed Proteobacteria and Streptococcus in 52 (100%) and 43 (83%) DFUs, respectively (Table 1). In only four DFUs did cultures capture an isolate that was not represented by 16S rRNA sequencing. Taken together, these results led us to rely on 16S rRNA sequence data for further analyses and to examine the relationship between DFU microbiome and clinical variables.
TABLE 1.
Comparison of the percentage of DFUs containing different types of bacteria as assessed using culture techniques vs. 16S rRNA gene sequencing
DFU microbiomes are heterogeneous.
To compare bacterial diversity colonizing individual DFUs, we calculated the Shannon index, an ecological measure of diversity that incorporates the total number of different OTUs and the relative proportions of those OTUs. Higher Shannon index values indicate greater diversity. The average Shannon index was 1.90, with a range of 0.25–3.43, suggesting great heterogeneity in the diversity colonizing individual DFUs (Fig. 2). Examination of the relative abundance of bacterial taxa was not immediately insightful, because there also appeared to be great heterogeneity in the taxa colonizing individual DFUs. We observed that, in general, DFUs were characterized by high relative abundance of Staphylococcus, Streptococcus, or neither. On closer examination of this third subset of DFUs with neither abundant Staphylococcus nor Streptococcus, taxonomic analysis at higher ranks revealed prevalent anaerobic bacteria or Proteobacteria in most of the samples (Fig. 2).
FIG. 2.

Heterogeneity of DFU microbiomes. Relative abundance (y-axis) of prevalent taxa colonizing DFUs (N = 52) are depicted in the bar chart. DFUs are grouped along the x-axis according to prevalent bacteria. Beneath the bar chart, Shannon diversity index for each respective sample is indicated, ranging between 0.25 and 3.43. Increasing index values represent greater bacterial diversity.
To consider the structure of the DFU bacterial community, we analyzed the 72 OTUs that were present in >10% of the samples and contained at least 100 sequences across all samples. Using normalized OTU counts, we calculated the Euclidean distance between DFUs. DFUs were then clustered by partitioning around medoids (PAM; see research design and methods) (Fig. 3A). This analysis favored partitioning DFUs into three clusters (N = 31, 15, and 6 DFUs each), with a silhouette score of 0.42. These clusters of DFUs, hereafter referred to as Euclidean ulcer clusters (EUCs), differed significantly in OTU richness, OTU diversity, bacterial load, and relative abundance of Proteobacteria, Staphylococcus, anaerobic bacteria, and Streptococcus (Fig. 3B–H; P = 0.018, 4.91 × 10−6, 0.05, 0.001, 1.60 × 10−7, 0.0018, and 0.0003, respectively). In particular, EUC1 contained significantly greater OTU richness than EUC2 (Fig. 3B; P = 0.006). EUC1 harbored the greatest OTU diversity as compared with EUC2 and EUC3 (Fig. 3C; P = 1.29 × 10−7 and 0.022, respectively). Interestingly, DFUs within EUC1 also contained significantly higher bacterial load than those in EUC2 (Fig. 3D; P = 0.025). EUC1 was characterized by significantly higher proportions of Proteobacteria than EUC2 or EUC3 (Fig. 3E; P = 0.0004 and 0.039, respectively). EUC2 was characterized by significantly higher relative abundance of Staphylococcus as compared with EUC1 and EUC3 (Fig. 3F; P = 6.20 × 10−8 and 3.69 × 10−5, respectively) and lower relative abundance of anaerobes as compared with EUC1 (Fig. 3G; P = 0.0003). EUC3 was characterized by the highest relative abundance of Streptococcus as compared with EUC1 and EUC2 (Fig. 3H; P = 0.0002 and 0.0005, respectively).
FIG. 3.

Clustering of DFUs according to Euclidean distance of the 72 species-level OTUs present in >10% of DFU samples and containing >100 sequence counts. A: Depicted is clustering of k = 3 medoids on the first two principle components, which together explain 83.19% of the point variability. The three clusters, or EUCs, are highlighted by different color ellipses, and the points within the clusters are represented by different symbols. The silhouette score for the clusters (average width) was 0.42. EUCs differed significantly by Kruskal-Wallis test (P ≤ 0.05) in Shannon diversity (B), OTU richness (C), log bacterial load (D), and relative abundance of Proteobacteria (E), Staphylococcus (F), anaerobic bacteria (G), and Streptococcus (H). Color of boxes in (B–H) correspond to color of ellipse encircling each EUC. B–H: The x-axis labels represent EUC clusters, boxes represent interquartile range, lines within the box depict median, and whiskers represent the lowest and highest values within 1.5-times the interquartile range. *Significantly different pair-wise comparison (P ≤ 0.05) by Wilcoxon rank-sum test.
