

Abstract
Multiple Sclerosis (MS) is an autoimmune disease that affects ~400,000 people in the US. It is a chronic, disabling disease with no cure, and the current treatment includes use of immunosuppressive drugs that often exhibit toxic side effects. Thus, there is a pressing need for alternate and more effective treatment strategies that target the components of inflammatory cells. In recent years, regulatory T-cells (Tregs) have been found to play an important role in preventing the development of autoimmunity. Thus, expansion of T regs in vivo has the therapeutic potential against autoimmune diseases. Because Tregs constitutively express IL-2 receptors (IL-2Rs), we tested the effect of administration of IL-2 on the development of experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (MS). We used IL-2 both before (pre-treatment) or after (post-treatment) immunization with myelin oligodendrocyte glycoprotein (MOG35-55) peptide to induce EAE. The data demonstrated that pre-treatment with a moderate dose of IL-2 caused significant amelioration of EAE. Tissue histopathology of the central nervous system also confirmed the effectiveness of IL-2 pre-treatment by decreasing cellular infiltration in the spinal cord and preserving tissue integrity. IL-2 pretreatment expanded Treg cells while preventing the induction of Th17 during EAE development. In contrast, post-treatment with IL-2 failed to suppress EAE despite induction of Tregs. Together, these studies demonstrate that while expansion of Tregs using IL-2, prior to immunization or the onset of disease, can suppress the immune response, their role is limited after the antigen-specific response is triggered. Because IL-2 is used to treat certain types of cancers, and Tregs have applications in preventing the rejection of transplants, our studies also provide useful information on the use and limitations of Tregs in such clinical manifestations.
Keywords: experimental autoimmune encephalomyelitis (EAE), multiple sclerosis (MS), recombinant interleukin-2 (rIL-2), regulatory T-cells (Treg), Th17 cells
Introduction
Multiple sclerosis (MS) is an inflammatory autoimmune disease that afflicts about 150 per 100,000 individuals in the world each year (Rosati, 2011). Patients diagnosed with MS suffer from impairments in both cognitive and motor functions (Compston, 2002). During the course of disease, the body’s immune system becomes sensitized to self-antigens, in this case, myelin based antigen. As a result, activated immune cells mount a response against myelinated tissues located in the brain and spinal cord of the CNS. Studies have shown that the cellular composition of the inflammatory legions consist of Th1 and Th17 T-cells, which produce a series of pro-inflammatory cytokines (Huang, 2000). The mouse model experimental autoimmune encephalomyelitis (EAE) has been used extensively to investigate the role of Th cells in disease development. For example, the adoptive transfer of myelin-specific CD4+ Th1 cells into naïve recipient mice was found to promote the induction of EAE (Pettinelli, 1981), (McDonald, 1988), (Ando, 1989), (Waldburger, 1996), (Yura, 2001), (Lovett-Racke, 2004), (Gocke, 2007). Additionally, important Th1 transcription factors STAT4 and T-bet have been shown to be elevated during EAE, driving T-cells towards Th1 differentiation and production of pro-inflammatory cytokines (Lovett-Racke, 2004), (Chitnis, 2001), (Bettelli, 2004), (Nath, 2006). Similarly, IL-17 producing T-cells, better known as Th17 cells, have also been reported to be a driving force in the development of EAE (Langrish, 2005). Furthermore, when attempts were made to inhibit Th17 differentiation, clinical symptoms in murine EAE as well as in MS patients were repressed (Chen, 2006), (Alexander, 2010), (Axtell, 2010).
In contrast to the pathogenic role of Th1 and Th17 cells, CD4+FoxP3+ regulatory T cells (Tregs) have been shown to down-regulate the immune response and have been shown to play a critical in preventing generalized multiorgan autoimmunity (Jeon, 2012), (Tran, 2012). Moreover, MS patients possess either a lower frequency of Tregs or impairment in their suppressor function, which promotes disease development (Bjerg, 2012). Thus, expansion of Tregs in vivo has the potential to treat autoimmune disease as well as prevent the rejection of organ transplants (Veenstra, 2012). While most T cells express IL-2 receptors (IL-2Rs) upon activation, Tregs express IL-2Rs constitutively and dependent on IL-2 for their growth and survival (Sakaguchi, 1995), (Thornton, 1998), (Thornton, 2004). Furthermore, IL-2 was found to be essential for the maintenance of Tregs (Kevin, 2005). Thus, as shown in our previous studies, Tregs can be expanded in vivo using IL-2 which in turn exhibit strong immunosuppression (Melencio, 2006).
In this study, we investigated how expansion of Tregs in vivo using recombinant (rIL-2) therapy could regulate Th17 differentiation and alter the course EAE development. Our results illustrated that pre-treatment with rIL-2 could expand Tregs and mitigate the clinical scores of EAE mice and prevent cellular infiltration of immune cells in the CNS. We also noted that administration of rIL-2, after the onset of the disease was not protective, thereby demonstrating that the Tregs were unable to suppress the disease process that had already been initiated by the pathogenic Th cells. Because IL-2 therapy is also used in the treatment of cancers such as melanomas and renal cell carcinomas, our studies provide useful clues on how such a treatment impacts the Tregs and potential anti-tumor immunity. Low dose IL-2 therapy that expands Tregs may also help in preventing transplant rejection.
