

Abstract
In invasive pulmonary aspergillosis, direct invasion and occlusion of pulmonary vasculature by Aspergillus hyphae causes tissue hypoxia, which is enhanced by secreted fungal metabolites that downregulate compensatory angiogenic signaling pathways. We assessed the effects of basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) on survival rates, fungal burden, and in situ angiogenesis in a murine invasive pulmonary aspergillosis model. bFGF and VEGF monotherapy significantly increased survival rates and potentiated the activity of amphotericin B. bFGF-containing regimens were associated with reduced tissue fungal burdens. bFGF and VEGF reversed the antiangiogenic activity of Aspergillus fumigatus; however, VEGF induced the formation of immature neovessels, providing an explanation for its lesser efficacy. Treatment with bFGF plus amphotericin B was associated with neutrophil influx into Aspergillus-infected pulmonary tissue, suggesting that this combination limits fungal growth through neutrophil trafficking. Vasculogenic pathways are unexplored targets for the treatment of invasive pulmonary aspergillosis and may potentiate both innate immunity and antifungal drug activity against A. fumigatus.
Keywords: angiogenesis inducing agents, vascular endothelial growth factor A, fibroblast growth factor 2, invasive pulmonary aspergillosis, animal model
(See the editorial commentary by Ito on pages 1031–3.)
Invasive aspergillosis is the most common opportunistic fungal infection in immunosuppressed patients with cancer. Despite the availability of new antifungal agents active against Aspergillus species in vitro and improved supportive care, mortality rates from invasive aspergillosis remain high [1–3]. Given the propensity of Aspergillus species to invade and occlude pulmonary vasculature, we and others hypothesized that vasculopathy may be a key contributor to treatment failure in invasive aspergillosis [4–6].
Angioinvasion, vascular thrombosis, and tissue infarction have long been recognized as central histopathological features of invasive pulmonary aspergillosis [7–9]. Moreover, tissue hypoxia has recently been demonstrated in Aspergillus-infected lung tissue of both neutropenic and nonneutropenic immunosuppressed mice [10]. Thus, both Aspergillus and host cells must adapt to an oxygen-limited microenvironment in the setting of invasive pulmonary aspergillosis. Indeed, an Aspergillus mutant deficient in a sterol regulatory element–binding protein required for hypoxia adaptation (ΔSrbA) was virtually avirulent in immunosuppressed mice [11]. For the host, angiogenesis represents a critical adaptive response to tissue hypoxia. We have previously shown that angiogenesis is suppressed, both in vitro and in vivo, by Aspergillus fumigatus secondary metabolites, specifically by the epipolythiodioxopiperazine gliotoxin [5]. Downregulation of host genes encoding for important mediators of angiogenesis, such as basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), and their respective receptors, occurred within 24 hours of infection in cyclophosphamide/cortisone-treated (neutropenic) mice but not corticosteroid-suppressed (nonneutropenic) mice [5]. Therefore, we hypothesized that, in the neutropenic host, the homeostatic angiogenic response is attenuated by A. fumigatus, leading to worsening tissue hypoxia and sequestration of infected tissue. Suppression of angiogenesis may represent a barrier to effective treatment of invasive pulmonary aspergillosis, as it limits the migration of immune effector cells and penetration of antifungal drugs into the site of infection. These findings suggested that repletion of proangiogenic growth factors might potentiate both innate antifungal defenses and the activity of antifungal drugs in vivo.
In the present study, we aimed to determine whether bFGF and VEGF, alone or in combination with amphotericin B, would reverse angiogenesis inhibition at the site of A. fumigatus infection and improve survival rates in an experimental model of invasive pulmonary aspergillosis.
MATERIALS AND METHODS
Aspergillus Culture
A. fumigatus strain Af293 was grown on yeast extract agar glucose (YAG; yeast extract, 5 g/L; glucose, 10 g/L; agar, 15 g/L; MgCl2 1 M, 10 mL/L; and trace elements, 1 mL/L) at 37°C for 48–72 hours. Conidia were collected in sterile saline, washed 3 times, and enumerated in a hemacytometer. Conidial suspensions were prepared at 2.5 × 108 cells/mL for intranasal inoculation (pulmonary model [5]) and at 5 × 107 cells/mL for subcutaneous inoculation (cutaneous model [12]). The inoculum suspensions were serially diluted in sterile saline and inoculated on YAG plates to verify <10% variation in viable conidia concentrations.
Drug Preparation
Amphotericin B deoxycholate (X-Gen Pharmaceuticals, Big Flats, NY) was prepared in sterile water at 5 mg/mL and further diluted 1:50 in 5% dextrose in water. Recombinant human VEGF, proximal splice site 165 (rh-VEGF165; R&D Systems, Minneapolis, MN), was prepared in sterile phosphate-buffered saline (PBS) with 0.1% bovine serum albumin at 10 µg/mL. Recombinant human bFGF (rh-bFGF; R&D Systems) was prepared in sterile PBS at 25 µg/mL.