To gain insight into bacterial community interactions in DFUs, we examined covariation of species-level OTUs. We calculated Spearman correlation coefficients for the relative abundance of the 72 OTUs that were present in >10% of the samples and contained at least 100 sequences across all samples (Supplementary Fig. 1). We detected significant positive correlation between relative abundance of multiple anaerobic bacteria, including those belonging to OTUs classified as Porphyromonas, Anaerococcus, Finegoldia, Peptoniphilus, Prevotella, and Incertae Sedis XI. The second prominent pattern we observed was that S. aureus abundance was negatively correlated with abundance of many anaerobes but positively correlated with relative abundance of a Corynebacterium OTU (Supplementary Fig. 1, Supplementary Table 1; ρ = 0.47; P < 0.001). These findings were consistent with clustering analyses of community structure, because EUC2 was characterized by high relative abundance of Staphylococcus and corresponding low relative abundance of anaerobes.
Ulcer duration, depth, and glycemic control are associated with DFU microbiome.
To determine if clinical factors were associated with different aspects of bioburden, we selected six variables measured in DFU subjects at time of enrollment: hemoglobin A1c (a measure of glycemic control over 6 weeks), mean tissue oxygenation (a measure of arterial perfusion), ulcer duration before sampling, ulcer depth, ulcer surface area, and necrotic tissue. Analysis of molecular variance (34) suggested that some of these clinical factors were associated with microbiome community structure (as measured by the Θ distance metric and the weighted UniFrac metric) and/or community membership (as measured by the Jaccard distance metric and the unweighted UniFrac metric) (Supplementary Table 2). We therefore proceeded with further analyses to validate these findings.
We calculated Spearman rank correlation coefficients to identify if these six clinical factors were associated with the three dimensions of bioburden (Fig. 4A and B; Supplementary Table 3). Ulcer duration was positively correlated with the number of species-level OTUs colonizing the DFU (ρ = 0.41; P = 0.0022), and with a higher Shannon diversity index (ρ = 0.32; P = 0.020) (Fig. 4A). Furthermore, ulcer duration was positively correlated with relative abundance of Proteobacteria (ρ = 0.38; P = 0.0059) and negatively correlated with relative abundance of Staphylococcus (ρ = −0.30; P = 0.0333) (Fig. 4B). Closer examination of species-level OTUs revealed that an OTU classified as Burkholderiaceae may be contributing to the positive correlation detected between Proteobacteria relative abundance and ulcer duration (ρ = 0.39; P = 0.005). Finally, whereas overall association of ulcer duration with EUC partitioning only came close to significance (P = 0.070), EUC1 was characterized by significantly higher average ulcer duration as compared with EUC2 (Fig. 4C; 40.6 days as compared with 25.9 days, respectively; P = 0.0258).
FIG. 4.

Association of clinical factors with various measures of bioburden. Depicted are (A) bacterial species-level richness and diversity, (B) dominant taxa colonizing DFUs, and (C–E) EUC. A and B: Columns correspond to clinical factors measured in patients with DFU at time of sampling. Spearman rank correlation coefficients are represented by the heat map, with red and white representing positive correlation and negative correlation, respectively. *Significant correlations with P ≤ 0.05. All Spearman correlation coefficient values are listed in Supplementary Table 3. C–E: Box plots of ulcer duration in weeks (C), ulcer depth in millimeters (D), and hemoglobin A1c in percentage (E) plotted against EUC (x-axis). Boxes represent interquartile range, lines within the box depict median, and whiskers represent the lowest and highest values within 1.5-times the interquartile range. Significant associations between EUC and clinical factors by the Kruskal-Wallis test were ulcer depth (D) and hemoglobin A1c (E), whereas ulcer duration only came close to significance (C). C–E: *Significant pair-wise comparisons between EUCs by the Wilcoxon rank-sum test (P ≤ 0.05).