Material and Methods
Mice
Female C57BL/6 mice (6-8 week of age) were purchased from National Institutes of Health (NIH). All animals were housed in the University of South Carolina Animal Facility (Columbia, SC). All animal procedures were performed according to the NIH guidelines under protocols approved by the Institute of Animal Care and Use Committee of the University of South Carolina.
Reagents and Monoclonal Antibodies (mAbs)
Recombinant IL-2 (rIL-2) was provided by the National Cancer Institute Biological Resources Branch (Rockville, MD). RPMI 1640, L-glutamine, HEPES, phosphate-buffered saline (PBS), and fetal bovine serum were purchased from VWR (West Chester, PA). ConA was purchased from Sigma-Aldrich (St. Louis, MO). The following mAbs were purchased from eBioscience (San Diego, CA): Fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD4 (L3T4) mAb (GK1.5; rat IgG2b), Phycoerythrin (PE) conjugated anti-mouse IL-17A (eBio17B7; rat IgG2a), PE-conjugated anti-mouse/rat FoxP3 (FJK-16s; IgG2a), Allophycocyanin (APC) anti-mouse CD8 (Ly-2) (53-6.7; rat IgG2a), and affinity purified anti-mouse CD16/32 (93; rat IgGa2a).
In addition, FoxP3 Tregulatory cell staining kit was purchased from Biolegend (San Diego, CA). For intracellular cytokine staining, BD Cytofix/Cyto Perm Fixation/Permeabilization Solution Kit was purchased from BD Biosciences (San Jose, CA).
Effect of IL-2 on Experimental Autoimmune Encephalomyelitis (EAE) in Mice
EAE was induced in female C57BL/6 mice (6-8 weeks old) as described in our previous studies (Singh, 2007), (Zhou, 2010), (Guan, 2011). Briefly, 100μL of 150 μg myelin oligodendrocyte glycoprotein (MOG35-55) peptide emulsified in complete Freund’s adjuvant (Difco, Detriot, MI) containing 4 mg/mL killed Mycobacterium tuberculosis (strain H37Ra; Difco) was injected subcutaneously. Following immunization, 200 ng of pertussis toxin (List Labs) was injected i.p. into mice on Day 0, followed by a 400 ng pertussis toxin i.p. injection on Day 2. IL-2 was administered either before immunization of mice with MOG (called pre-treatment) or after immunization (called post-treatment). In pre-treatment studies, mice were injected i.p. with 10,000 U of rIL-2 or PBS as a control, three times a day for three consecutive days followed by one additional injection of rIL-2 on day 4. On day 4, mice were immunized with MOG35-55 as described above. This day was considered day 0 for clinical evaluations and depiction of data. For studies involving post-treatment with IL-2, EAE was induced in mice on day 0 by immunization using MOG35-55 as described above. Next, on day 7 or day 11, mice were injected i.p. with 10,000 U of rIL-2 or PBS as a control, three times a day for three consecutive days. Clinical scores (0, no symptoms; 1, limp tail; 2, partial paralysis of hind limbs; 3, complete paralysis of hind limbs or partial hind and front limb paralysis; 4, tetraparalysis; 5, moribund; 6, death) were recorded on a daily basis. The mean score was calculated for each group every day.
Histological Analysis of Cell Infiltration
Spinal cords from PBS control or rIL-2 treated EAE mice were collected 15 days after immunization. The spinal cord sections were fixed with 10% formalin and paraffin blocks were prepared. Microtome sections were generated and tissue sections were stained using either hematoxylin & eosin (H&E). The sections were examined for inflammatory cell infiltrates under a microscope.
Cell Culture
Cell cultures were maintained in complete RPMI 1640 media supplemented with 10% heat inactivated fetal bovine serum, β-mercaptoethanol, 10 mM L-glutamine, 10mM HEPES, and 100 μg/mL penicillin/streptomycin at 37°C and 5% CO2.
Effects of IL-2 on Treg induction
Splenocytes and inguinal lymph node cells were isolated on Day 15 from naïve or EAE mice given vehicle (PBS) or rIL-2. Cells were harvested and prepared for the examination of cellular markers via flow cytometric analysis.
MOG35-55 Restimulation Assay
Splenocytes from normal or EAE mice treated with vehicle (PBS) or rIL-2 were isolated 15 days after immunization and cultured in a 24-well plate in the presence of 30 μg/mL MOG35-55 for three days. Cells were stimulated with PMA and calcium ionophore for four hours prior to harvesting, and then prepared for the examination of cellular markers via flow cytometric analysis.
