

In response to development of the euglycemic-hyperinsulinemic clamp technique in the 1980s, much emphasis was placed on skeletal muscle as the major site of insulin resistance in patients with type 2 diabetes (1). Following the discovery of the insulin receptor (IR) tyrosine kinase activity by Ron Kahn and colleagues (2) and the importance of IR substrate molecules in the 1990s (3,4), attention was directed on intrinsic alterations in the insulin signaling cascade in skeletal muscle and liver as fundamental causes of insulin resistance. In 1996, I joined Ron Kahn's laboratory as a participant in the gold rush to discover novel mechanisms responsible for insulin resistance. It was a time of intellectual and scientific awakening. With the advent of the Cre/loxP system, came the first surprise: muscle-specific IR knockout (MIRKO) mice showed no alterations in glucose homeostasis. Rather, they developed visceral adiposity (5). MIRKO mice exhibited impaired insulin activation of muscle glycogen synthase resulting in decreased muscle glycogen content (6,7). MIRKO mice also displayed reduced insulin-stimulated muscle glucose uptake during the euglycemic-hyperinsulinemic clamp (6). However, under physiological conditions of a glucose tolerance test, MIRKO mice had near normal skeletal muscle glucose uptake and did not display insulin resistance (7). Conversely, disruption of insulin action in the hepatocyte-specific IR knockout mouse produced severe resistance to the blood glucose–lowering effect of insulin (8). Together with the MIRKO, the hepatocyte-specific IR knockout mouse demonstrated the importance of the liver in postprandial glucose homeostasis and suggested that a considerable portion of the hypoglycemic effect of insulin was due to a suppression of hepatic glucose production rather than an increase in muscle glucose uptake (5,8). Did this mean that skeletal muscle was not necessary for glucose homeostasis? No, but although insulin is necessary for storage of glucose in the form of glycogen in skeletal muscle, IR signaling in muscle is not necessary to maintain postprandial glucose disposal in mice. Something else was needed independently of the muscle IR: noninsulin-dependent glucose uptake via skeletal muscle contraction and 5′-AMP-activated protein kinase. Indeed, exercise could still activate glucose transport in muscle in MIRKO mice independently of insulin (9), an observation that underscored the importance of AMP-activated protein kinase in muscle glucose uptake and “insulin” sensitivity (10). At the same time, the idea emerged that brain insulin action was also important for glucose homeostasis and that hypothalamic insulin resistance could contribute to altered suppression of hepatic glucose production and hyperglycemia in type 2 diabetes (11). Over time, the concept evolved that the common form of insulin resistance was not the result of genetic alteration in molecules in the insulin signaling cascade. Rather, it was secondary to impaired fuel homeostasis in insulin-sensitive tissues. Originally observed by Randle et al. (12), the inhibitory effect of lipid accumulation in skeletal muscle on glucose metabolism and, ultimately, insulin-sensitive glucose uptake, gained traction. This paradigm matured into a hypothesis that focused on the importance of inflammatory lipid metabolites (e.g., ceramides) that activate IκB kinase β, ultimately leading to serine phosphorylation and inhibition of insulin signaling molecules in various tissues (13). It also became clear that distant tissues such as fat were modulating muscle insulin sensitivity by releasing adipocytokines that promote insulin resistance—in the case of tumor necrosis factor-α (14)—or that promote insulin sensitivity as is the case with adiponectin (15). A parallel paradigm emerged that highlighted the importance of skeletal muscle blood flow and especially the integrity of vascular endothelial function in whole-body insulin sensitivity. Production of nitric oxide by endothelial nitric oxide synthase (eNOS) normally regulates blood pressure and increases muscle blood flow. Perhaps the first evidence of the importance of skeletal muscle blood flow in insulin sensitivity came from a study of eNOS-deficient mice (16). These mice developed hypertension and muscle insulin resistance. Further, endothelium-derived nitric oxide also mediates insulin-induced perfusion and substrate delivery to skeletal muscle. Not surprisingly, vascular endothelial elimination of IR in mice resulted in insulin resistance (17). In addition, mice with insulin resistance in the endothelium resulting from double IR substrates IRS1 and IRS-2 deficiencies had decreased insulin-stimulated glucose uptake in skeletal muscle (18). Nonetheless, the link among the metabolic syndrome, inflammation, and altered skeletal muscle blood flow was not firmly established. In this issue of Diabetes, Tanigaki et al. (19) close the loop on this link and reveal that C-reactive protein (CRP) causes muscle insulin resistance in mice. CRP is an acute-phase reactant synthesized in liver in response to acute inflammatory stimuli in humans. In contrast to most vertebrates, mice synthesize CRP in only trace amounts. Thus, it