

Abstract
A series of tetracyclic nitrofuran isoxazoline antituberculosis agents was designed and synthesized to improve the pharmacokinetic properties of an initial lead compound, which had potent antituberculosis activity but suffered from poor solubility, high protein binding and rapid metabolism. In this study, structural modifications were carried on the outer phenyl and piperidine rings to introduce solubilizing and metabolically blocking functional groups. The compounds generated were evaluated for their in vitro antitubercular activity, bacterial spectrum of activity, solubility, permeability, microsomal stability and protein binding. Pharmacokinetic profiles for the most promising candidates were then determined. Compounds with phenyl morpholine and pyridyl morpholine outer rings were found to be the most potent antituberculosis agents in the series. These compounds retained a narrow antibacterial spectrum of activity, with weak anti-gram positive and no gram negative activity, as well as good activity against non-replicating M. tuberculosis in a low oxygen model. Overall, the addition of solubilizing and metabolically blocked outer rings did improve solubility and decrease protein binding as designed. However, the metabolic stability for compounds in this series was generally lower than desired. The best three compounds selected for in vivo pharmacokinetic testing all showed high oral bioavailability, with one notable compound showing a significantly longer half-life and good tolerability supporting its further advancement.
Keywords: Tuberculosis, antibiotic, nitrofuran, isoxazoline, nitroaromatic
1. Introduction
On a global scale Tuberculosis is one of the leading causes of mortality from an infectious agent and a major AIDS-associated, opportunistic infection.1 The standard six-month drug treatment regime for tuberculosis is extremely long and often leads to patient non-compliance and consequent development of resistance.2 The long treatment time is believed to be caused by latent / slow growing bacterial subpopulations that are poorly susceptible to front line agents. These latent populations are thought to exist in part in an acidic low oxygen / microaerophilic state within the granuloma.3
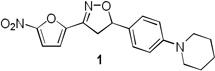
For this study, our efforts have focused on developing nitrofuran based anti-tuberculosis therapeutics that are active in latent TB models and thus have the potential to shorten tuberculosis therapy regimen.4–6,7–9 Initial series were based around nitrofuranyl amides, however the amide functionality proved to be a metabolic liability and it was replaced with a more stable 3,5-disubstituted isoxazoline functionality. Compounds in this series had excellent MIC activity, but suffered from overall poor solubility, high protein binding and poor microsomal stability. Herein we describe the synthesis of a new series of compounds designed to improve pharmacokinetic properties based on lead compound 1 (Figure 1A). The nitrofuran and isoxazoline A and B rings were kept intact to maintain antitubercular potency and the C and D rings were modified to increase solubility and metabolic stability while decreasing protein binding. Modification of the C and D rings employed a strategy similar to that used for second generation oxazolidinones. These antibacterial agents have a similar linear tetra cyclic structure with the outer rings playing a significant role in modulating their absorption, distribution and metabolic properties (Figure 1B).10–12
Figure 1.

A. 1 Initial lead anti-tubercular nitrofuran. In this study targeted changes were made to the outer C and D ring systems to improve ADME properties. B. Comparator oxazolidinone antibacterial agents Linezolid and Sutezolid with similar linear structures.
2. Chemistry
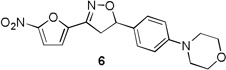
The target compounds of the present study were synthesized from their corresponding 4-bromo, 1-vinyl benzene C-ring precursors in two steps, using a modified Buchwald coupling of aryl halides with cyclic amines to add the substitute D rings followed by 3+2 cyclo addition reaction to the vinyl functionality simultaneously adding the nitrofuran A ring and isoxazoline B ring as described in Schemes 1–3. More specifically, compounds 6–8 were synthesized starting from their corresponding 4-bromo aryl iodide precursors 2 as depicted in Scheme 1. Iodobenzenes were converted into their vinyl analogues 3b-c by treating with tributyl(vinyl)tin in presence of Pd catalyst in DMF under microwave irradiation in 80–82% yields.13 The aryl amination reaction was carried out using morpholine, palladium acetate and phosphorous ligand in presence of sodium tert butoxide in toluene at 80 °C for overnight to afford 4a-c in 69–83 % yields.14 Finally, the isoxazoline ring was constructed by treating the olefins with nitrofuranyl chloroxime 5 in the presence of Et3N in CHCl3 at room temperature to give compounds 6–8 in 75–80 % yields.
Scheme 1.
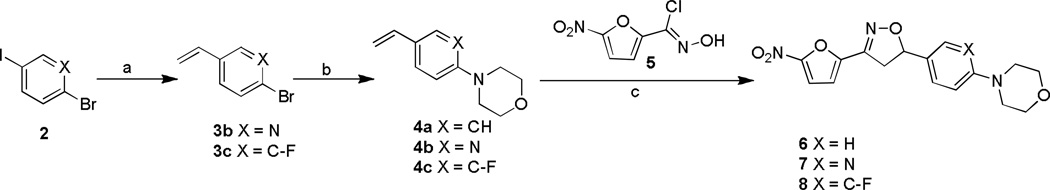
Reagents and conditions: a) tributyl (vinyl)tin, PdCl2(PPh3)2, DMF, MW, 160°C, 5 min; b) morpholine, Pd(OAc)2, 2-(Di-tert-butylphosphino) biphenyl, NaOtBu, toluene, 80°C, 12 h; c) Et3N, CHCl3, RT, 2h
Scheme 3.

Reagents and conditions: a) N,N'-dimethyl ethylene diamine, CuI, K2CO3, toluene, reflux, 6 h; b) 5, Et3N, CHCl3, RT, 2h
The thiomorpholine compound 11 was synthesized from 4-bromo styrene 9 by treating with thiomorpholine, palladium acetate and phosphorous ligand in presence of sodium tert butoxide in toluene at 80 °C for 4 h to afford 4-(4-vinylphenyl) thiomorpholine 10 in 77% yield (Scheme 2). Then the isoxazoline ring construction was carried out by treating 10 with nitrofuranyl chloroxime in the presence of Et3N in CHCl3 at room temperature for 2 h to give 11 in 63% yield. The thiomorpholine compound was oxidized to sulfoxide by treating with NaIO4 in methanol and water at room temperature for overnight to yield 12 in 58% yield.15
Scheme 2.
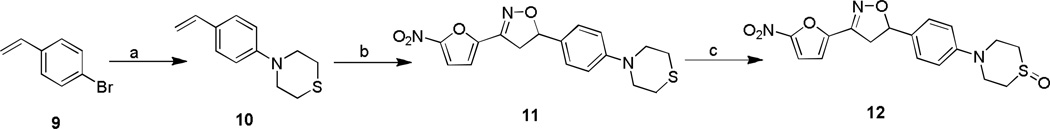
Reagents and conditions: a) thiomorpholine, Pd(OAc)2, 2-(Di-tert-butylphosphino) biphenyl, NaOtBu, toluene, 80 °C, 12 h; b) 5, Et3N, CHCl3, RT, 2h; c) NalO4, CH3CN, MeOH, H2O, RT, 12 h
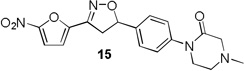
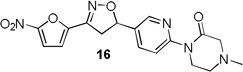
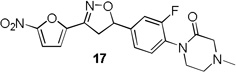
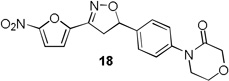
4-Methylpiperazin-2-one and morpholin-3-one analogues 15–20 were synthesized (Scheme 3) from appropriate aryl bromide precursors 3b-c by treating with cyclic amides 13 (Y = NCH3 for 15–17, Y = O for 18–20) in presence of N,N’-dimethyl ethylene diamine, CuI and K2CO3 in toluene at reflux temperature for 6 h.16 Then the isoxazoline ring was constructed using classical conditions as discussed in Scheme 1 to obtain compounds 15–20 in 57–73% yields.
3. MIC and in vitro pharmacokinetics properties
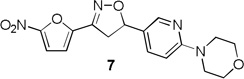
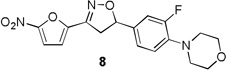
Table 1 summarizes the antituberculosis MIC activity and the in vitro pharmacokinetic profiling results. Across the whole series C ring substitutions did not have a major impact on measured parameters. Substitution with a pyridyl group (7, 16 and 19) resulted in a small decrease in protein binding overall. Compound 7 had slight improvements in solubility and microsomal stability over 6, however the efflux ratio suggested active efflux transport. As could be expected, fluorine substitution of the C ring (8, 17 and 20) did increase the lipophilicity and it decreased solubility in combination with morpholine and morpholinone D rings.
Table 1.
