

Abstract
Galectin-1 (gal-1), which binds β-galactoside groups on various cell surface receptors, is crucial to cell adhesion and migration, and is found to be elevated in several cancers. Previously, we reported on 6DBF7, a dibenzofuran (DBF)-based peptidomimetic of the gal-1 antagonist anginex. In the present study, we used a structure-based approach to optimize 6DBF7. Initial NMR studies showed that 6DBF7 binds to gal-1 on one side of the β-sandwich away from the lectin’s carbohydrate binding site. Although an alanine scan of 6DBF7 showed that the two cationic groups (lysines) in the partial peptide are crucial to its angiostatic activity, it is the hydrophobic face of the amphipath that appears to interact directly with the surface of gal-1. Based on this structural information, we designed and tested additional DBF analogs. In particular, substitution of the C-terminal Asp for alanine and branched alkyl side chains (Val, Leu, Ile) for linear ones (Nle, Nva) rendered the greatest improvements in activity. Flow cytometry with gal-1−/− splenocytes showed that 6DBF7 and two of its more potent analogs (DB16 and DB21) can fully inhibit fluorescein isothiocyanate–gal-1 binding. Moreover, heteronuclear single-quantum coherence NMR titrations showed that the presence of DB16 decreases gal-1 affinity for lactose, indicating that the peptidomimetic targets gal-1 as a noncompetitive, allosteric inhibitor of glycan binding. Using tumor mouse models (B16F10 melanoma, LS174 lung, and MA148 ovarian), we found that DB21 inhibits tumor angiogenesis and tumor growth significantly better than 6DBF7, DB16, or anginex. DB21 is currently being developed further and holds promise for the management of human cancer in the clinic.
Introduction
Management of angiogenesis is an attractive possibility for controlling cancer and metastasis. Consequently, antiangiogenic compounds have considerable potential as therapeutic agents. Many or most angiostatic compounds being developed and tested are inhibitors related to various components of growth factor pathways, e.g., anti-vascular endothelial growth factor antibodies and kinase inhibitors. However, because these agents have had limited success in the clinic, new compounds such as angiostatic agents that target different systems are sorely needed. Galectins provide one such novel molecular target for therapeutic intervention against cancer.
Galectins are a phylogenetically conserved family of carbohydrate binding lectins that share a conserved carbohydrate recognition domain (Barondes et al., 1994). They generally bind β-galactoside–containing glycoconjugates and promote cell–cell and cell–matrix interactions during cancer development and progression (Takenaka et al., 2004; Liu and Rabinovich, 2005). For example, galectin-1 (gal-1) interacts with various glycoconjugates of the extracellular matrix (e.g., laminin, fibronectin, the β1 subunit of integrins, and ganglioside GM1), as well as those on endothelial cells (e.g., integrins αvβ3 and αvβ5, ROBO4, CD36, and CD13) (Neri and Bicknell, 2005). Binding to cell surface glycoproteins can also trigger intracellular activity [e.g., elements of the rat sarcoma-methyl ethyl ketone-extracellular signal-regulated kinase pathway (Fischer et al., 2005)], and the presence of gal-1 in the cell nucleus promotes mRNA splicing (Liu et al., 2002). Differential stromal elevation of gal-1 over the tumor parenchyma has been reported in several cancers, including cancer of the brain, breast, colon, skin, prostate, and ovaries (Liu and Rabinovich, 2005; Lefranc et al., 2011).
Previously, we demonstrated that the designed peptide anginex targets gal-1 (Dings et al., 2003b, c; Thijssen et al., 2006; Dings and Mayo, 2007), and that this interaction inhibits tumor endothelial cell proliferation via anoikis and attenuates tumor angiogenesis and tumor growth (Dings et al., 2003b,c; Thijssen et al., 2006; Dings and Mayo, 2007). In addition, anginex synergistically enhances the effects of radiotherapy of several solid tumor types, presumably due to the antiangiogenic and tumor vascular damaging effects (Dings et al., 2005), as well as via induction of vascular normalization and reoxygenation of tumor tissue before radiation exposure (Dings et al., 2007). In addition, anginex can affect endothelial cell anergy and allow a normal immune response in tumors (Griffioen et al., 1999; Dings et al., 2011). We have also reported on the design of a dibenzofuran (DBF)-based peptidomimetic of anginex, 6DBF7, that is much smaller than anginex, yet maintains its β-sheet structure and functionally key amino acid residues (Fig. 1). 6DBF7 shows improved in vitro and in vivo activity profiles over parent anginex (Dings et al., 2003a; Mayo et al., 2003).
Fig. 1.

Sequence and general folding pattern of anginex and 6DBF7 are illustrated. The boxed sequences in anginex are those that have been preserved in 6DBF7 and related analogs as discussed in the text.
Because we had yet to validate that gal-1 is the molecular target of 6DBF7, the present study was designed in part to do just that. In this study, we used heteronuclear NMR spectroscopy to demonstrate that 6DBF7 and its analogs indeed target gal-1, and to determine the sites of the peptidomimetic interactions with the lectin. This structure-based information aided in optimization of 6DBF7. In vitro and in vivo activities of 6DBF7 were improved by replacing the C-terminal Asp residue with Ala, and by substituting specific branched alkyl side chains with linear ones. This work contributes to the development of novel therapeutic agents against cancer in the clinic.
Materials and Methods
Peptide Synthesis.
Peptides were synthesized using a Milligen/Biosearch 9600 peptide solid-phase synthesizer (Applied Biosystems, Inc., Foster City, CA) using fluorenylmethoxycarbonyl (Fmoc) chemistry. Lyophilized crude peptides were purified by preparative reversed-phase high-performance liquid chromatography (HPLC) on a C18 column with an elution gradient of 0–60% acetonitrile with 0.1% trifluoroacetic acid in water. Peptide purity and composition were verified by HPLC, N-terminal sequencing, and mass spectrometry.
Synthesis of DBF Analogs.