Drug Susceptibility Testing
We performed susceptibility testing of A. fumigatus strain Af293 to VEGF, bFGF, and the combination bFGF/VEGF by broth microdilution [13] at a range of relevant concentrations (0.015–4 µg/mL). We found no direct effect of bFGF, VEGF, or the combination on Af293 growth at these concentrations (data not shown).
Mouse Model of Invasive Pulmonary Aspergillosis
Six-week-old female BALB/c mice weighing 18–20 g (National Cancer Institute, Bethesda, MD) were used for the pulmonary aspergillosis model. All procedures were performed according to the highest standards for humane handling, care, and treatment of research animals and were approved by the M. D. Anderson Cancer Center Institutional Animal Care and Use Committee. Mice were kept in filter-topped cages in groups of 5 and fed autoclaved food and water. Immunosuppression was achieved by injection of cyclophosphamide monohydrate (100 mg/kg) intraperitoneally 4 days and 1 day before inoculation and injection of cortisone acetate (250 mg/kg) subcutaneously 1 day before inoculation. On inoculation day, 35 µL of A. fumigatus conidial suspension containing 8.75 × 106 freshly collected conidia were instilled intranasally under 2.5% isoflurane anesthesia. Additional doses of cyclophosphamide (100 mg/kg intraperitoneally) were given on days 2 and 5 after inoculation to maintain neutropenia. It should be noted that, because mice received a dose of cortisone acetate in addition to cyclophosphamide, the invasive pulmonary aspergillosis model described here is not a pure neutropenic model. We selected this protocol because we previously found it to be associated with rapid downmodulation of VEGF and bFGF gene expression in mouse lungs following infection with A. fumigatus [5]. Mortality was assessed daily until day 7 after inoculation. Animals displaying signs of morbidity were euthanized, and their death was recorded as occurring 12 hours later.
Treatment Regimens
We evaluated treatment with rh-bFGF, rh-VEGF165, and amphotericin B, individually and in combinations, for their effects on mortality, tissue fungal burden, and in situ angiogenesis in the murine invasive pulmonary aspergillosis model. Mice were allocated into the following 8 treatment arms: (1) amphotericin B, 1 mg/kg daily intraperitoneally for 5 days starting 3 hours after inoculation (AmB arm); (2) rh-VEGF165, 20 ng intravenous injection 3 hours after inoculation (VEGF arm); (3) rh-bFGF, 2 doses of 1.6 µg intravenously 3 hours and 5 hours after inoculation (bFGF arm); (4) amphotericin B plus rh-VEGF165 (doses as described above; AmB/VEGF arm); (5) amphotericin B plus rh-bFGF (AmB/bFGF arm); (6) rh-VEGF165 plus rh-bFGF (VEGF/bFGF arm); (7) amphotericin B plus rh-VEGF165 and rh-bFGF (AmB/VEGF/bFGF arm); and (8) mock treatment (intravenous saline injection). The rh-VEGF165 dose was based on a study showing that 20 ng rh-VEGF165 was sufficient to activate the phosphoinositol 3′-kinase/Akt signaling pathway in the lungs of mice, whereas a 10-fold higher dose was associated with toxicity due to interstitial edema [14]. The bFGF regimen was previously shown to protect mouse gut endothelial cells from radiation damage [15]. Mice were evaluated in each treatment arm in groups of 10, and experiments were repeated 3 times for a total of 30 mice per treatment arm.
Pulmonary Fungal Burden
Pulmonary fungal burden was determined in a subset of mice euthanized 72 hours after inoculation, as described previously [16, 17]. In brief, DNA was extracted from whole lung homogenates using the DNeasy blood and tissue kit (Qiagen, Valencia, CA). DNA samples were assayed in duplicate using an ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA). For each sample, real-time quantitative polymerase chain reaction (qPCR) was performed in duplicate, using primers and a dually labeled fluorescent hybridization probe complementary to a sequence from the A. fumigatus 18S ribosomal RNA gene (GenBank accession no. AB008401). The cycle threshold of each sample was interpolated from a 7-point standard curve prepared by spiking uninfected lungs with known amounts of A. fumigatus 293 conidia (101–107).
Histopathological and Immunohistochemical Analyses
Mouse lungs were excised 72 hours after infection, fixed in 10% formalin, embedded in paraffin wax, and sectioned. Specimens were stained with hematoxylin and eosin and with Gomori methenamine silver for semiquantitative assessment of fungal burden.