MIC and in vitro pharmacokinetic profile
| Compound | MIC µg/ml |
Solubility pH7.4 (mg/L) |
Microsomal stability t1/2 (h) |
Caco-2 | Protein binding (%) |
||||
|---|---|---|---|---|---|---|---|---|---|
| mouse | human | Papp A/B (nm/s) |
Papp B/A (nm/s) |
Efflux ratio |
mouse | human | |||
 |
0.006 | 1.1 (0.2) |
0.78 (0.03) |
0.47 (0.02) |
76.09 (38.6) |
103.04 (5.19) |
1.35 | 598.8 | ND |
 |
0.006 | 9.9 (0.2) |
1.06 (0.07) |
2.79 (0.18) |
171.8 (75.9) |
120.92 (19.8) |
0.70 | 96.4 (0.82) |
96.2 (1.06) |
 |
0.013 | 14.1 (0.3) |
1.23 (0.07) |
3.37 (0.26) |
64.73 (26.7) |
142.11 (98.5) |
2.20 | 95.3 (2.32) |
96.7 (0.63) |
 |
0.05 | 3.2 (0.2) |
0.92 (0.06) |
2.29 (0.10) |
200.66 (76.6) |
176.96 (54.6) |
0.88 | 93.9 (0.43) |
93.4 (0.75) |
 |
0.05 | 1.9 (0.1) |
0.04 (0.00) |
0.21 (0.00) |
96.15 (22.4) |
114.41 (12.6) |
1.19 | 98.7 (0.25) |
98.5 (0.21) |
 |
0.4 | 28.9 (0.2) |
0.87 (0.06) |
4.53 (0.17) |
241.45 (48.7) |
272.11 (3.27) |
1.13 | 82.2 (3.47) |
77.7 (0.69) |
 |
0.4 | 28.8 (0.3) |
0.39 (0.01) |
5.48 (0.48) |
283.99 (30.5) |
305.15 (40.2) |
1.07 | 76.1 (2.77) |
57.4 (8.49) |
 |
0.2 | 28.9 (0.6) |
0.35 (0.01) |
4.24 (0.24) |
365.97 (37.9) |
332.78 (28.9) |
0.91 | 59.8 (10.16) |
48.9 (8.80) |
 |
0.4 | 29.2 (0.3) |
0.62 (0.03) |
4.81 (0.31) |
269.81 (34.3) |
385.83 (36.2) |
1.43 | 82.8 (6.38) |
60.9 (14.80) |
 |
0.4 | 25.88 (0.16) |
1.40 (0.1) |
10.03 (2.8) |
126.36 (33.7) |
565.37 (46.8) |
4.47 | 66.0 (2.2) |
64.3 (2.9) |
 |
0.2 | 21.94 (0.63) |
1.01 (0.1) |
1.52 (0.1) |
86.05 (13.5) |
457.33 (37.9) |
5.31 | 58.9 (4.7) |
60.2 (1.6) |
 |
0.8 | 12.58 (1.49) |
1.38 (0.2) |
8.31 (1.7) |
154.14 (5.36) |
424.05 67.3) |
2.75 | 79.7 (1.0) |
70.8 (1.1) |
Standard deviations are in parentheses; Papp: The apparent permeability coefficient
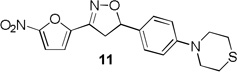
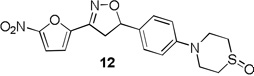
More pronounced SAR was observed for the D ring substitutions. Morpholines (6–8) retained good MIC, had improved solubility and microsomal stability, and a slight reduction in protein binding. Thiomorpholine (11) retained good MIC activity, and its S-oxide (12) has increased solubility and Caco-2 permeability, and reduced protein binding. However, the metabolic stability was very poor for 11, which is presumably rapidly S oxidized to 12 and then on to further metabolites. It was also noted that S-oxide 12 has an 8-fold weaker MIC value than 11, which is undesirable. The solubility and protein binding of the 4-Methylpiperazinones (15–17) was much improved and the Caco-2 permeability was the highest observed in the series. However, the MIC increased significantly (0.4 µg/ml) for these compounds and they were rapidly metabolized, perhaps due the introduction of a new site of metabolism via N-demethylation of the piperazine ring. The morpholinones (18–20) had a similar profile to the 4-methylpiperazinones with improved solubility and protein binding, but higher MICs. Microsomal stability was much better for these compounds and the best in the series. However, a comparatively high and undesirable efflux ratio was observed in this assay.
Based on the retention of a good MIC and modest improvements in solubility, microsomal stability and protein binding compounds 6 and 7 were advanced for further testing. Despite its higher MIC and short half-life 4-methylpiperazinone 15 was selected for comparison purposes due to its greater improvements in solubility and protein binding as well as its availability at the time of testing. Thiomorpholines 11 and 12 were excluded from further testing due to concerns over metabolic processing and observed toxicity. Morpholinones 18–20 were also excluded primarily due to the high efflux ratios as well as higher MIC values.
Previous compounds in the series of nitrofurans have demonstrated a narrow range of activity with selectivity for M. tuberculosis and activity against non-replicating TB.17 To ensure this series retained that narrow spectrum of activity, the MIC of compounds was determined by microbroth dilution for a representative panel of bacterial pathogens (Table 2). Weak to moderate MIC activity was observed against some gram positive bacteria most notably S. aureus, B. subtilis, B. anthracis and an efflux pump deficient strain of E. coli. No activity was observed against any of the following gram negative strains: Burkholderia cepacia, Proteus mirabilis, Proteus vulgaris, Klebsiella pneumoniae, Acinetobacter baumannii, Strenotrophomonas maltophilia, and Pseudomonas aeruginosa (data not shown). To ensure this series retained activity against non-replicating bacteria, compound 7 was tested in an oxygen depletion model developed by Voskuil and coworkers from the original Wayne model.18 It killed 99.9% of bacteria at 50 µg/ml and 66.7% at 10 µg/ml demonstrating 7 retained bactericidal activity against this important persistent population of TB.
Table 2.
Antibacterial spectrum of activity (µg/ml)
| Compd |
S. aureus |
S. pyogenes |
S. pneumonia |
E. faecalis |
B. subtilis |
B. anthracis |
E. coli K12 |
E. coli ΔtolC |
|---|---|---|---|---|---|---|---|---|
| 1 | 12.5 | 100 | 200 | >200 | 50 | >200 | >200 | 50 |
| 6 | 6.25 | 25 | 50 | >200 | 12.5 | 12.5 | >200 | 6.25 |
| 7 | 12.5 | 25 | 200 | 200 | 50 | 25 | >200 | 6.25 |
| 8 | 25 | 200 | 200 | 200 | 200 | 12.5 | >200 | 6.25 |
| 11 | 12.5 | >200 | 12.5 | 100 | 12.5 | 12.5 | >200 | 12.5 |
| 12 | 6.25 | 50 | 50 | 100 | 6.25 | 12.5 | 100 | 6.25 |
| 15 | 6.25 | 50 | 50 | 100 | 6.25 | 12.5 | 50 | 12.5 |
| 16 | 12.5 | 50 | 50 | 100 | 25 | 25 | 50 | 6.25 |
| 17 | 6.25 | 50 | 50 | 100 | 25 | 25, | 200 | 25 |
| 18 | 6.25 | 100 | 200 | >200 | 6.25 | 25 | 25 | 6.25 |
| 19 | 12.5 | 1005 | 50 | >200 | 12.5 | 25 | 25 | 12.5 |
| 20 | 6.25 | 100 | 200 | 200 | 12.5 | 25 | >200 | 12.5 |
Organisms abbreviated above are as follows:
S. aureus ATCC 29213; Strep. pyogenes ATCC 700294;Strep. pneumoniae DAW27; E. faecalis ATCC3186; B. subtilis ATCC 23857; B. anthracis Sterne 34F2; E. coli K12; E. coli K12 ΔtolC
4. In vivo pharmacokinetics
For the prioritized set of compounds (1, 6, 7 and 15), the in vivo pharmacokinetics were evaluated and the results are shown in Table 3. After intravenous administration of 10 mg/kg in rats, compounds 1, 6, 7 and 15 showed concentration-time profiles with areas under the curve ranging from 0.96 to 3.36 mg hr/L, and peak plasma concentrations of 0.83 to 2.73 mg/L. These peak plasma concentrations when converted to unbound concentrations were 4.6 times the MIC for compound 1, 5.0 times the MIC for compound 6, 9.8 times the MIC for compound 7, and 0.71 times the MIC for compound 15. Compounds 7 and 15 showed a similarly short elimination half-life of 0.61–0.72 hr, while compounds 1 and 6 exhibited a longer half-life largely due to their larger volume of distribution. Clearance was high in all four compounds close to or higher than hepatic blood flow, indicating potential extrahepatic metabolism. The fraction of dose excreted unchanged by the kidneys (fe) was negligible for all compounds. Both of these observations indicate rapid in vivo metabolic degradation for all four compounds as the major elimination pathway, which is consistent with their limited microsomal stability (Table 1). Their relatively large volumes of distribution and non-renal elimination are probably a reflection of their high log P values. The oral bioavailability was high for all compounds 29.4% for compound 1, nearly 100% for compound 6, 47.9% for 7 and 58.3% for 15. The lower bioavailability for compound 1 may be due to its poor solubility and 7 may be the consequence of active efflux counteracting absorption as indicated by the Caco-2 efflux ratio of 2.2 (Table 1).
Table 3.