We also detected associations between ulcer depth and bioburden. Ulcer depth was negatively associated with Staphylococcus relative abundance (ρ = −0.47; P = 0.0005) and positively associated with anaerobic bacteria relative abundance (ρ = 0.33; P = 0.0182) (Fig. 4B). Consistent with this finding, ulcer depth was associated with EUC (Fig. 4D; P = 0.017). EUC1 contained significantly deeper ulcers than EUC2 at an average depth of 0.297 cm as compared with 0.097 cm, respectively (P = 0.005). This finding is not surprising because EUC1 contained higher relative abundance of Proteobacteria and anaerobes and overall higher bacterial diversity, confirming our previous findings that these dimensions of bioburden are associated with greater ulcer depth. We also examined DFU surface area, finding a weak but significant positive correlation with OTU richness (Fig. 4A; ρ = 0.27; P = 0.051). However, surface area was not associated with EUC.
Finally, hemoglobin A1c values and, thus, glycemic control were linked to EUC partitioning of DFUs (Fig. 4E; P = 0.037). Highest hemoglobin A1c levels partitioned with EUC2 and EUC3 (median = 9.20 and 9.45, respectively), significantly higher than the EUC1 hemoglobin A1c (median = 8.01; P = 0.050 and 0.0413, respectively).
DISCUSSION
To our knowledge we are the first to show that the microbiome colonizing DFUs is associated with clinical factors. In addition, we demonstrate that neuropathic DFUs cluster into groups distinguished by the dimensions of bioburden that historically have been believed to be of importance (microbial load, diversity, and presence of pathogens). Nonetheless, these findings need to be validated in larger independent samples of neuropathic DFUs because this study included only 52 DFUs. Finally, like others, we found that quantitative cultures do not fully represent the microbiome of DFUs when compared with genomic techniques.
We found that ulcer depth and duration are associated with microbial diversity and the abundance of specific pathogens. Deep ulcers and those of longer duration have a more diverse microbiota, containing higher levels of anaerobes and Proteobacteria. Superficial ulcers and those of shorter duration were associated with higher relative abundance of Staphylococcus. Pathare et al. (1998) found similar results in their study of DFU that used tissue cultures (35). Poor glycemic control was associated with ulcers containing high relative abundance of Staphylococcus and Streptococcus. No significant associations between overall bacterial diversity and individual abundant species were detected, suggesting that unidentified dimensions of DFU bioburden, which are captured in EUC clustering, are responsive to glycemic control. Wound tissue oxygen was not associated with DFU bioburden or community structure. However, enrollment excluded ischemic DFUs, which undoubtedly have lower wound tissue oxygen levels and therefore may have a different bioburden and/or microbial community structure. Similarly, necrotic tissue in the wound bed was not associated with bioburden. Most of the ulcers in the sample were free of necrotic tissue, which decreased variability and the ability to detect differences. Neuropathic DFUs are frequently free of necrotic tissue, so the sample in this study was typical of this type of chronic wound. Our findings of the association of clinical factors with several measures of microbial community membership and structure suggest that metrics of this type may be most important in determining the role of DFU microbiota on ulcer outcomes, such as healing or amputation, because of the complex nature of the chronic wound microbiota. However, longitudinal studies that examine the dynamic relationship between microbial community membership and structure with ulcer outcomes are needed to support this assertion.
Comparisons of traditional quantitative cultures with 16S rRNA gene sequencing raise some points of interest with regard to the utility of using cultures in the clinical setting as a diagnostic tool to identify problematic bioburden. These comparisons are needed because cultures remain more widely available around the globe than DNA sequencing technology, and their utility needs to be identified in directing the management of DFUs. Historically, clinical practice for treating DFUs is based on assumptions that implicate the dominant and/or culturable bacteria as the pathogenic/destructive bacteria. Similar to other studies (7,12,36), we found that cultures do not fully represent bacterial diversity as compared with 16S rRNA gene sequencing. Microbial load is widely viewed as the reference standard for determining problematic bioburden in chronic wounds that may not express robust clinical signs (37). We demonstrated for the first time that cultures underrepresent the microbial load of the ulcer as compared with 16S rRNA gene approaches. These findings may have important clinical implications justifying the clinical use of molecular approaches rather than traditional cultures.
However, it is unclear if all organisms identified by 16S gene sequencing are important. Unlike cultures, dormant or dead bacteria, which may or may not be contributing to an altered wound environment, are identified by 16S gene sequencing without distinction between viable and nonviable organisms. Another limitation of this approach is that 16S gene sequencing only identifies bacteria. It is quite possible that fungi, viruses, and/or other microeukaryotes may be important components of the DFU microbiome. Third, profiling 16S rRNA genes only can tell us what is there and will not address the question, what is it doing? Future studies will need to address the functional and mechanistic implications of colonization by specific microbes or microbial communities.