For CD31 immunostaining, slides were rehydrated to water, and protein was digested with proteinase K for 10 minutes. Next, the slides were incubated with rat anti-mouse CD31 antibody (1:50; BD Pharmingen, San Jose, CA) for 60 minutes at room temperature, followed by incubation with anti-rat biotinylated secondary antibody for 30 minutes. Detection was performed by incubation with streptavidin-horseradish peroxidase solution for 30 minutes, followed by 3,3′-diaminobenzidine chromogen substrate for 5 minutes. Slides were counterstained with Mayer's hematoxylin, dehydrated, and covered by a coverslip.
In Vivo Matrigel Assay
To determine the effect of treatment with rh-VEGF165 and rh-bFGF on angiogenic activity around A. fumigatus–infected tissue, we induced cutaneous aspergillosis in cyclophosphamide-treated mice and analyzed angiogenesis in Matrigel plugs implanted subcutaneously near the site of infection [5]. Female BALB/c mice weighing 18–20 g (National Cancer Institute) were treated with cyclophosphamide (100 mg/kg) intraperitoneally 4 days and 1 day before infection. Infection was performed by injecting 100 µL of suspension containing 1.75 × 106 conidia subcutaneously into the right thigh of each mouse, under 2.5% isoflurane–induced anesthesia. Control mice received subcutaneous injection of sterile normal saline (sham infection). Additional cyclophosphamide (100 mg/kg intraperitoneally) was given 2, 4, and 6 days after inoculation, to maintain neutropenia. Mice (5 per treatment group) were treated with rh-VEGF165, rh-bFGF, or saline control, using the same protocol as the invasive pulmonary aspergillosis model.
Matrigel (BD Biosciences, San Jose, CA) was thawed and mixed with heparin (Fisher Scientific, Pittsburgh, PA) at 64 U/mL. On day 3 after inoculation of A. fumigatus conidia, 500 µL of Matrigel was injected between the A. fumigatus infection site and the dorsal midline, 10 mm from the infection site. Five days later mice, were killed by CO2-induced asphyxiation, and Matrigel plugs were excised, fixed in 10% formalin, embedded in paraffin wax, sectioned, and stained with Masson trichrome stain. Slides were observed with an Olympus BX41 microscope fitted with an Olympus DP71 camera. Images of approximately 10 randomly selected fields per specimen were captured at ×100 magnification, using CellA imaging software (Olympus). Endothelial cell density was measured with ImageJ software (available at: http://rsb.info.nih.gov/ij/ [National Institutes of Health, Bethesda, MD]).
Amphotericin B Pulmonary Tissue Concentrations
Lung tissue concentrations of amphotericin B were measured in a subset of mice killed 72 hours after inoculation (3 per treatment arm). The concentration of amphotericin B was assayed in lung homogenates by high-performance liquid chromatography at a reference laboratory (Fungal Testing Laboratory, University of Texas Health Science Center, San Antonio, TX).
Statistical Analysis
Survival curves were plotted using the Kaplan-Meier method, and comparisons among different treatment groups were performed with the log-rank test. Pulmonary fungal burdens (conidial equivalent rank sums) and tissue amphotericin B concentrations were analyzed using the nonparametric Kruskal-Wallis test, and the Dunn posttest was used to compare the rank sums of each pair of treatment arms. A P value threshold of < .05 was used to determine statistical significance. Statistics were calculated in GraphPad Prism 5.0 (GraphPad Software, San Diego, CA).
RESULTS
rh-bFGF and rh-VEGF165 Synergize With Amphotericin B to Reduce Mortality in Invasive Pulmonary Aspergillosis
Our murine invasive pulmonary aspergillosis model resulted in acute infection and almost 100% mortality within 5 days after infection (Figure 1). Treatment with amphotericin B alone (1 mg/kg per day for 5 days) had no significant effect on the mortality rate (P = .11). Treatment with rh-VEGF165 alone resulted in a modest but statistically significant increase in the survival rate as compared to saline treatment (hazard ratio [HR], 0.53; 95% confidence interval [CI], 0.33–0.86; P = .011). However, treatment with AmB/VEGF significantly increased the survival rate as compared to saline treatment (HR, 0.31; 95% CI, .19–.50; P < .0001), AmB alone (HR, 0.41; 95% CI, .25–.65; P = .0002), and VEGF alone (HR, 0.44; 95% CI, .27–.73; P = .0016). Of note, although treatment with AmB/VEGF prolonged the median survival period from 3 days (with saline treatment) to 5 days, the survival rate at 7 days was not significantly different between the 2 treatment arms (8.5% for saline control and 10% for AmB/VEGF).
Figure 1.

Survival curves of immunosuppressed mice with invasive pulmonary aspergillosis treated with various combinations of proangiogenic growth factors and amphotericin B (AmB). BALB/c mice were immunosuppressed with cyclophosphamide and cortisone acetate and infected intranasally with Aspergillus fumigatus conidia, as described in Methods. Kaplan-Meier survival curves are shown for 8 different treatment arms (30 mice per treatment arm). Survival curves that differed significantly from the saline treated control group by the log-rank test are marked as *P < .05 and **P < .001. Abbreviations: bFGF, recombinant human basic fibroblast growth factor; VEGF, recombinant human vascular endothelial growth factor, proximal splice site 165.