Effects of IL-2 in Primary T-cells
Primary T-cells from naïve C57BL/6 mice were isolated and cultured in the absence or presence of ConA (2.5 μg/mL) for 24 hours. Cultures were washed twice with complete RPMI and treated with either vehicle (PBS) or rIL-2 (60 units/mL) for an additional 24 hours. Cells were harvested and prepared for the examination of cellular markers using flow cytometric analysis.
Statistics
For EAE clinical studies, we used groups of 10 mice. Clinical scores were evaluated using the Mann-Whitney test. All in vitro assays were performed in triplicate. The means ± standard errors of the means (SEM) are shown for experiments that are applicable. The statistical difference in such experiments was calculated with Student’s t- test where applicable. A p value of ≤0.05 based on ANoVA was considered to be statistically significant. The data that are shown in this paper represent at least three independent experiments.
Results
Pre-treatment with rIL-2 suppresses the onset of EAE
Tregs have been well characterized to suppress the inflammatory and autoimmune response during EAE (Aharoni, 2012), (Fransson, 2012). Moreover, previous studies from our laboratory have shown that administration of IL-2 into mice can trigger CD4+CD25+ Tregs with immunosuppressive properties (Melencio, 2006). In the current study, therefore, we tested the hypothesis that IL-2 treatment may trigger Tregs and thereby ameliorate EAE. To this end, naïve C57BL/6 female mice were administered vehicle or 10,000 U rIL-2 three times a day for three consecutive days before immunizing them with MOG to induce EAE. Clinical scores from these experiments illustrated that pre-treatment with rIL-2 was effective at not only delaying the onset of disease but also diminishing the severity of clinical symptoms (Fig 1A). Histopathology of the spinal cord confirmed that vehicle-treated EAE mice had increased cellular infiltration and tissue damage within myelinated regions when compared to naive mice (Fig 1B). Also, mice pre-treated with rIL-2 exhibited minimal traces of cell infiltration and maintained normal tissue architecture of the spinal cord.
Figure 1. Pre-treatment with rIL-2 leads to the amelioration of EAE.

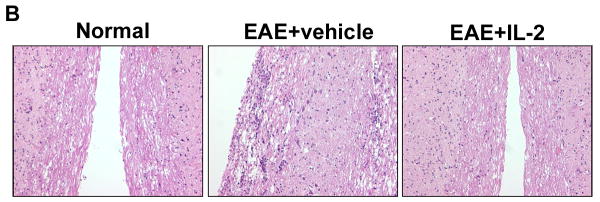
C57BL/6 mice were immunized with MOG35-55 peptide emulsified in complete Freund’s adjuvant along with 200 ng of pertussis toxin (day 0) and 400 ng pertussis toxin (Day 2). Mice pre-treated with rIL-2 received an i.p. injection with 10,000 U of rIL-2 three times a day for three days and once on day 4 prior to EAE induction (day 0 of immunization). (A) Clinical scores were recorded daily after EAE induction. (B) H&E spinal cord histopathology collected 15 days after immunization.
Effect of pre-treatment with rIL-2 on Tregs in EAE mice
After witnessing the protective effects of pre-treatment with rIL-2, we wanted to investigate its effects on immune cells in EAE mice at the peak of the disease. When we took a closer look at secondary lymphoid organs, such as spleen and inguinal lymph nodes, we observed no distinct differences in total cell number between vehicle and IL-2 pre-treated EAE mice (data not shown). However, analysis of CD4 and FoxP3 markers revealed that IL-2 pre-treated EAE mice showed a significant increase in the frequency of CD4+/FoxP3+ Tregs in both the spleens and LNs (Fig 2). When we calculated the absolute numbers of the Tregs based on the total cellularity of the spleen and LN, and the percentage of Tregs, we found no difference between vehicle or IL-2 treated EAE mice (data not shown). When splenocytes from these mice were restimulated in culture with MOG35-55, cells from vehicle and rIL-2 treated EAE mice displayed similar levels of Tregs, although both were significantly greater than naïve mice (Fig 3). However, when we examined CD4+/IL-17+ Th17 cells after restimulation, vehicle-treated EAE mice possessed a higher frequency than naïve mice. rIL-2 treated mice, on the other hand, showed decreased Th17 percentages back down to naïve levels. These data suggested that upon restimulation, while IL-2 treatment did not have a significant impact on Treg generation, it caused decreased differentiation of MOG-specific Th17 cells.
FIGURE 2. rIL-2 increases Tregs in the spleen and inguinal lymph nodes of EAE mice.
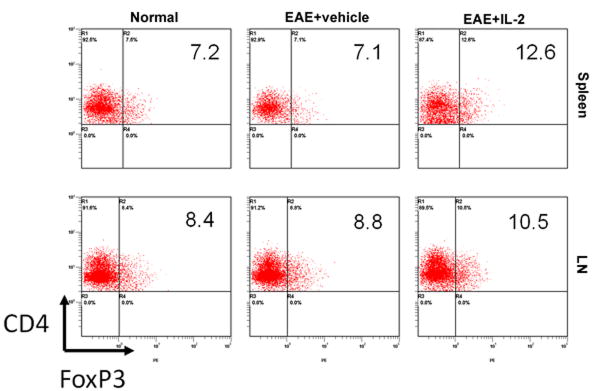
Spleen and inguinal lymph node (LN) cells were isolated on Day 15 from naïve and EAE mice pre-treated with vehicle or rIL-2. Cells were processed and stained for CD4+/FoxP3+ regulatory T-cells.