Unexceptional phases of solid-phase peptide synthesis were carried out on an Applied Biosystems, Inc. 431 peptide synthesizer using Fmoc methodology and BOP/HOBT as coupling reagents. Fmoc-DBF-CO2H was prepared by slight modification of the reported method (Bekele et al., 1997). In this nine-step synthesis, the intermediates and final product were characterized by thin layer chromatography and 1H NMR with gas chromatography couple to low resolution mass spectrometry, 13C NMR, m.p., and infrared analyses, as appropriate. Coupling of Fmoc-DBF-CO2H to I20 and coupling of L11 to peptide-DBF-NH2 was performed on the synthesizer. Coupling of Fmoc-K10-CO2H to the peptide-DBF-L11-NH2 sequence was difficult and required manual solid-phase peptide synthesis using the more reactive (O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate) reagent (Carpino, 1993). The remaining couplings required for production of 2DBF7 through 11DBF7 were carried out using benzotriazol-1-yloxytris(dimethyl amino)phosphonium hexafluorophosphate/1-Hydroxybenzotriazole conditions on the peptide synthesizer. After the final Fmoc deprotection, each of the DBF peptides was released frosinam the resin with simultaneous removal of all acidolyzable trityl and tert-butyl side-chain protecting groups using Reagent K (King et al., 1990). A Rink amide or similar resin was used to provide the primary amide form of the C-terminal D24 unit. Lyophilized crude peptides were purified by HPLC as described earlier. Peptide purity and composition were verified by analytical HPLC, matrix-assisted laser desorption ionization mass spectrometry using a Hewlett-Packard (Palo Alto, CA) G2025A system and sinapinic acid as matrix, and analysis of amino acid composition of hydrolysates (6 N HCl, 110°C, 24 hours, under argon).
Galectin-1 Preparation.
Unlabeled and uniformly 15N-labeled human gal-1 was expressed in BL21(DE3)-competent cells (Novagen; Millipore, Billerica, MA), grown in standard enriched media or in minimal media with 15N-ammonium chloride for 15N labeling, purified over a B lactose affinity column, and further fractionated on a gel filtration column, as described previously by Nesmelova et al. (2008b). Typically, 200 mg (unlabeled) or 44 mg (15N-enriched) of purified protein were obtained from 1 l of cell culture. The purity of the final sample was quantified using the Bio-Rad protein assay (Bio-Rad Labs, Hercules, CA) and was checked for purity using SDS-PAGE and mass spectrometry. Functional activity of the purified protein was assessed by a T cell death assay.
Fluorescein Isothiocyanate–Galectin-1 Preparation.
Gal-1 was conjugated with fluorescein isothiocyanate (FITC) using a FITC:protein molar ratio of 10:1. Gal-1 (2 mg/ml) was dissolved in 500 ml of 20 mM potassium phosphate buffer, pH 7.4, followed by the addition of ∼50 µl FITC (10 mg/mL) that had been dissolved in 0.1 mM sodium bicarbonate. The resulting lower pH (∼pH 3) was used to achieve selective labeling at the N-terminal amine group of the protein (Hungerford et al., 2007; Hermanson, 2008). This solution was then mixed thoroughly and incubated at room temperature (22°C) for 18 hours in the dark. During the course of the reaction, the mixture was gently vortexed three or four times. The resulting FITC-labeled protein was separated from unbound dye by filtration using an Amicon Ultra cellulose filter (Millipore; 10 kDa cutoff). Matrix-assisted laser desorption ionization–time-of-flight mass spectrometry demonstrated the addition of the 389 Da FITC label to galectin-1 and suggested >90% labeling efficiency.
NMR Spectroscopy.
For NMR measurements, solutions in which both uniformly 15N-labeled recombinant gal-1 (15N-gal-1) and a 6DBF7 analog were dissolved contained 20 mM potassium phosphate buffer (95% H2O/5% D2O), pH 7, 1% trifluoroethanol (TFE), 1% glycerol, 2% NP40 (nonionic detergent), and 150 mM NaCl, and were with or without 1 mM lactose. In other samples, 15N-gal-1 was dissolved in 20 mM potassium phosphate buffer (95% H2O/5% D2O), pH 7, with or without 1 mM lactose. The pH was adjusted to pH 7 by adding microliter quantities of NaOD or HCl to the peptide sample.
1H-15N heteronuclear single-quantum coherence (HSQC) NMR experiments were used to investigate binding of DB16 to 15N-labeled gal-1. 1H and 15N resonance assignments for gal-1 were previously reported (Nesmelova et al., 2008a,b). NMR experiments were carried out at 30°C on a Varian Unity Inova 600-MHz spectrometer (Agilent Technologies, Palo Alto, CA) equipped with an H/C/N triple-resonance probe and x/y/z triple-axis pulse field gradient unit. A gradient sensitivity-enhanced version of two-dimensional 1H-15N HSQC was applied with 256 (t1) × 2048 (t2) complex data points in nitrogen and proton dimensions, respectively. Raw data were converted and processed by using NMRPipe (Delaglio et al., 1995) and were analyzed using NMRview (Johnson, 2004).
Pulse Field Gradient NMR Self-Diffusion Measurements.
For NMR measurements, compounds were dissolved in 0.6 ml of unbuffered D2O, and the pH was adjusted by adding microliter quantities of NaOD or DCl. Pulse field gradient (PFG) NMR self-diffusion measurements were determined on a Varian INOVA-600 using a GRASP gradient unit (Agilent Technologies), as previously described (Mayo et al., 1996; Ilyina et al., 1997). NMR spectra for measurement of diffusion coefficients, D, were acquired using a 5-mm triple-resonance probe equipped with an actively shielded z-gradient coil. The maximum magnitude of the gradient was calibrated using the manufacturer’s standard procedure based on the frequency spread of the applied gradient, and was found to be 100 Gauss/cm. This was consistent with the value of 98 Gauss/cm obtained from analysis of PFG data on water using its known diffusion constant (Mills, 1973). The linearity of the gradient was checked by performing diffusion measurements on water over different ranges of the gradient. The PFG longitudinal eddy-current delay pulse sequence was used for self-diffusion measurements (Gibbs and Johnson, 1991).