No studies of purely neuropathic etiology were found to compare our findings. Although Dowd et al. (12) studied 40 DFUs using 16S rRNA sequencing, the location of ulcers reported in the study suggest a mixture of neuropathic, ischemic, and mixed DFUs, and other ulcers that may be primarily arterial. This study found Corynebacterium to be the most prevalent bacterial taxa, followed by Bacteroides and Peptoniphilus. Price et al. (7) reported that Streptococcus was more prevalent in the wounds of persons with diabetes (n = 12) than those free of diabetes. Reasons for the discrepancy in findings between our study and these may be attributable to differences in the ulcer samples.
Inconsistency in these findings highlights the need to describe and delineate the microbiome of wounds with homogenous etiology and other pathophysiological mechanisms to determine differences in microbiome that may be driven by the wound environment (36). In this way, the significance of the chronic wound microbiota can be compared and contrasted among chronic wounds of different types. Furthermore, studies that take advantage of clinical metadata to appropriately stratify patient populations ultimately will have the most potential to reveal causal links between microbiome variation and chronic wound outcomes using longitudinal designs.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by National Institutes of Health (NIH), National Institute of Nursing Research (S.E.G., NINR R01 NR009448), National Institute of Arthritis and Musculoskeletal and Skin Diseases (E.A.G., NIAMS R00 AR060873), and the NIH Intramural Research Program (J.A.S). It was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH, through grant UL1-RR-024979 (S.E.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
No potential conflicts of interest relevant to this article were reported.
S.E.G., S.L.H., J.A.S., and E.A.G. designed the study. S.E.G., K.H., and E.A.G. performed experiments. S.E.G., K.H., and E.A.G. analyzed the data. S.E.G. and E.A.G. performed statistical analyses and wrote the manuscript. S.L.H. analyzed the data and performed statistical analyses. All authors contributed to editing the manuscript. S.E.G. and E.A.G. are the guarantors of this work and take full responsibility for the integrity and accuracy of the data.
The authors thank the NIH Intramural Sequencing Center for their effort on this project. The authors thank David Margolis (University of Pennsylvania), Heidi Kong (NIH), Sean Conlan (NIH), Sandra Daack-Hirsch (University of Iowa), Jeff Murray (University of Iowa), Brian Schutte (University of Iowa), and members of the Grice laboratory (University of Pennsylvania) and Segre laboratory (NIH) for their underlying contributions, useful discussions, and/or critical review of the manuscript.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0771/-/DC1.
See accompanying commentary, p. 679.
REFERENCES
- 1.Reiber G. The Diabetic Foot. In The Diabetic Foot. Bowker J, Pfeifer M, Eds. St. Louis, Mosby, 2001, p. 13–32 [Google Scholar]
- 2.Howell-Jones RS, Wilson MJ, Hill KE, Howard AJ, Price PE, Thomas DW. A review of the microbiology, antibiotic usage and resistance in chronic skin wounds. J Antimicrob Chemother 2005;55:143–149 [DOI] [PubMed] [Google Scholar]
- 3.Gardner SE, Frantz RA, Doebbeling BN. The validity of the clinical signs and symptoms used to identify localized chronic wound infection. Wound Repair Regen 2001;9:178–186 [DOI] [PubMed] [Google Scholar]
- 4.Bowler PG, Duerden BI, Armstrong DG. Wound microbiology and associated approaches to wound management. Clin Microbiol Rev 2001;14:244–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA, Kong HH, Conlan S, et al. NISC Comparative Sequencing Program Topographical and temporal diversity of the human skin microbiome. Science 2009;324:1190–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, Liechty KW, Segre JA, NISC Comparative Sequencing Program Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci USA 2010;107:14799–14804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price LB, Liu CM, Melendez JH, et al. Community analysis of chronic wound bacteria using 16S rRNA gene-based pyrosequencing: impact of diabetes and antibiotics on chronic wound microbiota. PLoS ONE 2009;4:e6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hugenholtz P, Pace NR. Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol 1996;14:190–197 [DOI] [PubMed] [Google Scholar]
- 9.Pace NR. A molecular view of microbial diversity and the biosphere. Science 1997;276:734–740 [DOI] [PubMed] [Google Scholar]