In vivo pharmacokinetic parameters in rats [mean (SD)]
| Compound | Cmax [mg/L] |
AUCo0-∞ [hr mg/L] |
t½ [hr] |
CL [L/hr/kg] |
Vd [L/kg] |
fe [%] |
F [%] |
Cytotoxicity (in vitro) IC50ug/ml |
|---|---|---|---|---|---|---|---|---|
| 1 | 2.28 (0.43) |
2.75 (0.58) |
2.79 (0.36) |
3.77 (0.77) |
10.9 (3.7) |
0.00 | 29.4 | 2.5 (0.7) |
| 6 | 0.83 (0.19) |
1.63 (0.43) |
2.39 (0.74) |
6.50 (1.81) |
35.0 (24.2) |
0.00 | 100 | 2.9 (0.4) |
| 7 | 2.73 (0.41) |
3.36 (0.44) |
0.72 (0.17) |
3.02 (0.42) |
2.86 (0.62) |
0.021 (0.01) |
47.9 | 3.2 (0.3) |
| 15 | 1.19 (0.34) |
0.96 (0.37) |
0.61 (0.41) |
11.61 (4.04) |
3.1 (1.6) |
0.68 (0.44) |
58.3 | 20.3 (2.5) |
| Linezolid†21,22 | 15* | 15.5* | 1.0 | 0.63 | 0.72 | 73 | 109 | - |
| Ofloxacin†23,24 | 10.9* | 6.1–11.8* | 1.5–1.8 | 0.86– 1.62 |
0.99 | 46 | 78 | - |
data shown taken from previously published reports as cited.
Based on a 10 mg/kg IV dose.
In these in vivo pharmacokinetic studies, however, toxicity was observed in rats after IV dosing with 15, leading to seizures and death in one animal. No animals died after oral administration of this compound, probably because of the lower peak plasma concentrations compared to the IV route. Compounds 1, 6 and 7 were found to be non-toxic in both administration modes.
5. Conclusions
A novel series of tetra cyclic isoxazolines was synthesized to improve the pharmacokinetic and pharmacodynamic properties over an initial lead antituberculosis agent. Introduction of solubilizing and metabolically blocked outer rings was successful in improving the overall solubility and decreasing protein binding. However, this resulted in compounds with overall in vitro microsomal stability that was lower than desired. When comparing ring substitutions it was noted that introduction of more nonpolar-groups gave rise to better MIC values but also generally worse solubility and metabolic stabilities, a problem we have observed in other tuberculosis drug development projects.19 This created a necessary tradeoff between these values when selecting lead candidates for advancement into in vivo pharmacokinetic testing in rats. Thus, compounds 6, 7 and 15 were put forward as they possessed the best overall improvements in in vitro pharmacokinetic parameters tested while retaining excellent to good MIC activity. Encouragingly the pharmacokinetic profile of our compounds showed all compounds to have high oral bioavailability with the exception of initial compound 1 which was likely limited by poor solubility. However, compounds 7 and 15 were rapidly cleared in vivo and judged unsuitable for further advancement. Compound 6 demonstrated a significantly longer half-life, higher volume of distribution and good tolerability. This compound has now advanced forward into a in vivo efficacy testing phase that includes formulation optimization studies to boost its solubility and absorption, dose optimization using time kill experiments20 and efficacy testing in a rapid mouse model of tuberculosis infection.
To gain further insight the pharmacokinetic parameters of these agents were compared to the reported values of synthetic anti-bacterial agents linezolid and ofloxacin both known for their excellent bioavailability.21–24 As can be seen in Table 3 linezolid and ofloxacin have a substantially lower clearance and smaller volume of distribution. Thus, despite of similarly short half-lives in rats, their peak concentrations and systemic exposure are substantially higher than for the investigated nitrofuran isoxazolines, suggesting that further stabilization against metabolic degradation may be a promising pathway to further optimize the current lead compound.
Experimental
Reagents and Instrumentation
All anhydrous solvents and starting materials were purchased from Aldrich Chemical Co. (Milwaukee, WI). All reagent grade solvents used from chromatography were purchased from Fisher Scientific (Suwanee, GA) and flash column chromatography silica cartridges were obtained from Biotage Inc. (Lake Forest, VA). The reactions were monitored by thin-layer chromatography (TLC) on pre-coated Merch 60 F254 silica gel plates and visualized using UV light (254 nm). A Biotage FLASH column chromatography system was used to purify mixtures. All NMR spectra were recorded on a Bruker-400 spectrometer. Chemical shifts (δ) are reported in parts per million relative to the residual solvent peak or internal standard (tetramethylsilane), and coupling constants (J) are reported in hertz (Hz). High resolution mass spectra were recorded on a Waters Xevo G2 QTOF LCMS using ESI. Purity of the products was confirmed before testing by analytical RP-HPLC on a Shimadzu HPLC system, and all final compounds had a purity of 95% or greater as determined by RP-HPLC. Gradient Conditions M1: solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in MeOH): 0–1.00 min 95% A, 1.00–6.00 min 0–95% B (linear gradient), 6.00–9.50 min 100% B, 9.50–9.75 min 0–95% A, 9.75–10.0 min 95% A, detection by UV at 254 nm and by ELSD. Gradient Conditions M2: same as M1 except solvent B is (0.1% formic acid in acetonitrile).
General procedure I, for preparation of 3b-c
4-Bromo aryl iodide (1 mmol), tributyl(vinyl)stannane (1 mmol) and PdCl2(PPh3)2 (0.05 mmol) was disolved in anhydrous DMF and heated to 160 °C in microwave for 5 min. The reaction mass was cooled to room temperature, diethyl ether and water were added to form a partition and the organic layer was washed with water, brine, dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by flash chromatography to afford 3b-c in 80–82 % yields.
2-Bromo-5-vinylpyridine (3b)
2-Bromo-5-iodopyridine (2 g, 7.04 mmol), tributyl(vinyl)stannane (2.23 g, 7.04 mmol) and PdCl2(PPh3)2 (0.24 g, 0.35 mmol) was disolved in anhydrous DMF (6 mL) and the reaction was carried out as discribed in general procedure I to afford 3b (1.06 g) in 82% yield. 1H NMR (400 MHz, CDCl3): δ 8.35 (d, J = 2.4 Hz, 1H), 7.60 (dd, J = 8.3, 2.6 Hz, 1H), 7.44 (d, J = 8.2 Hz, 1H), 6.65 (dd, J = 17.7, 11.0 Hz, 1H), 5.83 (d, J = 17.6 Hz, 1H), 5.43 (d, J = 11.0 Hz, 1H); LCMS: 186 (M++2).
1-Bromo-2-fluoro-4-vinylbenzene (3c)
1-Bromo-2-fluoro-4-iodobenzene (2 g, 6.65 mmol), tributyl(vinyl)stannane (2.10 g, 6.65 mmol) and PdCl2(PPh3)2 (0.23 g, 0.33 mmol) was disolved in anhydrous DMF (6 mL) and the reaction was carried out as discribed in general procedure I to afford 3c (1.06 g) in 80% yield. 1H NMR (400 MHz, CDCl3): δ 7.46–7.55 (m, 1H), 7.16 (dd, J = 9.8, 2.0 Hz, 1H), 7.05 (dd, J = 8.2, 2.0 Hz, 1H), 6.63 (dd, J = 17.6, 10.9 Hz, 1H), 5.76 (d, J = 17.6 Hz, 1H), 5.34 (d, J = 10.9 Hz, 1H); LCMS: 203 (M++2).
General procedure II, for preparation of 4a-c
A mixture of aryl bromide (1.0 mmol), morpholine (2.0 mmol), diacetoxypalladium (0.2 mmol), sodium butan-1-olate (2.4 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.4 mmol) was dissolved in anhydrous toluene and heated to 80 °C for 12 h, then the reaction mass was concentrated under reduced pressure and purified directly by flash chromatography to afford 4a-c in 69–83 % yields.
4-(4-Vinylphenyl)morpholine (4a)
1-Bromo-4-vinylbenzene 9 (0.2 g, 1.09 mmol), morpholine (0.19 g, 2.18 mmol), diacetoxypalladium (0.05 g, 0.21 mmol), sodium butan-1-olate (0.25 g, 2.62 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.13 g, 0.43 mmol) was dissolved in anhydrous toluene and the reaction was carried out as discribed in general procedure II to afford 4a (0.16 g) in 77% yield. 1H NMR (400 MHz, CDCl3): δ 7.30–7.34 (m, 2H), 6.82–6.86 (m, 2H), 6.64 (dd, J = 17.6, 10.9 Hz, 1H), 5.59 (dd, J = 17.6, 1.0 Hz, 1H), 5.09 (dd, J = 10.9, 1.0 Hz, 1H), 3.77–3.84 (m, 4H), 2.70–2.77 (m, 4H); LCMS: 190 (M++1).