For unrestricted diffusion of a molecule in an isotropic liquid, the PFG NMR signal amplitude, A, normalized to the signal obtained in the absence of gradient pulses, is related to D by
| (1) |
where γ is the gyromagnetic ratio of the observed nucleus; g and δ are the magnitude and duration of the magnetic field gradient pulses, respectively; and Δ is the time between the gradient pulses (Stejskal and Tanner, 1965). For these experiments, δ = 4 ms, g = 1 – 75 G/cm, Δ = 34.2 ms, and the longitudinal eddy-current delay Te = 100 ms. Each diffusion constant, D, was determined from a series of 12 one-dimensional PFG spectra acquired using different g values.
NMR Solution Conditions for DBF Compounds and Galectin-1.
Because DB compounds have relatively low solubility in 20 mM potassium phosphate buffer at pH 7.0, the solvent system used for NMR studies of gal-1, we needed to identify solution conditions that allowed both DBs and gal-1 to remain in solution at concentrations that provided for good Signal:Noise ratio in the NMR spectra. DBs were soluble in low dielectric solvents, whereas gal-1 was either not soluble or became unfolded (random coil NMR spectrum). For this reason, we assessed the solubility limits, assessed by Beer’s law (Ingle and Crouch, 1988), of all DB compounds under various solution conditions. We found that, under most conditions, DB16 showed the best overall solubility. Therefore, to prepare NMR samples, we first dissolved DB16 in a small amount of TFE, and then slowly added Kphos (potassium phosphate) buffer (20 mM, pH 7.0) to achieve the desired concentration. However, when we added DB16 to the gal-1 solution, we observed precipitation within ∼1 hour, and the precipitation occurred more rapidly when the ratio of DB16 to gal-1 was increased to more than 1:1. To circumvent this problem, we tried many buffer cosolvents, such as TFE, hexafluoroisopropanol, SDS, NP40, glyerol, KCl, NaCl, etc., known in the literature to prevent precipitation and aggregation (Bondos and Bicknell, 2003; Chae et al., 2004). To study protein-peptide interaction, it is imperative to keep the protein in its native fully folded state. The challenge was to find suitable buffer cosolvents and to keep the protein in its native state and yet not be deleterious to the NMR experiment. After several attempts, we identified a combination of cosolvents that prevented precipitation and maintained native folding of gal-1. The best solution conditions were 20 mM Kphos buffer, pH 7.0, containing 1% TFE, 1% glycerol, 2% NP40, 150 mM NaCl, and 8 mM dithiothreitol (to maintain gal-1 in the Cys reduced state). In the absence of any one of these components, the lectin and DB16 precipitated over the time course of the NMR experiments.
Cells, Cultures, and Reagents.
Human umbilical vein endothelial cells (HUVECs) were harvested from normal human umbilical cords by perfusion with 0.125% trypsin/EDTA. HUVECs were cultured in gelatin-coated tissue-culture flasks (0.2%) in culture medium [RPMI 1640 with 20% (v/v) human serum, supplemented with 2 mM glutamine, 100 units/ml penicillin, and 0.1 mg/ml streptomycin]. LS174T (human prostate adenocarcinoma), MA148 (human ovarian carcinoma), and B16F10 (murine melanoma) were cultured on noncoated flasks using 10% fetal bovine serum and 1% penicillin/streptomycin in RPMI 1640. Cultures were split 1:3 every 3 days (Dings et al., 2006, 2008, 2011).
Proliferation Measurements.
HUVECs were seeded in a 96-well culture plate coated with 0.2% gelatin for 2 hours at 20°C (Sigma-Aldrich, St. Louis, MO). MA148 cells were seeded in noncoated 96-well plates (Corning, Lowell, MA). All cell types were seeded at a concentration of 3000 cells per well and allowed to adhere for at least 3 hours at 37°C in 5% CO2/95% air before treatments were initiated. The cells were then exposed to complete medium containing 20 ng/ml of basic fibroblast growth factor (Sigma-Aldrich), with or without various concentrations of anginex, for 72 hours or as indicated otherwise. A cell counting kit (CCK-8; Dojindo, Gaithersburg, MD) was used to assess cell proliferation rates relative to untreated cells, as described earlier (Dings et al., 2006, 2008, 2011). All measurements were performed in triplicate, and the experiments were conducted at least three times.
Flow Cytometry.
Male and female gal-1–null mice (Jackson Laboratory, Bar Harbor, ME) were provided water and standard chow ad libitum, and were maintained on a 12-hour light/dark cycle prior to experiments that were approved by the University of Minnesota Research Animal Resources Ethical Committee. For fluorescence-activated cell sorter experiments, spleens from these mice (6–10 weeks old) were harvested and nonenzymatically disrupted by shear force to yield single-cell suspensions (Dings et al., 2011). Cell suspensions were prepared in Hanks’ balanced solution. Red blood cells were lysed in ACK (Lonza, Walkersville, MD) for 5 minutes on ice, and suspensions were filtered through nylon mesh. Spleen cells were then washed and incubated with monoclonal antibodies, as indicated, for 40 minutes on ice. After an additional washing step, 0.1 µM FITC-gal-1 was added to these cells in the absence or presence of various concentrations of 6DBF7 or one of its analogs, DB16 or DB21. Mixtures were then incubated for 30 minutes on ice. Prior to fluorescence-activated cell sorter analysis, cell suspensions were washed once more and analyzed by multiparameter flow cytometry on an LSR II flow cytometer (BD Biosciences, San Jose, CA) using Flowjo software (Tree Star, Inc., Ashland, OR) (Dings et al., 2011).
Tumor Mouse Models.
LS174T (2 × 105) and MA148 cells (1 × 106) were inoculated in male and female nude mice, respectively. B16F10 cells (2 × 105) were inoculated into male C57/BL6 mice, as described previously (Brandwijk et al., 2006; Dings et al., 2006, 2008, 2011). All mice (5–6 weeks old) were purchased from the National Cancer Institute, except for the female gal-1–null mice, which were purchased from the Jackson Laboratory. After at least 1 week of acclimatization to local conditions, animals were given water and standard chow ad libitum, and were kept on a 12-hour light/dark cycle. Experiments were approved by the University of Minnesota Research Animal Resources Ethical Committee. Studies were carried out in a therapeutic intervention model with established tumors to test the capacity of the compounds to inhibit tumor growth. Tumors were allowed to grow to the size of approximately 75 mm3 prior to initiation of treatment, and growth was halted when tumor sizes reached 1000 mm3.