FIGURE 3. rIL-2 pre-treatment suppresses antigen specific Th17 cell expansion after MOG35-55 restimulation.

Splenocytes were isolated on Day 15 from naïve and EAE mice pre-treated with vehicle or rIL-2. Cells were cultured in the presence of MOG35-55 for three days. Flow cytometric analysis was used to identify (A) Tregs and (B) Th17 cells.
Post-treatment with rIL-2 exacerbates EAE progression
Based on the potential therapeutic effects observed with pre-treatment of rIL-2, we wanted to know if similar results could be achieved if IL-2 treatment was provided after the induction of EAE. To that end, two sets of rIL-2 regimens were given to mice after EAE induction, one starting at Day 7 and the other at Day 11, post-immunization with MOG. In both post-treatment regimens, vehicle or 10,000 U rIL-2 was given three times a day for three consecutive days. EAE mice that received rIL-2 therapy at Day 7 initially displayed similar disease progression as vehicle-treated EAE mice. However, over the course of the study, these mice demonstrated a greater increase in clinical scores (Fig 4A). EAE mice given rIL-2 treatment at Day 11 also demonstrated enhanced progression and severity of disease as time passed. H&E staining of spinal cord tissue taken at the peak of the disease was found to correlate very closely to clinical scores (Fig 4B). While vehicle-treated EAE mice did present with increased cellular infiltration and tissue damage compared to naive mice, EAE mice post-treated with rIL-2 exhibited far greater tissue destruction due to increased immune cell infiltration.
Figure 4. Post-treatment with rIL-2 exacerbates development of EAE mice.

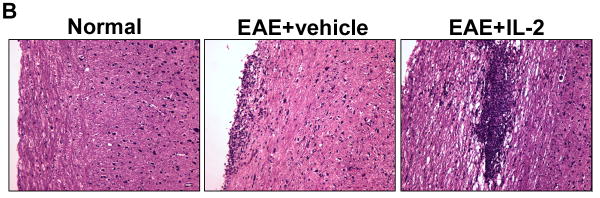
(A) Clinical scores of EAE mice post-treated with rIL-2 starting at Day 7 or Day 11 post-immunization. (B) Spinal cord H&E histopathology taken at day 15 of naïve or EAE mice post-treated with vehicle or rIL-2 (Day 7).
When we examined the secondary lymphoid organs, once again, we observed no notable differences in total cell number between vehicle or rIL-2 treated EAE mice (data not shown). Despite similar clinical scores and histopathology, EAE mice post-treated with rIL-2 were still able to show in most instances (except in the spleen with day 7 regimen), slightly higher frequencies of Tregs in both spleen and inguinal lymph nodes compared to vehicle (Fig 5). Even when the splenocytes from these mice were restimulated with MOG35-55, cells taken from rIL-2 treated mice, either from the Day 7 or Day 11 regimen, expressed greater proportions of Tregs than vehicle treated mice (Fig 6). In addition, restimulated cells from rIL-2 post-treated mice also illustrated reduced frequency of Th17 cells. These data together suggested that rIL-2 post-treatment does trigger Tregs and decreases the induction of Th17 cells although such treatment is not effective in ameliorating EAE.
Figure 5. Post-treatment with rIL-2 modulates Treg induction in EAE mice.
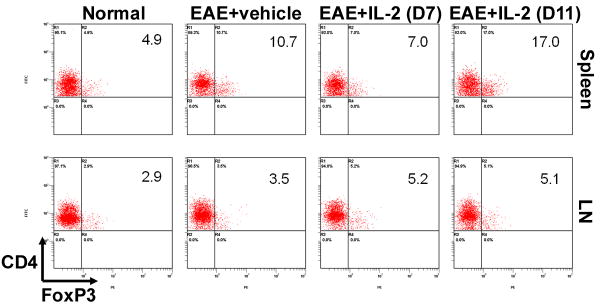
Spleen and inguinal lymph node cells were isolated on Day 15 from naïve and EAE mice post-treated with vehicle or rIL-2. Cells were processed and stained for CD4+/FoxP3+ regulatory T-cells.
Figure 6. rIL-2 post-treatment promotes antigen specific Tregs, while suppressing Th17 cells after MOG35-55 restimulation.