Treatment with rh-bFGF alone significantly increased the survival rate as compared with saline treatment (HR, 0.47; 95% CI, .24–.92; survival rate at day 7, 25.6% vs 8.6%, respectively; P = .028). Combined treatment with rh-bFGF plus amphotericin B increased the survival rate, compared with saline treatment (HR, 0.18; 95% CI, .10–.35; P < .0001), treatment with AmB alone (HR, 0.21; 95% CI, .11–.40; P < .0001), and treatment with bFGF alone (HR, 0.26; 95% CI, .09–.70; P = .008). The survival rate at 7 days was 62.9% in the bFGF/AmB arm, which was significantly higher than in any of the other treatment arms (Figure 1).
Combined treatment with rh-VEGF165 and rh-bFGF increased the survival rate as compared to the saline treatment arm (HR, 0.42; 95% CI, .22–.81; P = .009) but not more so than treatment with rh-bFGF or rh-VEGF165 alone (P = .9). Similarly, treatment with rh-VEGF165/rh-bFGF plus AmB increased the survival rate as compared to the saline treatment arm (HR, 0.27; 95% CI, .14–.51; P < .0001) and the AmB arm (HR, 0.34; 95% CI, .18–.64; P = .0008). However, the survival rate in the rh-VEGF165/rh-bFGF/AmB arm was nonsignificantly lower than that in the rh-bFGF/AmB arm (HR, 2.3, P = .07; Figure 1). Thus, we found no evidence of synergistic interaction between rh-VEGF165 and rh-bFGF.
rh-bFGF in Combination With Amphotericin B Reduces the Pulmonary Fungal Burden
Consistent with the survival analyses, pulmonary fungal burden (measured by qPCR and expressed as conidial equivalents) was significantly reduced in the bFGF/AmB arm and, to a lesser degree, in the bFGF arm, relative to other treatment arms. Specifically, the median pulmonary fungal burden was 1.0 log lower in the bFGF arm (9.6 × 107) and 1.7 log lower in the bFGF/AmB arm (2.4 × 107) as compared with no treatment (1.1 × 109; P < .001; Figure 2).
Figure 2.
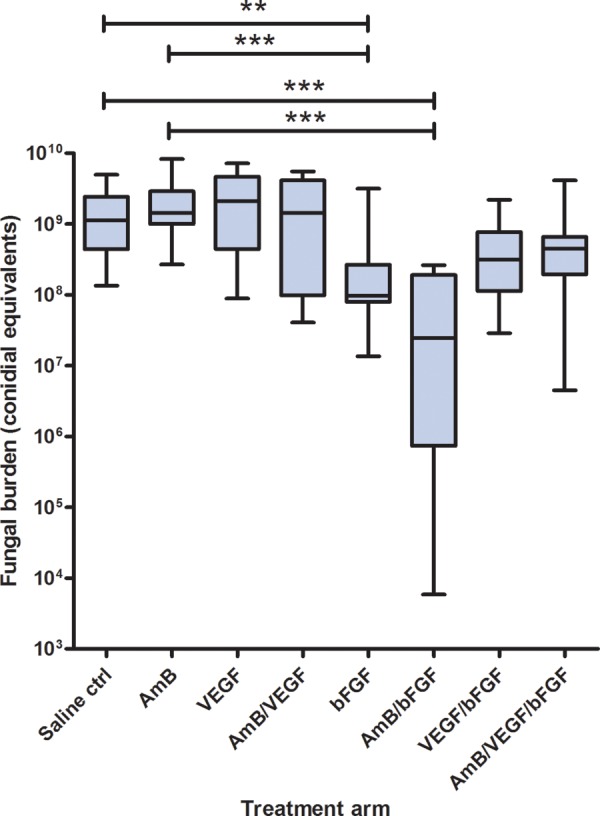
Tissue fungal burdens in the lungs of mice with invasive pulmonary aspergillosis 72 hours after infection. Tissue fungal burden was determined by real-time quantitative polymerase chain reaction with primers and probe specific for a sequence from the Aspergillus fumigatus 18S ribosomal RNA gene and expressed as conidial equivalents. Comparison of all treatment arms (30 mice per treatment arm) with the nonparametric Kruskal-Wallis test showed significant heterogeneity (P < .0001). Treatment arms that differed significantly from the saline control (Ctrl) or amphotericin B (AmB) treatment arms by the Dunn posttest are marked as **P < .001 and ***P < .0001. Abbreviations: bFGF, recombinant human basic fibroblast growth factor; VEGF, recombinant human vascular endothelial growth factor, proximal splice site 165.