Tumor volume was determined by measuring the size of the tumors on the flanks of the mice. The diameters of tumors were measured using calipers (Scienceware, Pequannock, NJ), and the volume was calculated using the equation to determine the volume of a spheroid [(a2 × b × π)/6], where a is the width of the tumor and b is the length of the tumor. As an indirect measurement of general toxicity, body weights and hematocrit levels were monitored (Brandwijk et al., 2006; Dings et al., 2006, 2008, 2011).
Immunohistochemistry.
Untreated MA148 tumors were embedded in tissue-freezing medium (Miles Inc., Elkart, IN), snap frozen in liquid nitrogen, and subsequently cut into 5-µm sections. Preparation and procedures for the frozen tumor sections were performed as described earlier, with some slight modifications (Brandwijk et al., 2006; Dings et al., 2006, 2008, 2011). Briefly, sections were fixed for 10 minutes in acetone. Subsequent incubation for 30 minutes with phosphate-buffered saline (PBS)/5% bovine serum albumin/3% fetal bovine serum prevented aspecific binding. After washing with PBS, the tissue was incubated with antimouse CD31 (1:50, PECAM-1; Pharmingen, San Diego, CA). After 1-hour incubation, the slides were washed thoroughly with PBS and developed with a biotinylated secondary antibody, streptavidin-horseradish peroxidase, and 3,3'-diaminobenzidine solution. Images of the sections were acquired on an Olympus BX-60 microscope (Olympus Inc., Center Valley, PA) at 200× magnification and digitally analyzed and differentially quantified by morphometric analysis (Schumacher et al., 2007).
Toxicity Assays.
As an indirect measurement of general toxicity, body weights of mice were monitored twice weekly, using a digital balance (Ohaus, Florham Park, NJ). To determine hematocrit levels, blood samples were extracted by tail vein bleedings one day after terminating treatment, and blood was collected in heparinized microhematocrit capillary tubes (Fisher, Pittsburgh, PA). For hematocrit levels, samples were spun down for 10 minutes in a microhematocrit centrifuge (Clay-Adams, New York, NY), and the amount of hematocrit was determined using an international microcapillary reader (International Equipment Company, Needham, MA) (Dings et al., 2008, 2010).
Statistical Analysis.
The Student’s t test was used where indicated to determine the validity of the differences between control and treatment data sets. A P value of 0.05 or less was considered significant.
Results
DBF Binding Site on Gal-1.
In our previous report on the NMR structure of 6DBF7 (Mayo et al., 2003), we found it necessary to dissolve the DBF-based compound in a mixture of dodecylphosphocholine and dimethylsulfoxide (DMSO), because it was relatively insoluble in aqueous solution. Because gal-1 displayed relatively poor solubility and native folding characteristics under these same solution conditions in the present study, we first had to define solution conditions under which both gal-1 and the DBF-based compound were soluble, and at concentrations reasonable for NMR studies. As explained in Materials and Methods, acceptable solution conditions were found with 20 mM potassium phosphate buffer, pH 7.0, containing 1% TFE, 1% glycerol, 2% NP40 (a nonionic detergent), 150 mM NaCl, and 8 mM DTT to maintain gal-1 in its Cys-reduced state.
In this solution, the maximum concentration of gal-1 that we could attain was about 200 µM, without apparent precipitation. However, the maximum solubility of 6DBF7 was less than 50 µM. To achieve reasonable ligand concentrations for 15N-gal-1 HSQC titration studies, we first needed to increase the solubility of 6DBF7 in this solution. To achieve this, we modified 6DBF7 to adjust the hydrophobic and hydrophilic properties of its peptide segments. We started by making variants of 6DBF7 in which amino acid residues were substituted individually with alanine, as shown with the series DB1 to DB13 in Table 1. Solubilities were assessed by measuring UV absorbance at 280 nm for four concentrations (5, 20, 50, and 100 µM). Because a number of these DBF-based compounds followed Beer’s law (Ingle and Crouch, 1988) up to 100 µM, their solubility is reported as >100 µM in Table 1. Most deviated from Beer’s law when the concentration reached 50 µM, and therefore are reported as having a solubility <50 µM. Improved solubility over 6DBF7 was noted especially for analogs with alanine substitutions of bulky alkyl groups such as valine, leucine, and isoleucine, as well as of the C-terminal aspartate residue. Solubility was improved further by making double substitutions of a few of these residues (M4, I7, and/or D13) as shown in the series DB14 to DB17 (Table 1).
TABLE 1.
DBF-based compounds
Solubility was assessed at 5, 20, 50, and 100 µM compound in 20 mM Kphos buffer, pH 7.0, with 1% TFE, 1% glycerol, 2% NP40, and 150 mM NaCl. Solubilities are reported as >100 µM or <50 µM, depending upon whether a given compound followed Beer’s law up to 100 µM or only up to 50 µM, respectively. Proliferation of endothelial cells is reported in terms of IC50 values (µM) for each compound. Bold letters indicate amino acids in the sequence that have been modified/changed.