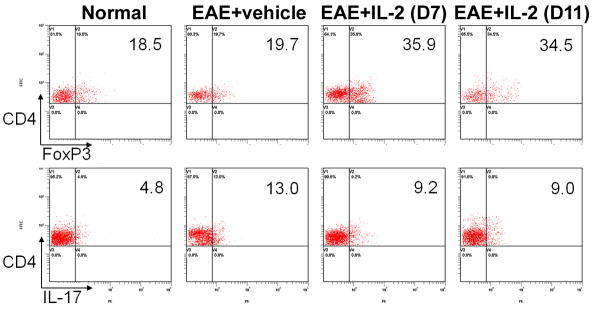
Splenocytes were isolated on Day 15 from naïve and EAE mice post-treated with vehicle or rIL-2. Cells were cultured in the presence of MOG35-55 for three days. Flow cytometric analysis was used to study Tregs and Th17 cells.
Effects of rIL-2 on naïve and ConA-activated T-cells in vitro
Due to the contrasting results in EAE progression between pre- and post-treatment with rIL-2, we wanted to determine how rIL-2 influenced T-cell differentiation in naïve and activated conditions. Moreover, because MOG-specific T cell frequency is low, we used ConA to polyclonally activate T cells and then tested the effect of IL-2 in the culture on the differentiation of Tregs and Th17cells. To this end, primary T-cells were isolated from naïve mice and cultured as naïve or activated with ConA for 24 hrs. Afterwards, cells were washed and exposed to vehicle or rIL-2 in culture for an additional 24 hrs. We found that rIL-2 treatment in naïve conditions increased Tregs frequency 3-fold compared to vehicle (Fig 7A). Vehicle-treated T-cells after ConA-activation also illustrated a considerable increase in Tregs compared to vehicle-treated naïve T-cells. However, when rIL-2 was added to ConA-activated T-cells, the frequency of Tregs was enhanced even further. Similar results were also noted when we measured the total number of cells generated (Fig 7B). These data demonstrated that IL-2 can not only promote the growth of naïve Tregs but upon T cell activation, IL-2 can cause a more robust increase in Tregs.
Figure 7. rIL-2 causes expansion of Tregs in vitro.


Naïve primary T-cells were cultured in the absence or presence of ConA for 24 hrs and treated with rIL-2 (60 U/mL). Cell were stained for (A) CD4+/FoxP3+ Tregs and (B) total cell number was calculated. Statistical significance in the data between the different groups has been indicated. (* p < 0.001, + p < 0.01)
In comparison, we also wanted to determine how rIL-2 could affect Th17 differentiation in culture. Results demonstrated both vehicle and rIL-2 treatment displayed low percentages and numbers of Th17 cells under naïve conditions (Fig 8A, B). Activation with ConA, on the other hand, led to significant increase in Th17 frequency and number, which was further increased with r-IL-2. (Fig 8A, B). Together, these data suggested that addition of IL-2 to polyclonally activated T cells leads to a dramatic increase in both Tregs and Th17cells.
Figure 8. rIL-2 triggers the expansion of Th17 cells in activated, but not naïve conditions.


Naïve primary T-cells were cultured in the absence or presence of ConA for 24 hrs and treated with rIL-2 (60 U/mL). Cells were stained for (A) CD4+/IL-17+ Th17 and (B) total cell number was calculated. Statistical significance in the data between the different groups has been indicated. (* p < 0.005)
Discussion
IL-2 is a well-known cytokine responsible for being a vital member of the immune system and a strong promoter of T-cell expansion. When T-cells bind antigen, there is a substantial increase in both IL-2 production as well as expression of the IL-2 receptor. As a result, this autocrine positive feedback loop helps drive the growth and differentiation of antigen specific T-cells (Stern, 1986), (Beadling, 1993), (Beadling, 2002). Furthermore, IL-2 has been shown to be necessary for development of memory T-cells after exposure to antigen. In contrast to most T cells that express IL-2R upon activation, naïve Tregs constitutively express IL-2R and therefore can be expanded by IL-2. Previous studies from our laboratory demonstrated that IL-2 administration in vivo led to significant expansion of Tregs with immunosuppressive functions (Melencio, 2006). Such studies helped us further address the role of IL-2 driven Tregs in the prevention and treatment of EAE. Overall, data from the current study indicated that while IL-2 pretreatment prior to the activation of MOG-specific T cells was effective in preventing the development of EAE, IL-2 post-treatment, after MOG immunization, failed to ameliorate EAE.