- 10.Gardner SE, Frantz RA. Wound bioburden and infection-related complications in diabetic foot ulcers. Biol Res Nurs 2008;10:44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowd SE, Sun Y, Secor PR, et al. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 2008;8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS ONE 2008;3:e3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adler AI, Boyko EJ, Ahroni JH, Smith DG. Lower-extremity amputation in diabetes. The independent effects of peripheral vascular disease, sensory neuropathy, and foot ulcers. Diabetes Care 1999;22:1029–1035 [DOI] [PubMed] [Google Scholar]
- 14.Barnett A, Dave B, Ksander GA, Vistnes LM. A concentration gradient of bacteria within wound tissues and scab. J Surg Res 1986;41:326–332 [DOI] [PubMed] [Google Scholar]
- 15.Margolis DJ, Allen-Taylor L, Hoffstad O, Berlin JA. Diabetic neuropathic foot ulcers: predicting which ones will not heal. Am J Med 2003;115:627–631 [DOI] [PubMed] [Google Scholar]
- 16.Reiber GE, Pecoraro RE, Koepsell TD. Risk factors for amputation in patients with diabetes mellitus. A case-control study. Ann Intern Med 1992;117:97–105 [DOI] [PubMed] [Google Scholar]
- 17.Christman AL, Selvin E, Margolis DJ, Lazarus GS, Garza LA. Hemoglobin A1c predicts healing rate in diabetic wounds. J Invest Dermatol 2011;131:2121–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Margolis DJ, Kantor J, Santanna J, Strom BL, Berlin JA. Risk factors for delayed healing of neuropathic diabetic foot ulcers: a pooled analysis. Arch Dermatol 2000;136:1531–1535 [DOI] [PubMed] [Google Scholar]
- 19.Gardner SE, Frantz RA, Saltzman CL, Hillis SL, Park H, Scherubel M. Diagnostic validity of three swab techniques for identifying chronic wound infection. Wound Repair Regen 2006;14:548–557 [DOI] [PubMed] [Google Scholar]
- 20.Gardner SE, Hillis SL, Frantz RA: A prospective study of the PUSH tool in diabetic foot ulcers. Journal of wound, ostomy, and continence nursing: official publication of The Wound, Ostomy and Continence Nurses Society / WOCN 2011;38:385-393 [DOI] [PMC free article] [PubMed]
- 21.Gardner SE, Frantz RA, Saltzman CL. Diabetes and inflammation in infected chronic wounds. Wounds 2005;17:203–205 [Google Scholar]
- 22.Gardner SE, Frantz RA, Hillis SL, Blodgett TJ, Femino LM, Lehman SM. Volume measures using a digital analysis system are reliable in diabetic foot ulcers. Wounds 2012;24:146–151 [PMC free article] [PubMed] [Google Scholar]
- 23.Bates-Jensen BM: The pressure sore status tool a few thousand assessments later. Adv Wound Care 1997;10:65-73 [PubMed]
- 24.Kong HH, Oh J, Deming C, et al. NISC Comparative Sequence Program Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 2012;22:850–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011;27:2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haas BJ, Gevers D, Earl AM, et al. Human Microbiome Consortium Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 2011;21:494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol 2011;77:3219–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cole JR, Chai B, Farris RJ, et al. The Ribosomal Database Project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 2007;35(Database issue):D169–D172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 2010;11:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grice EA, Kong HH, Renaud G, et al. NISC Comparative Sequencing Program A diversity profile of the human skin microbiota. Genome Res 2008;18:1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rousseeuw PJ. Silhouettes: a graphical aid to the interpretation and validation of cluster-analysis. J Comput Appl Math 1987;20:53–65 [Google Scholar]
- 33.Fleiss JL. Statistical methods for rates and proportions. New York, Wiley, 1981 [Google Scholar]
- 34.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol 2001;26:32–46 [Google Scholar]
- 35.Pathare NA, Bal A, Talvalkar GV, Antani DU. Diabetic foot infections: a study of microorganisms associated with the different Wagner grades. Indian J Pathol Microbiol 1998;41:437–441 [PubMed] [Google Scholar]
- 36.Han A, Zenilman JM, Melendez JH, et al. The importance of a multifaceted approach to characterizing the microbial flora of chronic wounds. Wound Repair Regen 2011;19:532–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heggers JP, Robson, MC. Variations on a theme. In Quantitative bacteriology: Its role in the armamentarium of the surgeon Heggers JP, Robson MD, Eds. Boca Raton, CRC Press, 1991, p. 15-23 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.