| Name | Sequence | Solubility | IC50 |
|---|---|---|---|
| µM | µM | ||
| Anginex | ANIKLSVQMKLFKRHLKWKIIVKLND | >100 | 4 |
| 6DBF7 | SVQMKL-[DBF]-IIVKLND | <50 | 22 |
| Single alanine substitution of 6DBF7 | |||
| DB1 | AVQMKL-[DBF]-IIVKLND | >100 | 19 |
| DB2 | SAQMKL-[DBF]-IIVKLND | <50 | 64 |
| DB3 | SVAMKL-[DBF]-IIVKLND | <50 | 28 |
| DB4 | SVQAKL-[DBF]-IIVKLND | >100 | 13 |
| DB5 | SVQMAL-[DBF]-IIVKLND | >100 | 202 |
| DB6 | SVQMKA-[DBF]-IIVKLND | >100 | 27 |
| DB7 | SVQMKL-[DBF]-AIVKLND | <50 | 19 |
| DB8 | SVQMKL-[DBF]-IAVKLND | >100 | 29 |
| DB9 | SVQMKL-[DBF]-IIAKLND | >100 | 31 |
| DB10 | SVQMKL-[DBF]-IIVALND | >100 | 74 |
| DB11 | SVQMKL-[DBF]-IIVKAND | <50 | 28 |
| DB12 | SVQMKL-[DBF]-IIVKLAD | <50 | 22 |
| DB13 | SVQMKL-[DBF]-IIVKLNA | <50 | 10 |
| Double alanine substitution of 6DBF7 | |||
| DB14 | SVQAKL-[DBF]-IIVKLNA | >100 | 8 |
| DB15 | SVQAKL-[DBF]-AIVKLND | <50 | 9 |
| DB16 | SVQMKL-[DBF]-AIVKLNA | >100 | 5 |
| Triple alanine substitution of 6DBF7 | |||
| DB17 | SVQAKL-[DBF]-AIVKLNA | <50 | 9 |
| Norleucine substitution for Lys in 6DBF7 | |||
| DB18 | SVQMNleL-[DBF]-IIVKLNA | <50 | 9 |
| DB30 | SVQMKL-[DBF]- NleIVKLNA | <50 | 3 |
| DB19 | SVQMKL-[DBF]-IIVNleLNA | <50 | 15 |
| DB29 | SVQMNleL-[DBF]-IIVNleLNA | <50 | 20 |
| Norvaline substitution for Val, Met, Leu, or Ile (branched to linear alkyl) in 6DBF7 | |||
| DB25 | SNvaQMKL-[DBF]-IIVKLNA | >100 | 10 |
| DB21 | SVQNvaKL-[DBF]-IIVKLNA | >100 | 3 |
| DB22 | SVQMKNva-[DBF]-IIVKLNA | <50 | 4 |
| DB23 | SVQMKL-[DBF]- NvaIVKLNA | <50 | 3 |
| DB26 | SVQMKL-[DBF]-IIVKNvaNA | <50 | 5 |
| DB24 | SVQMKL-[DBF]-IINvaKLNA | <50 | 3 |
| DB27 | SVQMKL-[DBF]-IIVKLNNva | <50 | 4 |
| DB28 | SVQNvaKNva-[DBF]- NvaINvaKLNA | <50 | 5 |
We next investigated the ability of 6DBF7 analogs to inhibit EC proliferation, for assurance that biologic activity had been preserved. Dose-response curves for a few DBF-based compounds are exemplified in Fig. 2A, and IC50 values taken from the midpoints of these curves are given in Table 1. Although most DBF analogs were found to inhibit EC proliferation similar to 6DBF7, activities did vary, providing useful structure activity relationship information. For example, an alanine scan of 6DBF7 (DB1–DB13) showed that hydrophobic Val2 and positively charged Lys5 and Lys10 were crucial to 6DBF7 function, whereas bulky hydrophobic Met4 and negatively charged Asp13 actually contributed to functional attenuation (Fig. 2B). Interestingly, some multiple substitutions that improved solubility also led to enhanced inhibition of EC proliferation. From Table 1, it appeared that either DB16 or DB21 would be a good candidate for NMR studies with gal-1. However, upon increasing concentrations further, we found that DB16 was somewhat more soluble than DB21, and therefore, we chose DB16 for NMR studies with gal-1.
Fig. 2.

(A) EC proliferation data are exemplified with dose-response curves for DBF-based compounds 6DBF7, DB16, and DB21 as labeled. Proliferation of basic fibroblast growth factor–stimulated (10 ng/ml) HUVEC cultures was measured by colorimetric quantification. (B) EC proliferation results from alanine scanning of 6DBF7 are shown in bar graph format as the ratio of IC50 values (from Table 1) for DB analogs (DB1–DB13) divided by that of the parent 6DBF7. For reference, the chemical structure of 6DBF7 is shown as an inset.
Binding of DB16 to Gal-1.
Prior to NMR studies with gal-1, we first needed to know whether the lectin retained its native fold in the solution of 20 mM potassium phosphate buffer, pH 7, with 1% TFE, 1% glycerol, 2% NP40 (nonionic detergent), and 150 mM NaCl. Overlays of HSQC spectra of 15N-gal-1 (100 µM) acquired in this solution (red cross peaks) and that in 20 mM potassium phosphate buffer, pH 7 (black cross peaks), are presented in Supplemental Fig. 1. Note that although cross peaks of gal-1 in both solution conditions are chemically shifted, they are similarly dispersed, indicating that the new solvent mixture does not significantly affect native folding of gal-1. The most shifted resonances are associated with residues on the surface of the folded protein, whereas the more hydrophobic residues within the interior of the β-sandwich are minimally perturbed. Moreover, residues at the dimer interface are also minimally perturbed, suggesting preservation of the gal-1 dimer state. This is supported by PFG NMR self-diffusion measurements (Miller et al., 2009a), where the diffusion coefficient for gal-1 dimer (1.05 × 10−6 cm2/s) remains the same in either solution (unpublished data).
We next acquired 15N-1H HSQC spectra of 15N-gal-1 in the presence and absence of DB16. From our solubility studies, we found that the limit for DB16 solubility under optimal conditions was ~0.4 mg/ml (200 µM). Therefore, we used 30 µM gal-1 to achieve the highest DB16:gal-1 molar ratio of 6:1 during the titration. Above this ratio, we observed some precipitation, which worsened as more DB16 was added to the solution. Overlaid expansions of HSQC spectra of 15N-gal-1 in the presence (red cross peaks) and absence (black cross peaks) of DB16 at the DB16:gal-1 molar ratio of 6:1 are shown in Supplemental Fig. 2. In the presence of DB16, gal-1 resonances are differentially broadened and chemically shifted, an observation that, by itself, indicates interaction of DB16 with the lectin. The presence of resonance broadening, combined with minimal chemical shift changes, indicates that DB16 binding to gal-1 occurs in the intermediate exchange regimen on the NMR chemical shift time scale (Keeler, 2005), suggesting that the equilibrium ligand dissociation constant, Kd, should fall approximately in the 5–100 µM range. However, because ligand binding also resides in this exchange regimen, we cannot accurately determine the Kd value.