Numerous studies have demonstrated the importance of IL-2 and Tregs in eliciting immune response and preventing autoimmunity (Sakaguchi, 1995). Also, Tregs and Th17 cells have been shown to be reciprocally regulated (Gallagher, 2007). Therefore, we wanted to investigate the effects of using IL-2 therapy in regulating the balance between Tregs and Th17 cells in the autoimmune disease model, EAE. Previous studies from our laboratory have shown that administration of high dose of IL-2 (75,000 U/mouse, three times a day for 3 days), as used in cancer therapy, into BL/6 wild-type mice triggered significant vascular leak syndrome (VLS) in the lungs, liver, and spleen (Guan, 2012), (Hao, 2011), a toxic effect also seen in humans (Gallagher, 2007). It is for this reason, we performed pilot studies and lowered the dose of rIL-2 to 10,000 U/mouse 3 times a day for 3 days, which does not cause VLS. In this study, we were able to demonstrate that this moderate treatment regimen of IL-2 was effective in delaying the onset and reducing the overall severity of EAE when administered before disease induction. Histopathology of the spinal confirmed the effectiveness of rIL-2 pre-treatment in which cellular infiltration and subsequent tissue damage was blocked compare to vehicle-treated EAE mice. Also, the results suggested the protective effects of pre-treatment with rIL-2 were due to an increase in Treg frequency within second lymphoid organs. Further analysis demonstrated rIL-2 pre-treatment was also able to inhibit the differentiation of MOG-specific Th17 cells.
Our studies raise the interesting question on why IL-pre-treatment but not post-treatment is effective in the amelioration of EAE. Several possibilities exist: First, naïve Tregs express IL-2R while effector Th1 or Th17 cells, involved in EAE, express IL-2R only after activation with MOG (Cerdan, 1992). Thus, pretreatment with IL-2 may expand only Tregs but not antigen- specific Th17 cells as noted in our study. Moreover, such Tegs may then inhibit the differentiation of Th17 cells upon MOG immunization. Secondly, IL-2 administration post-treatment may not only activate Tregs but also effector Th17 cells that begin to express IL-2R, which may account for the slight exacerbation of EAE as seen in this study. However, we wish to point out that EAE mice post-treated with rIL-2 were still able to show higher frequencies of Tregs in both spleen and inguinal lymph nodes. Moreover, when the splenocytes from these mice were restimulated with MOG35-55, there was increased generation of Tregs and reduced frequency of Th17 cells. These data together suggested that rIL-2 post-treatment does trigger Tregs and decreases the induction of Th17 cells although such treatment is not effective in ameliorating EAE. The third possibility as to why IL-2 post-treatment is not effective may be because Tregs have been shown to suppress the generation of effector cells prior to their generation and that they are less effective in suppressing effector T cells that are already induced (Sojka, 2008). It should be noted that because the in vivo generation of MOG-specific Th17 cells is difficult to demonstrate due to low frequency, we also used ConA to polyclonally expand T cells and found that the expansion of Th17 cells in the presence of IL-2 was markedly increased. Thus, additional studies are necessary to address if the failure of post-treatment with IL-2 is due to increased expansion of Th17 cells or the inability of Tregs to suppress such Tregs or a combination of both.
Previous studies have also demonstrated IL-2 can induce the activation and expansion of cytotoxic T-cells and natural killer (NK) cells during an immune response (Morisaki, 2012), (Balsamo, 2012). Subsequently, the expansion of these cells leads to the destruction and elimination of virally infected cells and cancer cells (Andrus, 1984), (Malek, 2002). As a result, IL-2 began being investigated as a potential therapy against various forms of cancer. Based on its effectiveness in clinical tries, high dose IL-2 was approved by the Food and Drug Administration (FDA) in the 1990s as a mode of treatment for metastatic kidney cancer and melanoma (Rosenburg, 2007). As a result, high dose IL-2 is still commonly used to treat these cancers and is capable of reversing the disease in 20-30% of cases (Baluna, 1997). Such a treatment modality is known to work by activation of cytotoxic lymphocytes such as NK, LAK or CD8+ T cells leading to killing of virally infected or cancer cells. If IL-2 treatment can also trigger Tregs, which can potentially suppress anti-tumor immunity, how can the success of IL-2 therapy against cancer be explained? This can be explained from our current study that in cancer patients, the effector T cells or NK cells may already have been activated and therefore they may be less susceptible to the suppressive activity of Tregs. Also, IL-2 treatment clearly increases the NK cells and potentially activated tumor-antigen specific T cells that express IL-2R thereby promoting anti-cancer activity. Because Tregs also have the therapeutic potential against transplant rejection, our studies suggest that IL-2pretreatment to expand Tregs prior to transplantation may constitute another therapeutic area with potential benefits. Together, our studies open new avenues for the clinical application of Treg expansion using IL-2.