rh-bFGF and rh-VEGF165 Modulate Angiogenesis and Inflammatory Response in Invasive Pulmonary Aspergillosis
Different patterns of histopathological findings were observed in lung specimens from various treatment groups. In untreated controls and AmB-treated mice, typical features of invasive pulmonary aspergillosis in neutropenic hosts were seen (Figure 3). Specifically, lung tissue was extensively invaded with branching hyphae, and there was scant polymorphonuclear leukocyte (PMNL) infiltration. Immunostaining for the endothelial cell surface marker CD31 (PECAM-1) showed little-to-no angiogenic activity in the vicinity of Aspergillus-infected lung tissue, although CD31-positive blood vessels could be observed in peripheral lung zones where no Aspergillus hyphae were present (Figure 3).
Figure 3.

Angiogenic and inflammatory responses in mouse lungs. Mouse lungs were removed 3 days after infection with Aspergillus fumigatus and stained with Gomori methenamine silver (GMS) or hematoxylin and eosin (H&E) combined with CD31 immunostaining, as described in Methods. Lungs of saline-treated mice (A) and amphotericin B (AmB)–treated mice (B) showed typical features of invasive aspergillosis in neutropenic hosts, consisting of extensive mycelial invasion of lung parenchyma and scant neutrophilic infiltrate. CD31-positive microvessels were absent from the periphery of fungal lesions. In both recombinant human vascular endothelial growth factor, proximal splice site 165 (VEGF)–treated mice and recombinant human basic fibroblast growth factor (bFGF)–treated mice, fungal hyphae were more widely dispersed, and networks of CD31-positive microvessels were observed around and inside Aspergillus-infected zones. Angiogenesis was disordered in AmB/VEGF-treated lungs, and large areas of hemorrhagic necrosis (HN) were observed. In AmB/bFGF-treated mice, the angiogenic response seen in bFGF-treated mice was augmented by neutrophilic infiltration of Aspergillus-infected zones, inflammatory necrosis (IN), and reduced fungal burden. Magnification: GMS, ×100; H&E/CD31, ×400; bottom panel, ×100.
In contrast, in both rh-VEGF165 and rh-bFGF–treated mice, a network of CD31-positive vessels was seen throughout the lung parenchyma, including around and among fungal hyphae. Moreover, hyphae were loosely dispersed, as opposed to the vasculocentric hyphal “sunburst” pattern seen in untreated controls. In bFGF/AmB-treated mice, enhanced angiogenic activity was accompanied by striking PMNL infiltration of fungal lesions, inflammatory necrosis of lung tissue, and reduced fungal burden (Figure 3). However, disordered angiogenesis and multiple areas of hemorrhagic necrosis were evident in VEGF- and VEGF/AmB-treated mouse lungs, with no apparent reduction in tissue fungal load (Figure 3).
rh-bFGF and rh-VEGF165 Reverse the Antiangiogenic Activity of A. fumigatus In Vivo
To provide quantitative assessment of the effects of rh-bFGF and rh-VEGF165 treatment on angiogenesis at the site of invasive aspergillosis, we performed in vivo Matrigel assays in mice with cutaneous aspergillosis. As previously reported [5], endothelial network formation was significantly reduced in Matrigel plugs implanted proximal to cutaneous Aspergillus lesions. Treatment with rh-bFGF reversed the inhibitory activity of Aspergillus infection and restored angiogenic activity to uninfected control levels (Figure 4). Surprisingly, treatment with rh-VEGF165 increased the Matrigel cell fraction above that of rh-bFGF treatment. However, inspection of Matrigel plugs from these mice revealed a disordered endothelial network and multiple blood-filled lacunae, consistent with the formation of immature and leaky microvessels and similar to our observations in CD31-stained mouse lungs (Figure 4).
Figure 4.

Angiogenesis at the site of invasive aspergillosis. Angiogenesis was measured in vivo, using Matrigel plugs implanted proximal to cutaneous aspergillosis lesions. All images were obtained 5 days after Matrigel plug implantation (Masson trichrome stain, ×100 magnification). Extensive endothelial cell migration and capillary network formation was seen in mice that received sham infection (A). In contrast, angiogenesis was almost completely inhibited in Aspergillus fumigatus–infected mice (B and E). Treatment with recombinant human basic fibroblast growth factor (bFGF; C and E) restored angiogenesis to uninfected control levels. Treatment with recombinant human vascular endothelial growth factor, proximal splice site 165 (VEGF; D and E), induced endothelial cell migration into Matrigel plugs and the formation of erythrocyte-filled lacunae, likely reflecting the formation of immature leaky neovessels.
bFGF and VEGF165 Treatment Does Not Alter Amphotericin B Lung Tissue Concentrations
To determine whether rh-bFGF and rh-VEGF165 treatment affects the accumulation of amphotericin B in lung tissue, we measured amphotericin B tissue concentrations in whole lung homogenates from each of the treatment arms. Amphotericin B tissue levels were not significantly different between mice treated with AmB alone and those treated with AmB in combination with either rh-bFGF or rh-VEGF165 (data not shown; P = .28).