Figure 3A shows a resonance broadening map (Miller et al., 2009b) in which signal intensities between apo and bound states are compared. Changes in gal-1 resonance intensities induced by the presence of DB16 were calculated as fractional changes by taking the intensity of a given HSQC cross peak divided by that from the sample of gal-1 alone and subtracting that ratio from one (Miller et al., 2009b). A value of +1 indicates that resonance is no longer apparent, a value of zero indicates no change in resonance intensity, and a value of –1 indicates an increase in resonance intensity. The degree of resonance broadening or change in resonance intensity depends on several factors, which include the strength of ligand binding, chemical shift differences between deconvunbound and bound states, and changes in internal motions or conformational exchange induced by ligand binding. It is this last factor that provides the most reasonable explanation as to why several gal-1 resonances exhibit an increase in resonance intensity upon binding DB16, i.e., intensity changes are negative. Interestingly, residues affected in this fashion are all proximal, and most are hydrophobic and within the β-sandwich. Leu17, Val19, and Glu22 are part of β-strand 2 of the 5-stranded β-sheet at the back side of gal-1 opposite the carbohydrate binding face of the sandwich. β-Strands 9 (Cys88, Ile89), 8 (Leu96), and 11 (Asp134) are also part of that same β-sheet, and Gly4 (β-strand 1) and Ala121 (β-strand 10) are part of the opposing β-sheet in the sandwich. In these instances, we interpret negative changes in intensities to indicate that DB16 binding modulates conformational fluctuations primarily within the hydrophobic core of gal-1. However, because intensity changes are a weighted sum of the various factors mentioned previously, other sites are likely also similarly affected. We simply cannot deconvolute the net effect, because we do not know the weighting of each factor at each site.
Fig. 3.

(A) Plot of intensity changes (apparent resonance broadening) versus the gal-1 amino acid sequence. Changes in resonance intensities (ΔI) were calculated as fractional changes by subtracting from one the intensity of a given HSQC cross peak divided by that from the sample of gal-1 alone, as described by Miller et al. (2009b). A value of +1 indicates that that resonance is no longer apparent, a value of zero indicates no change in resonance intensity, and a value of –1 indicates an increase in resonance intensity. (B) Gal-1 structure (PDB 1GZW) showing DB16 interacting residues highlighted in color.
That said, it is reasonable to assume that the most broadened resonances most likely belong to residues with which DB16 interacts. This is especially true for a resonance broadening map developed at the initial stages of the titration (as plotted in Fig. 3A for a 1:2 gal-1:DB16 molar ratio). Gal-1 has a β-sandwich structure comprising 11 β-strands, with the front-face β-sheet (antiparallel β-strands 1, 3, 10, 4, 5, and 6) containing the lactose binding site and the back-face β-sheet comprising β-strands 7, 8, 9, 2, and 11. β-Strands 1 and 11 interact to form the gal-1 dimer interface. Figure 3B highlights sequences in the gal-1 dimer (PDB 1GZW) where the most broadened resonances are found and that therefore contain residues that most likely interact with DB16. For orientation, bound lactose molecules are shown in red. Two regions or sites (highlighted in yellow) in particular appear to be significantly affected by DB16 binding. The region with the most changes (labeled site 1 in Fig. 3B) includes residues in segments 10–14, 33–42, 70–80, 90–95,and 106–116 on one edge of the gal-1 sandwich. The second region (labeled site 2 in Fig. 3B) includes segments 23–30, 50–56, and 125–129 on the opposite edge of the sandwich. Note there are two such sites on each gal-1 dimer, one site each per monomer subunit, as shown in Fig. 3B. Although we cannot rule out that DB16 interacts to varying degrees at both sites, we favor site 1 as the DB16 binding region for several reasons. Site 1 includes most of the highly perturbed residues; site 1 is larger and more contiguous than site 2, and the amino acid composition within site 1 is more consistent with SAR information on 6DBF7 analogs (Table 1), where it appears that both positive change and hydrophobic character are important to 6DBF7 activity. Sites 1 and 2 contain three and one negatively charged residues, respectively, and site 1 has more surface-exposed hydrophobic residues than site 2. In this instance, effects observed at site 2 would be the result of conformational perturbations induced by binding of DB16 at site 1.
We should also note that residues at the gal-1 dimer interface are minimally perturbed by the presence of DB16, suggesting that DB16 binding does not significantly affect the gal-1 dimer state. This conclusion is supported by the observation that PFG NMR-derived diffusion coefficients (Miller et al., 2009a) for the gal-1 dimer (~1.05 × 10−6 cm2/s) are the same in the presence or absence of DB16 (unpublished data).
However, DB16 binding does appear to affect some residues at the gal-1 carbohydrate binding site (residues ~40–70), suggesting that DB16 may influence carbohydrate binding. In fact, we find that lactose binding affinity to gal-1 is attenuated in the presence of DB16. Figure 4 shows 15N-gal-1 (100 µM) HSQC-derived lactose binding curves acquired in the absence and presence of DB16 (gal-1:DB16 molar ratio of 1:6). For each of the four residues shown (Val76, Gln72, His52, and Cys16), curves acquired with gal-1 in the presence of DB16 (compared with those in the absence of DB16) are shifted to higher concentrations of lactose, indicating weaker ligand binding to the lectin. When corrected for the gal-1–bound ligand, the midpoint of these curves indicates the Kd value (free ligand concentration). When we then average individual Kd values for the top 20 lactose-induced gal-1 shifted resonances, we find that the average Kd value for binding of lactose to gal-1 alone is 405 ± 70 µM. This value is about 4 times larger than the Kd value in phosphate buffer alone (Nesmelova et al., 2010), indicating that the new solution itself reduces the affinity of lactose for gal-1. In the presence of DB16, the average Kd value is increased to 625 ± 80 µM, indicating an even further reduction in affinity of gal-1 for lactose due to binding of DB16.
Fig. 4.

Lactose binding to gal-1. 15N-gal-1 (100 µM) HSQC-derived lactose binding curves acquired in the absence and presence of DB16 (gal-1:DB16 molar ratio of 1:6) are shown for four residues [(A) Val76, (B) Gln72, (C) His52, and (D) Cys16]. When corrected for gal-1–bound ligand, the midpoint of these curves indicates the actual Kd value (free ligand concentration). Kd values for the top 20 lactose-induced gal-1–shifted resonances were averaged as discussed in the text.
Design of Improved DBF Analogs.