is not possible to study the effect of endogenous CRP in mice. Using a transgenic mouse overexpressing rabbit CRP to levels observed in insulin-resistant patients, Tanigaki et al. show that elevated CRP levels impair both insulin-induced skeletal muscle blood flow and muscle glucose delivery, ultimately resulting in insulin resistance. The effect of CRP is mediated by an isoform of the Fcγ receptors (FcγRs)—designated FcγRIIB—that binds CRP and is abundantly expressed in endothelial cells in skeletal muscle. Insulin signaling in endothelial cells normally stimulates the phosphorylation and activation of the eNOS on S1176 residue to increase muscle blood flow, thereby promoting glucose disposal (Fig. 1). CRP activation of FcγRIIB causes eNOS-S1176 dephosphorylation. Using a knock-in mouse with eNOS resistance to S1176 dephosphorylation, Tanigaki et al. show that CRP loses the power to induce muscle insulin resistance (19). CRP does not affect hepatic or adipose insulin action; it selectively produces skeletal muscle insulin resistance. Thus, during inflammatory states, CRP activation of FcγRIIB causes impaired insulin endothelial action in skeletal muscle, impaired muscle glucose delivery, and insulin resistance (Fig. 1). Because the mass of skeletal muscle is predominant relative to other tissues, muscle glucose uptake is clinically critical to whole-body glucose disposal. However, the importance of muscle glucose uptake is not due solely to IR signaling in muscle. Rather, it is the combination of muscle insulin action, muscle contraction, and vascular endothelium-induced muscle blood flow that together synergize to promote muscle glucose uptake. In demonstrating that muscle blood flow and therefore insulin sensitivity can be impaired by the production of CRP, Tanigaki et al. open a new avenue in the research for the treatment of insulin resistance.
FIG. 1.
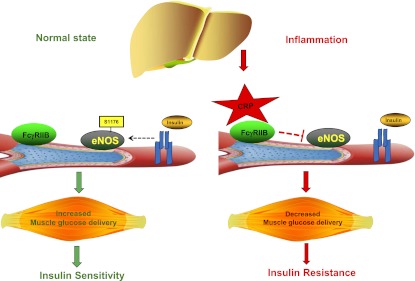
Role of CRP in skeletal muscle insulin resistance. In the normal state, insulin signaling in endothelial cells stimulates the phosphorylation and activation of the eNOS on S1176 residue to increase muscle blood flow, thereby promoting muscle glucose delivery and insulin sensitivity. During inflammation, CRP activation of FcγRIIB in muscle endothelial cells causes eNOS-S1176 dephosphorylation leading to impaired insulin endothelial action in skeletal muscle, impaired muscle glucose delivery, and insulin resistance.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (RO1 DK074970) and the American Heart Association (11IRG5570010).
No potential conflicts of interest relevant to this article were reported.
Footnotes
See accompanying original article, p. 721.
REFERENCES
- 1.DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest 1985;76:149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kasuga M, Zick Y, Blithe DL, Crettaz M, Kahn CR. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 1982;298:667–669 [DOI] [PubMed] [Google Scholar]
- 3.Araki E, Lipes MA, Patti ME, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994;372:186–190 [DOI] [PubMed] [Google Scholar]
- 4.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900–904 [DOI] [PubMed] [Google Scholar]
- 5.Brüning JC, Michael MD, Winnay JN, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 1998;2:559–569 [DOI] [PubMed] [Google Scholar]
- 6.Kim JK, Michael MD, Previs SF, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 2000;105:1791–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mauvais-Jarvis F, Virkamaki A, Michael MD, et al. A model to explore the interaction between muscle insulin resistance and beta-cell dysfunction in the development of type 2 diabetes. Diabetes 2000;49:2126–2134 [DOI] [PubMed] [Google Scholar]
- 8.Michael MD, Kulkarni RN, Postic C, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 2000;6:87–97 [PubMed] [Google Scholar]
- 9.Wojtaszewski JF, Higaki Y, Hirshman MF, et al. Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J Clin Invest 1999;104:1257–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 1998;47:1369–1373 [DOI] [PubMed] [Google Scholar]
- 11.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002;8:1376–1382 [DOI] [PubMed] [Google Scholar]
- 12.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;1:785–789 [DOI] [PubMed] [Google Scholar]
- 13.Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001;293:1673–1677 [DOI] [PubMed] [Google Scholar]
- 14.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91 [DOI] [PubMed] [Google Scholar]
- 15.Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001;7:941–946 [DOI] [PubMed] [Google Scholar]
- 16.Duplain H, Burcelin R, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001;104:342–345 [DOI] [PubMed] [Google Scholar]
- 17.Vicent D, Ilany J, Kondo T, et al. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest 2003;111:1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab 2011;13:294–307 [DOI] [PubMed] [Google Scholar]
- 19.Tanigaki K, Vongpatanasin W, Barrera JA, et al. C-reactive protein causes insulin resistance in mice through Fcγ receptor IIB–mediated inhibition of skeletal muscle glucose delivery. Diabetes 2013;62:721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]