DISCUSSION
Invasive aspergillosis remains a frequently fatal disease in immunocompromised patients [1–3]. Failure of amphotericin B treatment has been shown to occur despite full susceptibility to this drug [6], suggesting that impaired drug delivery may be an important factor. The introduction of novel antifungals, notably voriconazole and the echinocandins, represented significant progress in the treatment of invasive aspergillosis [2, 18]. Nevertheless, even with optimal medical management, as many as 30%–40% of patients with invasive aspergillosis die of the infection [2, 3]. Importantly, the efficacy of voriconazole, currently the drug of choice for invasive aspergillosis [18], is threatened by the emergence of azole-resistance among clinical and environmental Aspergillus isolates [19]. Therefore, new treatment strategies against invasive aspergillosis are urgently needed [20].
In the hyperacute/high-inoculum invasive pulmonary aspergillosis model used in this study, amphotericin B deoxycholate monotherapy was ineffective at reducing mortality rate and tissue fungal burden. In contrast, treatment with rh-VEGF165, rh-bFGF, or both significantly prolonged survival duration. rh-bFGF treatment had a greater effect on survival rate than rh-VEGF165 and, in addition, significantly reduced the tissue fungal burden. Moreover, rh-bFGF and rh-VEGF165 potentiated the in vivo activity of amphotericin B, leading to significantly improved survival rates and to lower pulmonary fungal burdens in mice receiving the bFGF/AmB combination. These effects correlated with enhanced angiogenic activity at the site of infection in both pulmonary and cutaneous models of invasive aspergillosis. Patterns of hyphal growth were also affected by bFGF and VEGF therapy; vasculocentric “sunburst” formations, considered to represent metastatic foci of infection in the lung [21], were absent in treated mice, suggesting that proangiogenic therapy limited the vascular spread of A. fumigatus.
An interesting feature of bFGF/AmB-treated mouse lungs was the presence of neutrophilic infiltrates in Aspergillus-infected tissue, suggesting that the beneficial effect of bFGF may be attributable, at least in part, to increased influx of PMNLs into the fungal lesion. Of note, amphotericin B has well-documented proinflammatory effects, including enhanced release of proinflammatory cytokines from monocytes [22] and additive antifungal effects with PMNLs against A. fumigatus hyphae [23]. Our results do not allow us to determine whether bFGF increases the delivery of AmB to Aspergillus-infected tissue. AmB concentrations in whole-lung homogenates were not affected by rh-bFGF or rh-VEGF165 treatment; however, these crude measurements do not necessarily reflect AmB concentrations within the fungal lesions, and this issue merits further study.
Treatment with rh-VEGF165 and VEGF/AmB was associated with modest effects on the survival rate of mice with experimental invasive aspergillosis and with no effect on fungal burden, compared with treatment with rh-bFGF and bFGF/AmB, respectively. In our previous work, downregulation of bFGF gene expression in mouse lungs during the acute phase of invasive aspergillosis was more prominent than that observed for VEGF gene expression [5]. Taken together, these findings suggest that the bFGF pathway is the more important target of A. fumigatus anti-angiogenic secondary metabolites. Another explanation for the relative lack of efficacy of rh-VEGF165 monotherapy is that this growth factor is known to induce the formation of immature and unstable microvessels that are hyperpermeable and leaky [24, 25]. In support of this hypothesis, analysis of pulmonary tissue specimens and Matrigel plugs implanted at the site of cutaneous invasive aspergillosis in VEGF-treated mice revealed disorganized angiogenesis, extruded erythrocytes, and hemorrhagic necrosis. Thus, VEGF-induced angiogenesis is ineffective at reversing invasive aspergillosis-associated vasculopathy.
Our observations allow us to propose a theoretical model for the role of vasculopathy in invasive aspergillosis pathogenesis. A. fumigatus invades pulmonary vessels by penetrating vascular endothelial cells from their abluminal side, precipitating tissue factor expression and intravascular thrombosis [26, 27]. These direct effects are compounded by the inhibition of compensatory angiogenesis by A. fumigatus secondary metabolites [5]. Ischemia induces necrotic cell death and the release of damage-associated molecular patterns (“alarmins”), which activate cellular signaling pathways that converge on nuclear factor kB (NF-kB) [28], a powerful proangiogenic signal [29]. Inhibition of NF-kB and HIF-1α–driven angiogenic response by A. fumigatus–secreted gliotoxin and, likely, by other, as yet uncharacterized secondary metabolites is expected to promote tissue hypoxia [5, 10]. This hypoxic/necrotic environment bears similarity in many respects to the natural ecosystem to which A. fumigatus is well adapted: the compost heap [30]. Specifically, high temperature, low oxygen tension, varying pH, and restricted supply of carbon sources and metal ions are environmental challenges that are met by A. fumigatus thermotolerance, nutrient-regulated stress responses, and highly efficient ion-scavenging systems [31, 32]. Moreover, vascular injury might sequester infected tissue, severely impairing access to antifungal agents [6] and immune effector cells and allowing invasive aspergillosis to progress unchallenged. Our results indicate that shifting the balance from A. fumigatus–driven antiangiogenic activity to proangiogenic signaling may facilitate fungal clearance and response to amphotericin B. However, our observations in the neutropenic invasive aspergillosis setting do not allow us to generalize this model to nonneutropenic corticosteroid-treated hosts.