4-(5-Vinylpyridin-2-yl)morpholine (4b)
2-Bromo-5-vinylpyridine 3b (0.6 g, 3.26 mmol), morpholine (0.56 g, 6.52 mmol), diacetoxypalladium (0.14 g, 0.65 mmol), sodium butan-1-olate (0.75 g, 7.82 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.38 g, 1.30 mmol) was dissolved in anhydrous toluene and the reaction was carried out as discribed in general procedure II to afford 4b (0.51 g) in 83% yield. 1H NMR (400 MHz, CDCl3): δ 8.15–8.21 (m, 1H), 7.62 (dd, J = 8.9, 2.4 Hz, 1H), 6.55–6.67 (m, 2H), 5.58 (d, J = 17.6 Hz, 1H), 5.13 (d, J = 11.0 Hz, 1H), 3.78–3.86 (m, 4H), 3.48–3.56 (m, 4H); LCMS: 191 (M++1).
4-(2-Fluoro-4-vinylphenyl)morpholine (4c)
1-Bromo-2-fluoro-4-vinylbenzene 3c (1.4 g, 6.96 mmol), morpholine (1.21 g, 13.92 mmol), diacetoxypalladium (0.31 g, 1.39 mmol), sodium butan-1-olate (1.60 g, 16.71 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.83 g, 2.79 mmol) was dissolved in anhydrous toluene and the reaction was carried out as discribed in general procedure II to afford 4c (1.0 g) in 69% yield. 1H NMR (400 MHz, CDCl3): δ 7.05–7.17 (m, 2H), 6.87 (t, J = 8.5 Hz, 1H), 6.55–6.70 (m, 1H), 5.63 (d, J = 17.6 Hz, 1H), 5.19 (d, J = 10.8 Hz, 1H), 3.83–3.91 (m, 4H), 3.07–3.12 (m, 4H); LCMS: 208 (M++1).
General procedure III, for preparation of 6–8
At room temperature, with vigorous stirring, a solution of Et3N (1.2 mmol) in anhydrous CHCl3 was slowly added to a solution of olefin (1.0 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (1.2 mmol) in anhydrous CHCl3. The reaction mixture was stirred at room temperature for 2 h and then diluted with excess CHCl3, washed with water, dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by flash chromatography to afford compounds 6–8 in 75.3–80 % yields.
4-(4-(3-(5-Nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)morpholine (6)
To a solution of 4-(4-vinylphenyl)morpholine 4a (0.2 g, 1.05 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.24 g, 1.26 mmol) in anhydrous CHCl3 (5 mL) was added Et3N (0.17 mL, 1.26 mmol) in CHCl3 (1 mL) and the reaction continued as described in general procedure III to afford 0.29 g of 6 in 80% yield. 1H NMR (500 MHz, CDCl3): δ 7.39 (d, J = 3.9 Hz, 1H), 7.27 (d, J = 9.2 Hz, 2H), 7.03 (d, J = 3.9 Hz, 1H), 6.91 (d, J = 8.5 Hz, 2H), 5.75 (dd, J = 11.2, 9.03 Hz, 1H), 3.86 (t, J = 4.6 Hz, 4H), 3.75 (dd, J = 17.0, 10.9 Hz, 1H), 3.39 (dd, J = 17.0, 8.7 Hz, 1H), 3.17 (t, J = 4.8 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 151.63, 147.81, 147.50, 129.97, 127.18, 115.61, 113.08, 112.43, 84.00, 66.78, 48.96, 41.20; HRMS m/z [M+H]+ calcd for C17H17N3O5: 344.125, found: 344.124.
4-(5-(3-(5-Nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)pyridin-2-yl)morpholine (7)
To a solution of 4-(5-vinylpyridin-2-yl)morpholine 4b (1.00 g, 5.26 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (1.20 g, 6.31 mmol) in anhydrous CHCl3 (15 mL) was added Et3N (0.87 mL, 6.31 mmol) in CHCl3 (3 mL) and the reaction continued as described in general procedure III to afford 7 (1.42 g) in 78.6% yield. 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 2.4 Hz, 1H), 7.51 (dd, J = 8.9, 2.5 Hz, 1H), 7.40 (d, J = 3.9 Hz, 1H), 7.05 (d, J = 3.9 Hz, 1H), 6.66 (d, J = 8.8 Hz, 1H), 5.74 (dd, J = 11.0, 9.0 Hz, 1H), 3.85 – 3.70 (m, 5H), 3.53 (t, 4H), 3.37 (dd, J = 17.2, 9.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 159.79, 147.90, 147.23, 146.47, 135.50, 123.63, 113.04, 112.57, 106.95, 82.16, 66.66, 45.44, 40.86; HRMS m/z [M+H]+ calcd for C16H16N4O5: 345.119; found: 345.119.
4-(2-Fluoro-4-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)morpholine (8)
To a solution of 4-(2-fluoro-4-vinylphenyl)morpholine 4c (0.38 g, 1.83 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.41 g, 2.20 mmol) in anhydrous CHCl3 (6 mL) was added Et3N (0.30 mL, 2.20 mmol) in CHCl3 (2 mL) and the reaction continued as described in general procedure III to afford 8 (0.5 g) in 75.3% yield. 1H NMR (400 MHz, CDCl3): δ7.40 (d, J = 3.9 Hz, 1H), 7.06 (m, 3H), 6.94 (t, J = 8.6 Hz, 1H), 5.76 (dd, J = 11.2, 8.4 Hz, 1H), 3.87 (t, J = 4.0 Hz 4H), 3.79 (dd, J = 17.2, 8.4 Hz, 1H) 3.38 (dd, J = 17.2, 8.3 Hz, 1H), 3.09 (t, J = 4.0 Hz 4H); 13C NMR (101 MHz, CDCl3) δ 156.81, 154.35, 147.72, 147.13, 140.34, 133.69, 122.13, 118.84, 114.04, 113.82, 113.03, 112.66, 83.00, 66.90, 50.73, 41.53, 29.70; HRMS m/z [M+H]+ calcd for C17H16FN3O5: 362.114; found: 362.113.
4-(4-vinylphenyl)thiomorpholine (10)
A mixture of 1-bromo-4-vinylbenzene 9 (1.0 g, 5.46 mmol), thiomorpholine (1.127 g, 10.93 mmol), sodium butan-1-olate (1.26 g, 13.11 mmol), Pd(OAc)2 (0.245 g, 1.09 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.652 g, 2.18 mmol) in anhydrous toluene was heated at 80 °C for 4 h. The reaction mass was then concentrated under reduced pressure and purified by flash chromatography to afford 10 (0.86 g) in 76.6% yield. 1H NMR (400 MHz, CDCl3): δ 7.30–7.34 (m, 2H), 6.82–6.86 (m, 2H), 6.64 (dd, J = 17.6, 10.9 Hz, 1H), 5.59 (dd, J = 17.6, 1.0 Hz, 1H), 5.09 (dd, J = 10.9, 1.0 Hz, 1H), 3.53–3.62 (m, 4H), 2.70–2.77 (m, 4H); LCMS: 206 (M++1).
3-(5-nitrofuran-2-yl)-5-(4-thiomorpholinophenyl)-4,5-dihydroisoxazole (11)
To a stirred solution of 4-(4-vinylphenyl)thiomorpholine 10 (0.65 g, 3.17 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.72 g, 3.80 mmol) in anhydrous CHCl3 (10 mL), Et3N (0.53 mL, 3.80 mmol) was added at room temperature and stirred at the same temperature for an additional 2 h. The reaction mass was then diluted with CHCl3 (20 mL), washed with water (2 × 20 mL), dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by flash chromatography to afford 11 (0.71 g) in 63% yield. 1H NMR (400 MHz, CDCl3): δ 7.41 (d, J = 3.9 Hz, 1H), 7.26 (d, J = 7.8 Hz, 2H), 7.05 (d, J = 3.9 Hz, 1H), 6.88 (d, J = 7.8 Hz , 2H), 5.76 (dd, J = 11.1, 8.9 Hz, 1H), 3.76 (dd, J = 17.2, 11.1 Hz, 1H), 3.63 – 3.55 (m, 4H), 3.40 (dd, J = 17.2, 11.1 Hz, 1H), 2.77 – 2.70 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 151.31, 147.83, 147.51, 129.52, 127.33, 116.77, 113.08, 112.43, 84.01, 51.69, 41.16, 26.45; HRMS m/z [M+H]+ calcd for C17H17N3O4S: 360.101; found: 360.101.
4-(4-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)thiomorpholine 1-oxide (12)
To a solution of 3-(5-nitrofuran-2-yl)-5-(4-thiomorpholinophenyl)-4,5-dihydroisoxazole 11 (0.05 g, 0.139 mmol) in MeOH (2 mL) and water (0.5 mL) was added NaIO4 (0.03 g, 0.15 mmol). The mixture was allowed to stir at room temperature for 12 h and filtered. The filtrate was concentrated under reduced pressure and purified by flash chromatography to afford 12 (0.03 g) in 58% yield; 1H NMR (400 MHz, CDCl3): δ 7.40 (d, J = 3.8 Hz, 1H), 7.29 (d, J = 8.0 Hz , 2H), 7.05 (d, J = 3.9 Hz, 1H), 6.96 (d, J = 8.0 Hz, 2H), 5.77 (dd, J = 11.1, 8.8 Hz, 1H), 4.07 – 4.00 (m, 2H), 3.77 (dd, J = 17.2, 11.1 Hz, 1H), 3.67 – 3.57 (m, 2H), 3.40 (dd, J = 17.2, 8.8 Hz, 1H), 2.94 – 2.79 (m, 4H); HRMS m/z [M+H]+ calcd for C17H17N3O5S: 376.095; found: 376.095.