Acknowledgments
Funding: The research was funded in part by NIH grants R01AT006888, P01AT003961, R01ES019313, R01MH094755 and VA Merit Award 1I01BX001357. The funding agency had no role in experimental design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- MS
multiple sclerosis
- EAE
experimental autoimmune encephalomyelitis
- Tregs
regulatory T-cells
- MOG
myelin oligodendrocyte glycoprotein
- rIL-2
recombinant interleukin-2
- IL-2R
interleukin-2 receptor
Footnotes
Competing Interests: The authors declare no competing interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aharoni R, Saada R, Eilam R, Hayardeny L, Sela M, Arnon R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012 doi: 10.1016/j.jneuroim.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Alexander JS, Harris MK, Wells SR, Mills G, Chalamidas K, Ganta VC, McGee J, Jennings MH, Gonzalez-Toledo E, Minagar A. Alterations in serum MMP-8, MMP-9, IL-12p40 and IL-23 in multiple sclerosis patients treated with interferon-beta1b. Mult Scler. 2010;16:801–809. doi: 10.1177/1352458510370791. [DOI] [PubMed] [Google Scholar]
- Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- Andrus L, Granelli-Piperno A, Reich E. Cytotoxic T cells both produce and respond to interleukin 2. J Exp Med. 1984;159(2):647–652. doi: 10.1084/jem.159.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsamo M, Vermi W, Parodi M, Pietra G, Manzini C, Queirolo P, Lonardi S, Augugliaro R, Moretta A, Facchetti F, Moretta L, Mingari MC, Vitale M. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur J Immunol. 2012 doi: 10.1002/eji.201142179. Ahead of Print. [DOI] [PubMed] [Google Scholar]
- Baluna R, Vitetta ES. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology. 1997;37:117–132. doi: 10.1016/s0162-3109(97)00041-6. [DOI] [PubMed] [Google Scholar]
- Beadling C, Johnson KW, Smith KA. Isolation of interleukin 2-induced immediate-early genes. Proc Nat Acad Sci USA. 1993;90(7):2719–2723. doi: 10.1073/pnas.90.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadling CB, Smith KA. DNA array analysis of interleukin-2-regulated immediate/early genes. Med Immunol. 2002;1(1):2. doi: 10.1186/1476-9433-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerg L, Brosbøl-Ravnborg A, Tørring C, Dige A, Bundgaard B, Petersen T, Höllsberg P. Altered frequency of T regulatory cells is associated with disability status in relapsing-remitting multiple sclerosis patients. J Neuroimmunol. 2012;249(1-2):76–82. doi: 10.1016/j.jneuroim.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Cerdan C, Martin Y, Courcoul M, Brailly H, Mawas C, Birg F, Olive D. Prolonged IL-2 Receptor α/CD25 expression after T-cell activation via the adhesion molecules CD2 and CD28. Demonstration of combined transcriptional and post-transcriptional regulation. J Immunol. 1992;149(7):2255–2261. [PubMed] [Google Scholar]
- Chen M, Chen G, Nie H, Zhang X, Niu X, Zang YC, Skinner SM, Zhang JZ, Killian JM, Hong J. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T Cells in MS. Eur J Immunol. 2009;39:2525–2536. doi: 10.1002/eji.200838879. [DOI] [PubMed] [Google Scholar]
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, Hunter CA, Kastelein RA, Cua DJ. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359(9313):1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, Loskog AS. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9(1):112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DC, Bhatt RS, Parikh SM, Patel P, Seery V, McDermott DF, Atkins MB, Sukhatme VP. Angiopoietin 2 is a potential mediator of high-dose interleukin 2-induced vascular leak. Clin Cancer Res. 2007;13(7):2115–2120. doi: 10.1158/1078-0432.CCR-06-2509. [DOI] [PubMed] [Google Scholar]
- Gocke AR, Cravens PD, Ben L, Hussain RZ, Northrop SC, Racke MK, Lovett-Racke AE. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–1348. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- Guan H, Nagarkatti PS, Nagarkatti M. CD44 Reciprocally regulates the differentiation of encephalitogenic Th1/Th17 and Th2/regulatory T cells through epigenetic modulation involving DNA methylation of cytokine gene promoters, thereby controlling the development of experimental autoimmune encephalomyelitis. J Immunol. 2011;186(12):6955–6964. doi: 10.4049/jimmunol.1004043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Singh NP, Singh UP, Nagarkatti PS, Nagarkatti M. Resveratrol Prevents Endothelial Cells Injury in High-Dose Interleukin-2 Therapy against Melanoma. PLoS One. 2012;7(4):e35650. doi: 10.1371/journal.pone.0035650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Campagnolo D, Liu R, Piao W, Shi S, Hu B, Xiang R, Zhou Q, Vollmer T, Van Kaer L, La Cava A, Shi FD. Interleukin-2/interleukin-2 antibody therapy induces target organ natural killer cells that inhibit central nervous system inflammation. Ann Neurol. 2011;69(4):721–734. doi: 10.1002/ana.22339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Han Y, Rani MR, Glabinski A, Trebst C, Sørensen T, Tani M, Wang J, Chien P, O’Bryan S, Bielecki B, Zhou ZL, Majumder S, Ransohoff RM. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol Rev. 2000;177:52–67. doi: 10.1034/j.1600-065x.2000.17709.x. [DOI] [PubMed] [Google Scholar]