With knowledge of how DB16 interacts with gal-1, and our biologic readouts on 6DBF7 analogs DB1–DB16 (Table 1), we were able to design DBF analogs with improved biologic activities. As mentioned earlier (Table 1), three 6DBF7 alanine variants (V2A in DB2, K5A in DB5, and K10A in DB10) showed dramatically decreased activity, whereas five others (M4A in DB4; D13A in DB13; and combinations of M4A, I7A, and D13A in DB14–DB16) showed increased activity. Although it is apparent that a positive charge is crucial to activity, the structural insight (Fig. 3B) into how DB16 interacts with gal-1 suggests that it is also the hydrophobic face of the peptidomimetic that interacts with the surface of the lectin.
With this in mind, we made additional DB analogs (DB18–DB28; Table 1). We started with DB13 because it lacked the activity-attenuating, negatively charged C-terminal aspartate, which removes all negative charge from the peptide because the C termini in all 6DBF7 mimetics are amidated. From our insight into how DB16 interacts with gal-1, it appeared that reducing the bulky character of branched alkyl groups of valine, leucine, and isoleucine in DB analogs may promote better interactions with the protein surface. Therefore, we initially substituted key residues in DB13, one at a time, with either norleucine (Nle, DB18, DB19, DB30) or norvaline (Nva, DB21–DB27) in DB peptides shown in Table 1. From this series, we discovered that Nva substitutions in particular increased antiproliferation activity against ECs. Although DB25 showed no significant improvement in activity, DB26 showed a 2-fold enhancement over DB13. The greatest effect was seen in DB21, DB22, DB23, DB24, and DB27, which showed an approximate 3-fold increase in activity over DB13.
In two other DB13 analogs (DB18 and DB19), lysines were substituted individually to assess how removal of the primary amine group alone affected activity. Whereas substitution of Lys5 (DB18) showed no change in activity, substitution of Lys10 (DB19) decreased activity slightly. This SAR information indicated that the four methylene groups in either lysine may indeed contribute to interactions with the lectin, as reflected by essentially no change in activity, whereas alanine substitution (DB5 and DB10) produced significant decreases in activity. Moreover, when this is done, only one charged lysine appears to be necessary for full bioactivity. Another analog, DB28, which is a tetra-substituted analog of DB13, showed no further improvement in activity.
Using flow cytometry, we found that binding of gal-1 to cell surface glycans is attenuated in the presence of 6DBF7, DB16, or DB21. FITC-labeled gal-1 (0.1 µM) binding affinity for natural glycans on splenocytes is significantly attenuated as the concentration of these three DBF-based compounds is increased (Fig. 5). The effect is deconvoluted with respect to leukocytes (CD4+/CD8-/CD31- and CD8+/CD4-/CD31- cells) and endothelial cells (CD31+/ CD8-/CD4-). In all instances, 6DBF7, DB16, and DB21 effectively attenuate binding of FITC-gal-1 (0.1 µM set at 100% in the absence of compounds) to cell surface glycans in a dose-dependent manner. Between 1 and 100 nM of each compound, gal-1 binding is least affected, whereas at 10 µM of compound, gal-1 binding is mostly, if not fully, inhibited. DB21 was the most effective of the three analogs. For all three cell types, apparent IC50 values (taken at 50% binding inhibition) for DB21, 6DBF7, and DB16 are 0.3–1.2, 2–3, and 4–5 µM, respectively. Moreover, all three compounds (especially DB21) demonstrated their best effects on ECs, which is interesting because these compounds, similar to parent anginex, function primarily as antiangiogenic agents.
Fig. 5.

Flow cytometry data for 6DBF7, DB16, and DB21 competitive binding to splenocytes. Multicolor flow cytometry was used to assess the effects of 6DBF7, DB16, and DB21 on FITC-gal-1 binding to splenocytes [CD4+ (A) and CD8+ (B) leukocytes and CD31+ (C) endothelial cells] derived from gal-1−/− mice. Binding of 0.1 µM FITC-gal-1 to these cells was assessed as a function of concentration of 6DBF7, DB16, or DB21. Experiments were performed on an LSR II flow cytometer (BD Biosciences) and data were analyzed using Flowjo software (Tree Star, Inc.).
Effects of DB16 and DB21 in Mouse Tumor Models.
Based primarily on EC proliferation activities of our substituted DB13 analogs and their relatively good solubility profiles in aqueous solution (Table 1), we selected DB16 and DB21 for activity assessment in vivo. Prior to this, we assessed binding of DB21 to gal-1 using the same NMR approach described earlier. However, because the solubility of DB21 vis-à-vis DB16 was not as good at the concentrations needed for the NMR work, HSQC results were only acquired at lower concentrations and molar ratios of 15N-Gal-1 and DB21. Moreover, some minimal precipitation was noted with DB21 upon interacting with gal-1, causing a diminished signal-to-noise ratio as compared with DB16. Because of this, these data are not shown. Nevertheless, analysis of HSQC data at initial titration points did show the same spectral trends as observed with DB16. Therefore, we conclude that DB21 and DB16 interact similarly with gal-1. Additionally, we tested the antiproliferative effects of 6DBF7, DB16, DB21, and anginex on the tumor cell lines to be used in the tumor mouse models. We found that only DB16 showed an inhibitory effect of 30% at 10 µM on MA148 cells (no effect on B16F10 or LS174T), whereas the other compounds showed no inhibitory effect on any of these three tumor cell lines.
We first tested the activities of DB16 and DB21 in vivo in the B16F10 melanoma mouse model, a syngeneic model in immunocompetent mice. In brief, 100 μl of tumor cell suspension (2 × 106 B16F10 cells/ml) was injected subcutaneously into the right hind flank of each mouse. Tumors were allowed to grow to approximately 50 mm3, and treatment (i.p. injection twice daily for a total dose of 10 mg/kg/day for 5 days), was initiated on day 7 (Fig. 6A). All compounds were first dissolved in DMSO and subsequently diluted to a final concentration of 3.5% (v/v) DMSO in commercial saline. The control mice were administered the vehicle (3.5% DMSO in saline). On day 12, we observed that DB21 had inhibited tumor growth on average by 71%, whereas anginex, 6DBF7, and DB16 all inhibited growth by about 50%. For DB21-, 6DBF7-, and anginex-treated mice, Fig. 6B also shows immunohistochemistry on tumor tissue sections stained for CD31 (endothelial cell marker) and 4′,6-diamidino-2-phenylindole (cell nuclei marker). Compared with control tissue, DB21 had the greatest effect on reducing vessel density in tumors, as quantified in Fig. 6C by pixilation and averaging of 20 images of at least three different tumors for each treatment regimen. We also observed no sign of toxicity from any compound, as assessed by unaltered behavior, normal weight gain, hematocrit levels, and organ morphology. Weight gain and hematocrit levels from treatment with DB21 are shown in Fig. 6, D and E.