General procedure IV, for preparation of 14a-f
A mixture of aryl bromide (1.0 mmol), 4-methylpiperazin-2-one (for compounds 15–17)/ morpholin-3-one (for compounds 18–20) (2.0 mmol), N, N' dimethylethylene diamine (0.1 mmol), K2CO3 (2.0 mmol) and CuI (0.05 mmol) in anhydrous toluene was heated to reflux with stirring for 6 h. Then the reaction mixture was cooled to room temperature, poured into water, stirred vigorously and extracted thrice with ethyl acetate, dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by flash chromatography to afford compounds 14a-f in 54–68% yields.
4-methyl-1-(4-vinylphenyl)piperazin-2-one (14a)
A mixture of 1-bromo-4-vinylbenzene 9 (1.0 g, 5.46 mmol), 4-methylpiperazin-2-one (1.24 g, 10.93 mmol), N, N' dimethylethylene diamine (0.04 g, 0.54 mmol), K2CO3 (1.51 g, 10.93 mmol) and CuI (0.05 g, 0.27 mmol) in anhydrous toluene was heated to reflux and the reaction was continued as described in general procedure IV to afford 14a (0.7 g) in 60% yield. 1H NMR (400 MHz, CDCl3): δ 7.39–7.48 (m, 2H), 7.24–7.27 (m, 2H), 6.70 (dd, J = 17.6, 10.9 Hz, 1H), 5.73 (dt, J = 17.6, 0.9 Hz, 1H), 5.26 (dt, J = 10.9, 0.9 Hz, 1H), 3.65–3.75 (m, 2H), 3.28 (s, 2H), 2.75–2.83 (m, 2H), 2.41 (s, 3H); LCMS: 217 (M++1).
4-Methyl-1-(5-vinylpyridin-2-yl)piperazin-2-one (14b)
A mixture of 2-bromo-5-vinylpyridine 3b (0.15 g, 0.81 mmol), 4-methylpiperazin-2-one (0.18 g, 1.63 mmol), N, N' dimethylethylene diamine (0.007 g, 0.08 mmol), K2CO3 (0.22 g, 1.63 mmol) and CuI (0.007 g, 0.04 mmol) in anhydrous toluene was heated to reflux and the reaction was continued as described in general procedure IV to afford 14b (0.11 g) in 62.8% yield. 1H NMR (400 MHz, CDCl3): δ 8.40 (d, J = 2.3 Hz, 1H), 8.12 (dd, J = 8.6, 1.0 Hz, 1H), 7.80 (dd, J = 8.7, 2.4 Hz, 1H), 6.72 (dd, J = 17.7, 11.0 Hz, 1H), 5.81 (dd, J = 17.7, 0.7 Hz, 1H), 5.38 (dd, J = 11.0, 0.8 Hz, 1H), 3.66–3.76 (m, 2H), 3.28 (s, 2H), 2.76–2.82 (m, 2H), 2.41 (s, 3H); LCMS: 218 (M++1).
1-(2-Fluoro-4-vinylphenyl)-4-methylpiperazin-2-one (14c)
A mixture of 1-bromo-2-fluoro-4-vinylbenzene 3c (0.2 g, 0.99 mmol), 4-methylpiperazin-2-one (0.22 g, 1.99 mmol), N, N' dimethylethylene diamine (0.008 g, 0.09 mmol), K2CO3 (0.27 g, 1.99 mmol) and CuI (0.009 g, 0.04 mmol) in anhydrous toluene was heated to reflux and the reaction was continued as described in general procedure IV to afford 14c (0.12 g) in 55% yield. 1H NMR (400 MHz, CDCl3): δ 7.18–7.24 (m, 3H), 6.66 (dd, J = 17.5, 10.8 Hz, 1H), 5.74 (d, J = 17.5 Hz, 1H), 5.32 (d, J = 10.8 Hz, 1H), 3.62–3.69 (m, 2H), 3.30 (s, 2H), 2.77–2.84 (m, 2H), 2.42 (s, 3H); LCMS: 235 (M++1).
4-(4-Vinylphenyl)morpholin-3-one (14d)
A mixture of 1-bromo-4-vinylbenzene 9 (0.2 g, 1.09 mmol), morpholin-3-one (0.22 g, 2.18 mmol), N, N' dimethylethylene diamine (0.01 g, 0.10 mmol), K2CO3 (0.30 g, 2.18 mmol) and CuI (0.01 g, 0.05 mmol) in anhydrous toluene was heated to reflux and the reaction was continued as described in general procedure IV to afford 14d (0.14 g) in 64% yield. 1H NMR (400 MHz, CDCl3): δ 7.44–7.47 (m, 2H), 7.28–7.32 (m, 2H), 6.71 (dd, J = 17.6, 10.9 Hz, 1H), 5.74 (dd, J = 17.6, 0.9 Hz, 1H), 5.27 (dd, J = 10.8, 0.9 Hz, 1H), 4.35 (s, 2H), 4.00–4.07 (m, 2H), 3.73–3.80 (m, 2H); LCMS: 204 (M++1).
4-(5-vinylpyridin-2-yl)morpholin-3-one (14e)
A mixture of 2-bromo-5-vinylpyridine 3b (0.2 g, 1.08 mmol), morpholin-3-one (0.22 g, 2.17 mmol), N, N' dimethylethylene diamine (0.01 g, 0.10 mmol), K2CO3 (0.30 g, 2.17 mmol) and CuI (0.01 g, 0.05 mmol) in anhydrous toluene was heated to reflux and the reaction was continued as described in general procedure IV to afford 14e (0.15 g) in 67.5% yield. 1H NMR (400 MHz, CDCl3): δ 8.42 (d, J = 2.3 Hz, 1H), 8.11 (dd, J = 8.5, 0.8 Hz, 1H), 7.80 (dd, J = 8.7, 2.4 Hz, 1H), 6.71 (dd, J = 17.7, 11.0 Hz, 1H), 5.81 (dd, J = 17.7, 0.7 Hz, 1H), 5.38 (dd, J = 11.0, 0.7 Hz, 1H), 4.38 (s, 2H), 4.18 – 4.02 (m, 4H); LCMS: 205 (M++1).
4-(2-fluoro-4-vinylphenyl)morpholin-3-one (14f)
A mixture of 1-bromo-2-fluoro-4-vinylbenzene 3c (0.2 g, 0.99 mmol), morpholin-3-one (0.20 g, 1.99 mmol), N, N' dimethylethylene diamine (0.008 g, 0.09 mmol), K2CO3 (0.27 g, 1.99 mmol) and CuI (0.01 g, 0.05 mmol) in anhydrous toluene was heated to reflux and the reaction was continued as described in general procedure IV to afford 14f (0.11 g) in 53.6% yield. 1H NMR (400 MHz, CDCl3): δ 7.18–7.24 (m, 3H), 6.66 (dd, J = 17.5, 10.8 Hz, 1H), 5.74 (d, J = 17.5 Hz, 1H), 5.32 (d, J = 10.8 Hz, 1H), 4.39 (s, 2H), 4.14 – 4.03 (m, 4H); LCMS: 222 (M++1).
4-methyl-1-(4-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)piperazin-2-one (15)
To a solution of 4-methyl-1-(4-vinylphenyl)piperazin-2-one 14a (0.82 g, 3.79 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.86 g, 4.55 mmol) in anhydrous CHCl3 (10 mL) was added Et3N (0.63 mL, 4.55 mmol) in CHCl3 (3 mL) and the reaction was continued as described in general procedure III to afford 15 (0.94 g) in 67.2% yield. 1H NMR (400 MHz, CDCl3): δ 7.44 – 7.29 (m, 5H), 7.03 (d, J = 3.8 Hz, 1H), 5.84 (dd, J = 11.1, 8.0 Hz, 1H), 3.82 (dd, J = 17.1, 11.2 Hz, 1H), 3.71 (dd, J = 6.2, 4.6 Hz, 2H), 3.40 (dd, J = 17.1, 8.1 Hz, 1H), 3.28 (s, 2H), 2.79 (t, J = 12.0 Hz, 2H), 2.41 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 166.88, 152.19, 147.66, 147.05, 142.22, 138.05, 126.67, 126.39, 112.87, 83.21, 59.77, 52.03, 50.03, 45.12, 41.76; HRMS m/z [M+H]+ calcd for C18H18N4O5: 371.134; found: 371.134.