Because most cases of invasive aspergillosis occur in patients with hematological malignancies, the effect of proangiogenic factors on cancer progression should be taken into account. The role of angiogenesis in solid tumor progression is well established, and similar associations have been recently described for various hematologic cancers. For example, the expression of VEGF and marrow microvessel density correlate with poor prognosis in acute myelocytic leukemia [33]. Future research will need to address concerns about the use of proangiogenic growth factors in such patients. Strategies targeted at selectively modulating angiogenesis at the site of infection are warranted, to avoid unwanted systemic proleukemic effects.
In summary, our results provide the first evidence that proangiogenic factors can favorably affect the course of invasive aspergillosis and its response to conventional antifungal treatment. Viewed in the context of our previous work [5], these findings indicate that overcoming the antiangiogenic activity of secondary metabolites secreted by A. fumigatus could represent a novel therapeutic strategy for this difficult infection. More broadly, recognition that the vasculopathy associated with invasive aspergillosis presents a barrier to successful fungal clearance may open a path to the discovery of novel drug targets.
Notes
Financial support. This work was supported by the National Institutes of Health (NIH; grant R03 AI083733-01 to D. P. K.), an NIH Cancer Center Support Grant, and the Frances King Black endowed professorship (to D. P. K.).
Potential conflicts of interest. R. B.-A. has received consulting fees and research support from Pfizer. R. E. L. has received research support from Merck. D. P. K. has received research grants from Merck, Pfizer, Astellas, and Gilead and serves on the advisory board of Merck and on the speakers bureau of Gilead. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Kontoyiannis DP, Marr KA, Park BJ, et al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin Infect Dis. 2010;50:1091–100. doi: 10.1086/651263. [DOI] [PubMed] [Google Scholar]
- 2.Herbrecht R, Denning DW, Patterson TF, et al. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347:408–15. doi: 10.1056/NEJMoa020191. [DOI] [PubMed] [Google Scholar]
- 3.Cornely OA, Maertens J, Bresnik M, et al. Liposomal amphotericin B as initial therapy for invasive mold infection: a randomized trial comparing a high-loading dose regimen with standard dosing (AmBiLoad trial) Clin Infect Dis. 2007;44:1289–97. doi: 10.1086/514341. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Ami R, Lewis RE, Kontoyiannis DP. Enemy of the (immunosuppressed) state: an update on the pathogenesis of Aspergillus fumigatus infection. Br J Haematol. 2010;150:406–17. doi: 10.1111/j.1365-2141.2010.08283.x. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Ami R, Lewis RE, Leventakos K, Kontoyiannis DP. Aspergillus fumigatus inhibits angiogenesis through the production of gliotoxin and other secondary metabolites. Blood. 2009;114:5393–9. doi: 10.1182/blood-2009-07-231209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paterson PJ, Seaton S, Prentice HG, Kibbler CC. Treatment failure in invasive aspergillosis: susceptibility of deep tissue isolates following treatment with amphotericin B. J Antimicrob Chemother. 2003;52:873–6. doi: 10.1093/jac/dkg434. [DOI] [PubMed] [Google Scholar]
- 7.Rankin NE. Disseminated aspergillosis and moniliasis associated with agranulocytosis and antibiotic therapy. Br Med J. 1953;1:918–9. doi: 10.1136/bmj.1.4816.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balloy V, Huerre M, Latge JP, Chignard M. Differences in patterns of infection and inflammation for corticosteroid treatment and chemotherapy in experimental invasive pulmonary aspergillosis. Infect Immun. 2005;73:494–503. doi: 10.1128/IAI.73.1.494-503.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stergiopoulou T, Meletiadis J, Roilides E, et al. Host-dependent patterns of tissue injury in invasive pulmonary aspergillosis. Am J Clin Pathol. 2007;127:349–55. doi: 10.1309/UJRV9DLC11RM3G8R. [DOI] [PubMed] [Google Scholar]