- Jeon EJ, Yoon BY, Lim JY, Oh HJ, Park HS, Park MJ, Lim MA, Park MK, Kim KW, Cho ML, Cho SG. Adoptive transfer of all-trans-retinal-induced regulatory T cells ameliorates experimental autoimmune arthritis in an interferon-gamma knockout model. Autoimmunity. 2012 doi: 10.3109/08916934.2012.682666. Ahead of Print. [DOI] [PubMed] [Google Scholar]
- Kevin J, Powrie M, Powrie F. Fueling regulation: IL-2 keeps CD4+ Treg cells fit. Nat Immunol. 2005;6:1071–1072. doi: 10.1038/ni1105-1071. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein M, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Racke AE, Rocchini AE, Choy J, Northrop SC, Hussain RZ, Ratts RB, Sikder D, Racke MK. Silencing T-bet defines critical role in the differentiation of autoreactive T lymphocytes. Immunity. 2004;21:719–731. doi: 10.1016/j.immuni.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Malek TR. T helper cells, IL-2 and the generation of cytotoxic T-cell responses. Trends Immunol. 2002;23(10):465–467. doi: 10.1016/s1471-4906(02)02308-6. [DOI] [PubMed] [Google Scholar]
- McDonald AH, Swanborg RH. Antigen-specific inhibition of immune interferon production by suppressor cells of autoimmune encephalomyelitis. J Immunol. 1988;140:1132–1138. [PubMed] [Google Scholar]
- Melencio L, McKallip RJ, Guan H, Ramakrishnan R, Jain R, Nagarkatti PS, Nagarkatti M. Role of CD4(+)CD25(+) T regulatory cells in IL-2-induced vascular leak. Int Immunol. 2006;18(10):1461–1471. doi: 10.1093/intimm/dxl079. [DOI] [PubMed] [Google Scholar]
- Morisaki T, Umebayashi M, Kiyota A, Koya N, Tanaka H, Onishi H, Katano M. Combining cetuximab with killer lymphocytes synergistically inhibits human cholangiocarcinoma cells in vitro. Anticancer Res. 2012;32(6):2249–2256. [PubMed] [Google Scholar]
- Nath N, Prasad R, Giri S, Singh AK, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology. 2006;118:384–391. doi: 10.1111/j.1365-2567.2006.02385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt1+2-T lymphocytes. J Immunol. 1981;127:1420–1423. [PubMed] [Google Scholar]
- Rosati G. The prevalence of multiple sclerosis in the world: an update. Neurol Sci. 2011;22(2):117–139. doi: 10.1007/s100720170011. [DOI] [PubMed] [Google Scholar]
- Rosenburg SA. Interleukin 2 for patients with renal cancer. Nat Clin Pract Oncol. 2007;4:497. doi: 10.1038/ncponc0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–1164. [PubMed] [Google Scholar]
- Singh NP, Hegde VL, Hofseth LJ, Nagarkatti M, Nagarkatti P. Resveratrol (trans-3,5,4’-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol Pharmacol. 2007;72(6):1508–1521. doi: 10.1124/mol.107.038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojka DK, Huang YH, Fowell FJ. Mechanisms of regulatory T-cell suppression – a diverse arsenal for a moving target. Immunology. 2008;124(1):13–22. doi: 10.1111/j.1365-2567.2008.02813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern J, Smith KA. Interleukin-2 induction of T-cell G1 progression and c-myb expression. Science. 1986;233(4760):203–206. doi: 10.1126/science.3523754. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172(11):6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188(2):287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran GT, Hodgkinson SJ, Carter NM, Verma ND, Plain KM, Boyd R, Robinson CM, Nomura M, Killingsworth M, Hall BM. IL-5 promotes induction of antigen-specific CD4+CD25+ T regulatory cells that suppress autoimmunity. Blood. 2012;119(19):4441–4450. doi: 10.1182/blood-2011-12-396101. [DOI] [PubMed] [Google Scholar]
- Veenstra RG, Taylor PA, Zhou Q, Panoskaltsis-Mortari A, Hirashima M, Flynn R, Liu D, Anderson AC, Strom TB, Kuchroo VK, Blazar BR. Contrasting acute graft-versus-host-disease effects of Tim-3/galectin-9 pathway blockade dependent upon the presence of donor regulatory T cells. Blood. 2012 doi: 10.1182/blood-2011-10-387977. Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldburger KE, Hastings RC, Schaub RG, Goldman SL, Leonard JP. Adoptive transfer of experimental allergic encephalomyelitis after in vitro treatment with recombinant murine interleukin-12. Preferential expansion of interferon-gamma-producing cells and increased expression of macrophage-associated inducible nitric oxide synthase as immunomodulatory mechanisms. Am J Pathol. 1996;148:375–382. [PMC free article] [PubMed] [Google Scholar]
- Yura M, Takahashi I, Serada M, Koshio T, Nakagami K, Yuki Y, Kiyono H. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE) J Autoimmun. 2001;17:17–25. doi: 10.1006/jaut.2001.0520. [DOI] [PubMed] [Google Scholar]
- Zhou J, Nagarkatti P, Zhong Y, Nagarkatti M. Immune modulation by chondroitin sulfate and its degraded disaccharide product in the development of an experimental model of multiple sclerosis. J Neuroimmunol. 2010;223(1-2):55–64. doi: 10.1016/j.jneuroim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]