4-methyl-1-(5-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)pyridin-2-yl)piperazin-2-one (16)
To a solution of 4-methyl-1-(5-vinylpyridin-2-yl)piperazin-2-one 14b (0.06 g, 0.27 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.06 g, 0.33 mmol) in anhydrous CHCl3 (3 mL) was added Et3N (0.04 mL, 0.33 mmol) in CHCl3 (0.5 mL) and the reaction was continued as described in general procedure III to afford 16 (0.05 g) in 56.7% yield. 1H NMR (400 MHz, CDCl3): δ 8.42 (d, J = 2.4 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.69 (dd, J = 8.7, 2.5 Hz, 1H), 7.40 (d, J = 3.9 Hz, 1H), 7.06 (d, J = 3.8 Hz, 1H), 5.85 (dd, J = 11.2, 8.3 Hz, 1H), 4.14 – 3.95 (m, 2H), 3.86 (dd, J = 17.2, 11.1 Hz, 1H), 3.40 (dd, J = 17.2, 8.3 Hz, 1H), 3.31 (s, 2H), 2.80 (t, J = 5.5 Hz, 2H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 167.79, 153.72, 152.23, 147.73, 146.74, 145.40, 134.82, 131.54, 119.74, 112.99, 81.27, 60.32, 52.03, 46.38, 45.00, 41.53; HRMS m/z [M+H]+ calcd for C17H17N5O5: 372.130; found: 372.129.
1-(2-fluoro-4-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)-4-methylpiperazin-2-one (17)
To a solution of 1-(2-fluoro-4-vinylphenyl)-4-methylpiperazin-2-one 14c (0.07 g, 0.29 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.06 g, 0.35 mmol) in anhydrous CHCl3 (3 mL) was added Et3N (0.05 mL, 0.35 mmol) in CHCl3 (0.5 mL) and the reaction was continued as described in general procedure III to afford 17 (0.07 g) in 61% yield. 1H NMR (400 MHz, CDCl3): δ 7.39 (dd, J = 3.9, 0.6 Hz, 1H), 7.33–7.29 (m, 1H), 7.24 – 7.14 (m, 2H), 7.04 (d, J = 3.9 Hz, 1H), 5.83 (dd, J = 11.2, 7.6 Hz, 1H), 3.85 (dd, J = 17.2, 11.2 Hz, 1H), 3.65 (t, J = 12 Hz, 2H), 3.39 (dd, J = 17.1, 7.6 Hz, 1H), 3.29 (s, 2H), 2.81 (t, J = 12 Hz, 2H), 2.42 (s, 3H); 13C NMR (101 MHz, CDCl3): δ 166.95, 159.21, 156.70, 147.61, 146.73, 141.38, 129.75, 129.29, 121.88, 114.35, 114.13, 112.95, 82.34, 59.44, 51.84, 49.75, 45.11, 41.89; HRMS m/z [M+H]+ calcd for C18H17FN4O5: 389.125; found: 389.125.
4-(4-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)morpholin-3-one (18)
To a solution of 4-(4-vinylphenyl)morpholin-3-one 14d (0.04 g, 0.19 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.04 g, 0.23 mmol) in anhydrous CHCl3 (3 mL) was added Et3N (0.03 mL, 0.23 mmol) in CHCl3 (0.5 mL) and the reaction was continued as described in general procedure III to afford 18 (0.05 g) in 72.5% yield. 1H NMR (400 MHz, CDCl3): δ 7.46 – 7.33 (m, 5H), 7.04 (d, J = 3.9 Hz, 1H), 5.85 (dd, J = 11.2, 8.1 Hz, 1H), 4.35 (s, 2H), 4.08 – 4.01 (m, 2H), 3.89 – 3.73 (m, 3H), 3.40 (dd, J = 17.1, 8.1 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 166.74, 147.67, 147.01, 141.60, 138.25, 126.73, 125.88, 112.87, 83.13, 68.58, 64.10, 49.54, 41.80; HRMS m/z [M+H]+ calcd for C17H15N3O6: 358.103; found: 358.103.
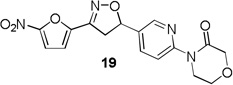
4-(5-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)pyridin-2-yl)morpholin-3-one (19)
To a solution of 4-(5-vinylpyridin-2-yl)morpholin-3-one 14e (0.08 g, 0.39 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.09 g, 0.47 mmol) in anhydrous CHCl3 (4 mL) was added Et3N (0.06 mL, 0.47 mmol) in CHCl3 (0.5 mL) and the reaction was continued as described in general procedure III to afford 19 (0.09 g) in 66.8% yield. 1H NMR (400 MHz, CDCl3): δ 8.43 (d, J = 2.4 Hz, 1H), 8.20 (d, J = 8.7 Hz, 1H), 7.71 (dd, J = 8.6, 2.5 Hz, 1H), 7.41 (d, J = 3.8 Hz, 1H), 7.07 (d, J = 3.9 Hz, 1H), 5.86 (dd, J = 11.2, 8.3 Hz, 1H), 4.36 (s, 2H), 4.19 – 4.01 (m, 4H), 3.87 (dd, J = 17.2, 11.2 Hz, 1H), 3.41 (dd, J = 17.2, 8.3 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 167.86, 152.96, 147.74, 146.73, 145.36, 135.09, 131.72, 119.09, 112.95, 81.22, 68.63, 64.29, 45.63, 41.58; HRMS m/z [M+H]+ calcd for C16H14N4O6: 359.098; found: 359.098.
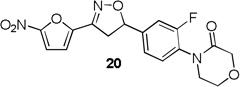
4-(2-fluoro-4-(3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazol-5-yl)phenyl)morpholin-3-one (20)
To a solution of 4-(2-fluoro-4-vinylphenyl)morpholin-3-one 14f (0.05 g, 0.22 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride (0.05 g, 0.27 mmol) in anhydrous CHCl3 (3 mL) was added Et3N (0.03 mL, 0.27 mmol) in CHCl3 (0.5 mL) and the reaction was continued as described in general procedure III to afford 20 (0.05 g) in 66.3% yield. 1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 4.0 Hz, 1H), 7.33–7.37 (m, 1H), 7.19–7.24 (m, 2H), 7.05 (d, J = 3.9 Hz, 1H), 5.84 (dd, J = 11.2, 7.6 Hz, 1H), 4.36 (s, 2H), 4.06 – 4.01 (m, 2H), 3.86 (dd, J = 17.2, 11.3 Hz, 1H), 3.75 – 3.68 (m, 2H), 3.40 (dd, J = 17.1, 7.6 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 166.75, 159.11, 156.60, 147.61, 146.68, 141.72, 129.61, 128.64, 121.94, 114.44, 114.23, 112.96, 99.98, 82.25, 68.52, 64.04, 49.68, 41.93; HRMS m/z [M+H]+ calcd for C17H14FN3O6: 376.093; found: 376.092.
MIC Determination
MICs were determined using the microbroth dilution method according to Clinical Laboratory Standards Institute (CLSI) standards25 and were read by visual inspection. Two-fold serial dilutions of antibiotic in 100 µl of the appropriate broth media were first prepared in 96-well round bottom microtiter plates (Nunc, USA). An equivalent volume (100µL) of bacterial broth inoculum (containing approximately 106 bacterial CFU/mL for M. tuberculosis and 105 cfu/mL for all other bacteria) was added to each well to give final concentrations of drug starting at 200 µg/mL and the plates were incubated aerobically at 37°C. M. tuberculosis microtiter plates were incubated for 7 or 14 days and all other strains were incubated overnight. The MIC was recorded as the lowest concentration of drug which prevented visible growth.
Cytotoxicity
Vero cells (kidney epithelial cells; ATCC CCL-81) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and maintained in a humidified incubator (37°C, 5% CO2). Monolayers were trypsinized, seeded at ~10% confluency in white-wall, clear-bottom 96-well microtiter plates, and allowed to adhere overnight. The next day, media was removed and replaced with fresh DMEM/FBS containing two-fold serial dilutions of test compounds. Following additional 72 hours of incubation, cell viability was evaluated using MTT (CellTiter96®, Promega) according to the manufacturer’s instructions, with overnight solubilization. Absorbance at 570nm was recorded and IC50 values calculated from corresponding dose response curves. Results reported are the average of at least 2 independent experiments.
Solubility
This assay is performed on Biomek FX ADME-TOX workstation (Beckman Coulter, Inc.; Fullerton, CA). 30 µL of 10 mM compounds solution in DMSO was applied to each well in a stock plate. In a reference plate, compounds were diluted 600 fold in system solution buffer (SSB, pH=7.4; pION INC, Woburn, MA) and iso-propanol (1:1, v/v). Concentrations were assessed by UV spectrometry (230–500 nm). In the sample plate, compounds were diluted 100 fold in system solution buffer, incubated at room temperature for 18 h to allow the compounds to be fully stable, and then filtered through a 96-well filter plate (pION Inc., Woburn, MA). Fractions were collected from the filtered sample plate, diluted with isopropanol by 1:1 (v/v), and determined concentration via UV spectrometry. Calculation was carried out by µSOL Evolution software and all compounds were tested in triplicates.