- 10.Grahl N, Puttikamonkul S, Macdonald JM, et al. In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PLoS Pathog. 2011;7:e1002145. doi: 10.1371/journal.ppat.1002145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willger SD, Puttikamonkul S, Kim KH, et al. A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog. 2008;4:e1000200. doi: 10.1371/journal.ppat.1000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Ami R, Kontoyiannis DP. A nonlethal murine cutaneous model of invasive aspergillosis. Methods Mol Biol. 2012;845:569–82. doi: 10.1007/978-1-61779-539-8_42. [DOI] [PubMed] [Google Scholar]
- 13.Clinical and Laboratory Standards Institute. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard M38-A2. Wayne, PA: National Committee for Clinical Laboratory Standards; 2008. [Google Scholar]
- 14.Doukas J, Wrasidlo W, Noronha G, et al. Phosphoinositide 3-kinase gamma/delta inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:19866–71. doi: 10.1073/pnas.0606956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paris F, Fuks Z, Kang A, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 16.Bowman JC, Abruzzo GK, Anderson JW, et al. Quantitative PCR assay to measure Aspergillus fumigatus burden in a murine model of disseminated aspergillosis: demonstration of efficacy of caspofungin acetate. Antimicrob Agents Chemother. 2001;45:3474–81. doi: 10.1128/AAC.45.12.3474-3481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiederhold NP, Kontoyiannis DP, Chi J, Prince RA, Tam VH, Lewis RE. Pharmacodynamics of caspofungin in a murine model of invasive pulmonary aspergillosis: evidence of concentration-dependent activity. J Infect Dis. 2004;190:1464–71. doi: 10.1086/424465. [DOI] [PubMed] [Google Scholar]
- 18.Walsh TJ, Anaissie EJ, Denning DW, et al. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:327–60. doi: 10.1086/525258. [DOI] [PubMed] [Google Scholar]
- 19.Snelders E, van der Lee HA, Kuijpers J, et al. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med. 2008;5:e219. doi: 10.1371/journal.pmed.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis. 2006;42:657–68. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 21.Kradin RL, Mark EJ. The pathology of pulmonary disorders due to Aspergillus spp. Arch Pathol Lab Med. 2008;132:606–14. doi: 10.5858/2008-132-606-TPOPDD. [DOI] [PubMed] [Google Scholar]
- 22.Sau K, Mambula SS, Latz E, Henneke P, Golenbock DT, Levitz SM. The antifungal drug amphotericin B promotes inflammatory cytokine release by a Toll-like receptor- and CD14-dependent mechanism. J Biol Chem. 2003;278:37561–8. doi: 10.1074/jbc.M306137200. [DOI] [PubMed] [Google Scholar]
- 23.Roilides E, Lyman CA, Filioti J, et al. Amphotericin B formulations exert additive antifungal activity in combination with pulmonary alveolar macrophages and polymorphonuclear leukocytes against Aspergillus fumigatus. Antimicrob Agents Chemother. 2002;46:1974–6. doi: 10.1128/AAC.46.6.1974-1976.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 25.Nagy JA, Feng D, Vasile E, et al. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest. 2006;86:767–80. doi: 10.1038/labinvest.3700436. [DOI] [PubMed] [Google Scholar]
- 26.Lopes Bezerra LM, Filler SG. Interactions of Aspergillus fumigatus with endothelial cells: internalization, injury, and stimulation of tissue factor activity. Blood. 2004;103:2143–9. doi: 10.1182/blood-2003-06-2186. [DOI] [PubMed] [Google Scholar]
- 27.Kamai Y, Chiang LY, Lopes Bezerra LM, et al. Interactions of Aspergillus fumigatus with vascular endothelial cells. Med Mycol. 2006;44(Suppl 1):S115–7. doi: 10.1080/13693780600897989. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 29.Maulik N. Redox signaling of angiogenesis. Antioxid Redox Signal. 2002;4:805–15. doi: 10.1089/152308602760598963. [DOI] [PubMed] [Google Scholar]
- 30.Millner PD, Marsh PB, Snowden RB, Parr JF. Occurrence of Aspergillus fumigatus during composting of sewage sludge. Appl Environ Microbiol. 1977;34:765–72. doi: 10.1128/aem.34.6.765-772.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhodes JC. Aspergillus fumigatus: growth and virulence. Med Mycol. 2006;44(Suppl 1):S77–81. doi: 10.1080/13693780600779419. [DOI] [PubMed] [Google Scholar]
- 32.Cooney NM, Klein BS. Fungal adaptation to the mammalian host: it is a new world, after all. Curr Opin Microbiol. 2008;11:511–6. doi: 10.1016/j.mib.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trujillo A, McGee C, Cogle CR. Angiogenesis in acute myeloid leukemia and opportunities for novel therapies. J Oncol. 2012;2012:128608. doi: 10.1155/2012/128608. [DOI] [PMC free article] [PubMed] [Google Scholar]