Metabolic stability
Sample preparation for microsomal stability was modified from the published method of Di.26 DMSO stock solutions of test compounds were prepared at 10 mM concentration. Mouse liver microsomal solution was prepared by adding 0.058 mL of concentrated Mouse liver microsomes (20 mg/mL protein concentration) to 1.756 mL of 0.1 M potassium phosphate buffer (pH 7.4) and 5 µL of 0.5 M EDTA to make a 0.6381 mg/mL (protein) microsomal solution. NADPH regenerating agent contained 0.113 mL of NADPH A, 0.023 mL of NADPH B, and 0.315 mL of 0.1 M potassium phosphate buffer (pH 7.4). 2.2 µL of each test compound diluted solution was each added directly to 1.79 mL of liver microsomal solution. This solution was mixed and 90 µL was transferred to 6 time points plates (each in triplicate wells). For the Time 0 plate, 225 µL of cold acetonitrile with internal standard (4 µg/ml warfarin) was added to each well, followed by addition of NADPH regenerating agent (22.5 µL) and no incubation. For other five time points’ plate, NADPH regenerating agent (22.5 µL) was added to each well to initiate the reaction, the plate was incubated at 37° C for required time, followed by quenching of the reaction by adding 225 µL of cold acetonitrile with internal standard (4 µg/ml warfarin) to each well. All of the plates were sealed and mixed well at 600 rpm for 10 min and were centrifuged at 4000 rpm for 20 min. The supernatants (120 µL) were transferred to analytical plates for analysis by LC–MS. Conditions for Waters ACQUITY-UPLC-MS-UV system were described separately. The metabolic stability is evaluated via the half-life from least-squares fit of the multiple time points based on first-order kinetics.
Caco-2 permeability
High throughput Caco-2 permeability determinations were performed in the 96-well Transwell system using a modified method.27 Caco-2 cells were maintained at 37 °C in a humidified incubator with an atmosphere of 5% CO2. The cells were cultured in eagles’s minimum essential medium containing 20% FBS in 75 cm2 flasks, 100 units/ml of penicillin, and 100 µg/ml of streptomycin. The Caco-2 cells were seeded onto inserts of a 96-well plate at a density of 0.165×105 cells/insert and cultured in the MEM containing 10% FBS for 7 days. Each cultured monolayer on the 96-well plate was washed twice with HBSS/HEPES (10 mM, pH 7.4). The permeability assay was initiated by the addition of each compound solution (50 µmol/L) into inserts (apical side, A) or receivers (basolateral side, B). The Caco-2 cell monolayers were incubated for 2 h at 37 °C. Fractions were collected from receivers (if apical to basal permeability) or inserts (if basal to apical permeability), and concentrations were assessed by UPLC/MS (Waters; Milford, MA). All compounds were tested in triplicates. The A → B (or B → A) apparent permeability coefficients (Papp, cm/s) of each compound were calculated using the equation, Papp=(dQ/dt)/(A×C0). The flux of drug across the monolayer is dQ/dt (µmol/s). The initial drug concentration on the apical side is C0 (µmol/L). The surface area of the monolayer is A (cm2).
Plasma protein binding
Solutions were prepared at 10 mM in DMSO. Dulbecco's phosphate buffered saline (DPBS; pH 7.4) was obtained from Invitrogen (Carlsbad, CA). Single-Use RED (rapid equilibrium dialysis) device was obtained from Thermo scientific (Rockford, IL). Mouse and human plasma was obtained Lampire Biological Laboratories (Pipersville, PA). Sample preparation for plasma protein binding was modified from the method of Waters.28 The Teflon base plate with the RED inserts (MWCO 8 K) were allowed to use without any pre-condition the membrane inserts. Mouse plasma was thawed and centrifuged at 1000 rpm for 2 min to remove any particulates. Each compound was prepared at 10 µM in mouse and human plasma. This was done by adding 1 µL of drug solution (10 mM in DMSO) to 1000 µL of mouse or human plasma (0.1% DMSO). Spiked plasma solutions (300 µL) were placed into the sample chamber (indicated by the red ring) and 500 µL of DPBS into the adjacent chamber. The plate was sealed and incubated at 37°C on an orbital shaker (100 rpm) for 4 hours. After incubation, the seal was removed from the RED plate and the volume of the insert confirmed—little to no volume change occurred. Aliquots (50 µL) were removed from each side of the insert and dispensed into a 96-well deep plate. An equal volume of blank plasma or DPBS was added to the required wells to create analytically identical sample matrices (matrix matching). To each sample 200 µL of ACN containing 4 µg/ml warfarin internal standard was added. All of the plates were sealed and mixed well at 600 rpm for 10 min and were centrifuged at 4000 rpm for 20 min. The supernatants (120 µL) were transferred to analytical plates for analysis by LC–MS. Conditions for Waters ACQUITY-UPLC-MS-UV system were described separately. The test compound concentrations were quantified in both buffer and plasma chambers via peak areas relative to the internal standard. The percentage of the test compound bound to plasma was calculated on following equations: %Free= (Concentration buffer chamber/concentration plasma chamber) × 100% and %Bound=100%-%Free.
In vivo pharmacokinetics
Catheterized male Sprague-Dawley rats (jugular vein alone for oral study and jugular vein and femoral vein for intravenous study) weighing approximately 225g were obtained from Harlan Bioscience (Indianapolis, IN). Animals were kept on a 12h light/ dark cycle with access to food and water ad libitum. Animal studies were conducted according to the guideline of Animal Welfare Act and the Public Health Service Policy on Humane Care and Use of Laboratory Animals. The study protocol was approved by the institutional animal care and use committee of the University of Tennessee Health Science Center. Test compounds (1, 6, 7 and 15) were administered to a group of rats (n= 5) either intravenously (IV) or per oral (PO) at a dose of 10 mg/kg or 100 mg/kg respectively. For oral administration, the animals were fasted overnight, but had access to water ad libitum.
For the IV studies, compound 6 (10 mg/kg) was prepared by dissolving the drug in 2% ethanol and saline, whereas the formulations of compounds 1, 7 and 15 (10 mg/kg) were prepared by dissolving the drug in 30% DMSO, 30% propylene glycol, 20% PEG 3000 and 20% saline. The formulations were administered via femoral vein catheter followed by flushing the catheter with heparinized locking solution. For the PO studies, compound 6 (100 mg/kg) was prepared by dissolving the drug in 2% methanol and sonicated for 15 minutes, while the formulation for compounds 7 and 15 (100 mg/kg) were prepared in 60:40 PEG 3000 water and for compound 1 in 10% vitamin E TPGS solution. The oral doses were administered using oral gavage needle fitted to a 1.0 mL syringe.
Serial blood samples (approx. 250 µL) were collected at 0 (pre-dose), 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 4.0, 6.0, 8.0, 12.0, 24.0, 36.0 and 48.0 h post-dose. Plasma was separated immediately by centrifugation (3000xg for 10 min at 4°C) and stored at −80°C until analysis. Urine samples were collected at an interval of 0–6, 6–12, 12–24, 24–36 and 36–48 h post-dose and stored at −80°C until analysis. Plasma and urine samples were analyzed for drug concentrations by LC-MS/MS assay.
LC-MS/MS assay for plasma and urine drug concentrations
Sample preparation involved a simple protein precipitation method using acetonitrile. Plasma/urine proteins were precipitated by the addition of three volumes of internal standard (IS) spiked acetonitrile to 50 µL aliquots of plasma/urine test samples. The samples were vortex mixed for 1 minute and centrifuged at 3,000×g for 10 min at 4°C and the supernatants were collected for LC-MS/MS assay.
Chromatographic separations were carried out using a Shimadzu liquid chromatograph (Shimadzu Corporation, USA) consisting of two pumps, online degasser, system controller and a CTC Leap auto sampler (Leap Technologies, Carrboro, NC). Mobile phase consist of acetonitrile and 10 mM ammonium acetate buffer pH ~3.8 was used at a flow rate of 0.3 mL/min in gradient mode. A Phenomenex® C18(2), 3 µm, 50 × 2.0 mm column (Phenomenex, Torrance, CA) protected with a guard column was used for the separation. The samples (20 µL) were injected onto the column and the eluate was led directly into a mass spectrometer. An API 3000 triple-quadruple mass spectrometer (Applied Biosystems ABI/MDS-Sciex, Foster City, CA) equipped with an electrospray ion source was operated in the positive ion mode. The resulting multiple reaction monitoring chromatograms were used for quantification using the Analyst software version 1.4.2 (Applied Biosystems ABI/ MDS-Sciex, Foster City, CA). A calibration curve ranging from 3.9–5000 µg/L was constructed for each test compound by spiking the test compound into 50 µL of blank rat plasma or urine. A structurally similar analogue to the test compounds, Lee 1106,29 was used as IS to all calibration standards and all plasma or urine specimens. Linearity for calibration standards in triplicates was assessed by subjecting the spiked concentrations and the respective peak areas to least-square linear regression analysis with and without intercepts, and a weighted least-square regression (1/x or 1/x2). A proper calibration model was chosen after examination of residuals and coefficient of correlation in each case.
Pharmacokinetic Data Analysis
The pharmacokinetic profile of the test compounds was analyzed from plasma concentration-time data after IV and PO administration by non-compartmental analysis using Phoenix-WinNonlin 6.2 (Pharsight Corporation, Mountain View, CA). The terminal half-life (t1/2) was calculated as 0.693/λz, where λz is the terminal phase rate constant. The peak plasma concentration (Cmax) was obtained by visual inspection of the plasma concentration–time curves. The area under the plasma concentration–time curve from time 0 to infinity (AUC0-∞) was calculated by the trapezoidal rule with extrapolation to time infinity. Volume of distribution (Vd) was calculated as ratio of the area under the first moment curve (AUMC0-∞) time dose divided by the square of AUC0-∞. The total body clearance (CL) was calculated using the equation CL=Doseiv/AUC0-∞, iv. Oral bioavailability (F) was calculated by using F= (AUC0-∞, oral × Doseiv) / (AUC0-∞,iv × Doseoral), where Doseoral, Doseiv, AUC0-∞,iv, and AUC0-∞, oral are the oral and IV dose and the corresponding areas under the plasma concentration-time curves from time 0 to infinity, respectively. The fraction (fe) of the test compound excreted in urine was calculated as the cumulative amount of dose excreted unchanged in urine divided by the administered dose of the test compound.
Acknowledgements
This study was supported by the National Institutes of Health grant AI062415 and the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bailer AJ. Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm. 1988;16:303–309. doi: 10.1007/BF01062139. [DOI] [PubMed] [Google Scholar]
- 2.Yee D, Valiquette C, Pelletier M, Parisien I, Rocher I, Menzies D. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am J Respir Crit Care Med. 2003;167:1472–1477. doi: 10.1164/rccm.200206-626OC. [DOI] [PubMed] [Google Scholar]
- 3.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., 3rd PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tangallapally RP, Sun D, Rakesh, Budha N, Lee REB, Lenaerts AJM, Meibohm B, Lee RE. Discovery of novel isoxazolines as anti-tuberculosis agents. Bioorg. Med. Chem. Lett. 2007;17:6638–6642. doi: 10.1016/j.bmcl.2007.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tangallapally RP, Yendapally R, Lee RE, Lenaerts AJM, Lee RE. Synthesis and evaluation of cyclic secondary amine substituted phenyl and benzyl nitrofuranyl amides as novel antituberculosis agents. J Med Chem. 2005;48:8261–8269. doi: 10.1021/jm050765n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tangallapally RP, Yendapally R, Lee RE, Hevener K, Jones VC, Lenaerts AJM, McNeil MR, Wang Y, Franzblau S, Lee RE. Synthesis and evaluation of nitrofuranylamides as novel antituberculosis agents. J Med Chem. 2004;47:5276–5283. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 7.Tawari NR, Bairwa R, Ray MK, Rajan MG, Degani MS. Design, synthesis, and biological evaluation of 4-(5-nitrofuran-2-yl)prop-2-en-1-one derivatives as potent antitubercular agents. Bioorg Med Chem Lett. 2010;20:6175–6178. doi: 10.1016/j.bmcl.2010.08.127. [DOI] [PubMed] [Google Scholar]
- 8.Moraski GC, Thanassi JA, Podos SD, Pucci MJ, Miller MJ. One-step syntheses of nitrofuranyl benzimidazoles that are active against multidrug-resistant bacteria. J Antibiot (Tokyo) 2011;64:667–671. doi: 10.1038/ja.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldman RC. Maximizing bactericidal activity with combinations of bioreduced drugs. Future Med Chem. 2010;2:1253–1271. doi: 10.4155/fmc.10.215. [DOI] [PubMed] [Google Scholar]
- 10.Gadwood RC, Shinabarger DA. Chapter 12. Progress in the oxazolidinone antibacterials. Annual Reports in Medicinal Chemistry. 2000;35:135–144. [Google Scholar]
- 11.Barbachyn MR, Cleek GJ, Dolak LA, Garmon SA, Morris J, Seest EP, Thomas RC, Toops DS, Watt W, Wishka DG, Ford CW, Zurenko GE, Hamel JC, Schaadt RD, Stapert D, Yagi BH, Adams WJ, Friis JM, Slatter JG, Sams JP, Oien NL, Zaya MJ, Wienkers LC, Wynalda MA. Identification of phenylisoxazolines as novel and viable antibacterial agents active against gram-positive pathogens. Journal of Medicinal Chemistry. 2003;46:284–302. doi: 10.1021/jm020248u. [DOI] [PubMed] [Google Scholar]
- 12.Shaw KJ, Barbachyn MR. The oxazolidinones: past, present, and future. Ann N Y Acad Sci. 2011;1241:48–70. doi: 10.1111/j.1749-6632.2011.06330.x. [DOI] [PubMed] [Google Scholar]
- 13.Liu H, Hancock AA, Cowart MD. Cyclobutyl amine derivatives and their preparation, pharmaceutical compositions and use as histamine-3 receptor ligands. WO2006132914A2. 2006
- 14.Yang BV, Doweyko LM, Vaccaro W, Huynh TN, Tortolani DR, Dhar Tg. M. N-Heteroaryl carboxamides as modulators of glucocorticoid receptor, AP-1, and/or NF-κB activity and their preparation, pharmaceutical compositions and use in the treatment of diseases. WO2008057862A2. 2008
- 15.Chaffee SC, Albrecht BK, Hodous BL, Martin MW, McGowan DC, Dimauro EF, Reddy G, Cee VJ, Olivieri PR, Reed A, Romero K. Heteroaryl-substituted alkyne compounds as protein kinase inhibitors, their preparation, pharmaceutical compositions, and use in therapy. WO2006044823A2. 2006
- 16.Iijima T, Yamamoto Y, Akatsuka H, Kawaguchi T. Preparation of benzoxazines and related nitrogen-containing heterobicyclic compounds as mineralocorticoid receptor modulators. WO2007089034A1. 2007
- 17.Hurdle JG, Lee RB, Budha NR, Carson EI, Qi J, Scherman MS, Cho SH, McNeil MR, Lenaerts AJ, Franzblau SG, Meibohm B, Lee RE. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J Antimicrob Chemother. 2008;62:1037–1045. doi: 10.1093/jac/dkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honaker RW, Leistikow RL, Bartek IL, Voskuil MI. Unique roles of DosT and DosS in DosR regulon induction and Mycobacterium tuberculosis dormancy. Infect Immun. 2009;77:3258–3263. doi: 10.1128/IAI.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee RE, Protopopova M, Crooks E, Slayden RA, Terrot M, Barry CE., 3rd Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J Comb Chem. 2003;5:172–187. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 20.Budha NR, Lee RB, Hurdle JG, Lee RE, Meibohm B. A simple in vitro PK/PD model system to determine time-kill curves of drugs against Mycobacteria. Tuberculosis (Edinb) 2009 doi: 10.1016/j.tube.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slatter JG, Adams LA, Bush EC, Chiba K, Daley-Yates PT, Feenstra KL, Koike S, Ozawa N, Peng GW, Sams JP, Schuette MR, Yamazaki S. Pharmacokinetics, toxicokinetics, distribution, metabolism and excretion of linezolid in mouse, rat and dog. Xenobiotica; the fate of foreign compounds in biological systems. 2002;32:907–924. doi: 10.1080/00498250210158249. [DOI] [PubMed] [Google Scholar]
- 22.FDA Briefing Package: Anti-Infective Drugs Advisory Committee; Zyvox Preclinical Summary, U.S. Food and Drug Administration. 2000 http://www.fda.gov/ohrms/dockets/ac/00/backgrd/3597b1bb.pdf.
- 23.Wang H, Liao ZX, Chen M, Hu XL. Effects of hepatic fibrosis on ofloxacin pharmacokinetics in rats. Pharmacological research : the official journal of the Italian Pharmacological Society. 2006;53:28–34. doi: 10.1016/j.phrs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Foote EF, Halstenson CE. Effects of probenecid and cimetidine on renal disposition of ofloxacin in rats. Antimicrobial Agents and Chemotherapy. 1998;42:456–458. doi: 10.1128/aac.42.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National, C.f. C. L. S. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically—Seventh Edition: Approved Standard M7-A7. Wayne, PA, USA: CLSI; 2006. [Google Scholar]
- 26.Di L, Kerns EH, Ma XJ, Huang Y, Carter GT. Applications of high throughput microsomal stability assay in drug discovery. Comb Chem High Throughput Screen. 2008;11:469–476. doi: 10.2174/138620708784911429. [DOI] [PubMed] [Google Scholar]
- 27.Uchida M, Fukazawa T, Yamazaki Y, Hashimoto H, Miyamoto Y. A modified fast (4 day) 96-well plate Caco-2 permeability assay. J Pharmacol Toxicol Methods. 2009;59:39–43. doi: 10.1016/j.vascn.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Waters NJ, Jones R, Williams G, Sohal B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci. 2008;97:4586–4595. doi: 10.1002/jps.21317. [DOI] [PubMed] [Google Scholar]
- 29.Hurdle JG, O'Neill AJ, Chopra I, Lee RE. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nature reviews. Microbiology. 2011;9:62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]