Fig. 6.
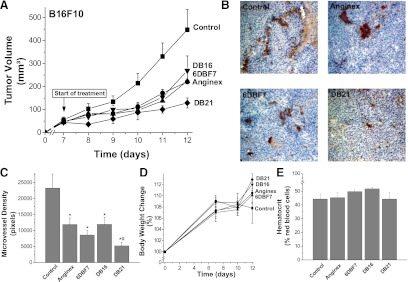
Effects of DB16 and DB21 in the B16F10 melanoma mouse model are shown. Tumors were allowed to grow to approximately 50 mm3, and treatment with DB16 and DB21, as well as with parent 6DBF7 and anginex as controls, was initiated on day 7 by i.p. injection twice daily for a total dose of 10 mg/kg/day for 5 days (A). General control groups of animals were treated with PBS alone. Tumor volumes (for all groups n = 10, ±S.E.M.) are plotted as mm3 versus days postinoculation. Tumor volumes were determined by measuring diameters of tumors using calipers, and using the equation for the volume of a spheroid, (a2 × b × Π) / 6, where “a” is the width and “b” the length of the tumor. Tumor weights correlated well with tumor volumes calculated in this way. For DB21-, 6DBF7-, and anginex-treated mice, (B) shows immunohistochemistry on tumor tissue sections stained for CD31 (endothelial cell marker) and DAPI (cell nuclei marker). Compared with control tissue, DB21 had the greatest effect on reducing vessel density in tumors, as quantified in (C) by pixilation and averaging of 20 images for each treatment regimen. Weight gain and hematocrit levels from treatment with DB21 are shown in panels (D) and (E), respectively.
To assess the effect of treatment on tumors growing in the absence of gal-1, we performed similar studies with DB21 in the B16F10 model using gal-1–null mice. However, tumor growth in these animals (control and treated) was greatly retarded, presumably due to the absence of gal-1, as we reported previously (Thijssen et al., 2006). Moreover, because of this, treatment with DB21 showed no significant effect in inhibiting tumor growth any further, also as essentially reported earlier for anginex treatment in gal-1–null mice (Thijssen et al., 2006).
We next tested DB16, DB21, and 6DBF7 in the LS174T lung cancer mouse model, another syngeneic model in immunocompetent mice. As with the B16F10 model, tumors were allowed to grow to approximately 50 mm3. In this model, treatment was initiated on day 8 by i.p. injection twice daily for a total dose of 10 mg/kg/day for 7 days (Fig. 7A). On day 20, we found that DB21 had inhibited tumor growth on average by about 80%, whereas 6DBF7 and DB16 inhibited growth by about 55 and 34%, respectively.
Fig. 7.

(A) DB16, DB21, and 6DBF7 were tested in the LS174T lung cancer mouse model. As with the B16F10 model, tumors were allowed to grow to approximately 50 mm3. Treatment was initiated on day 8 by i.p. injection twice daily for a total dose of 10 mg/kg/day for 7 days. A control group of animals was treated with PBS alone. (B) As a preliminary assessment of pharmacodynamics in mice, we used the slower-growing MA148 tumor model, in which tumors grew to about 70 mm3 in 35 days. Treatment was then initiated with DB21 in four groups of animals (n = 10 each): control; two injections on days 35 and 42; three injections on days 35, 40, and 45; and five injections on day 35, 38, 41, 44, and 47. Tumor volumes in (A) and (B) are plotted as cubic millimeters versus days postinoculation (for all groups n = 11, ±S.E.M.). Tumor volumes were determined by measuring diameters of tumors using calipers, and the equation for the volume of a spheroid, (a2 × b × Π) / 6, where “a” is the width and “b” the length of the tumor. Tumor weights correlated well with tumor volumes calculated in this way.
Because DB21 inhibited tumor growth the best in both syngeneic models, we wanted an initial assessment of the compound’s activity and pharmacodynamics in a xenograft model. For this study, we used the slower-growing human ovarian MA148 tumor model, in which tumors grew to about 70 mm3 in 35 days. Treatment was then initiated with DB21 in four groups of animals (n = 10 each): control; two injections on days 35 and 42; three injections on days 35, 40, and 45; and five injections on days 35, 38, 41, 44, and 47. Figure 7B shows that administration of DB21 every three or five days works equally well by inhibiting tumor growth on day 55 by about 75%. This level of inhibition is essentially the same as that from twice-daily injections in either the B16F10 or LS174T mouse model, suggesting that in vivo exposure and pharmacodynamic properties of DB21 are relatively good.
Discussion
Previously, we reported on 6DBF7, a DBF-based peptidomimetic of the angiostatic gal-1 antagonist anginex (Dings et al., 2003a; Mayo et al., 2003). 6DBF7, which is about one-third the mass of anginex, was shown to inhibit tumor growth in mice better than parent anginex. Although both anginex and 6DBF7 share the same spatial arrangements of functionally important amino acid residues in the context of antiparallel β-sheet structure (Dings et al., 2003a; Mayo et al., 2003; Arroyo and Mayo, 2007), it was unknown whether 6DBF7 targeted gal-1 similar to anginex. The previous results demonstrate that the DBF-based peptidomimetic indeed targets gal-1. Using heteronuclear HSQC NMR spectroscopy, we showed that DB16, a more aqueous soluble analog of 6DBF7, interacts with gal-1 primarily at one edge of the β-sandwich of the lectin. Moreover, by perturbing this network of residues, DB16 binding reduces the affinity of lactose to gal-1.
We also reported on structure-activity relationships and the design of 6DBF7 analogs with the enhanced ability to inhibit EC proliferation. For one, we found that positive charge, especially from Lys5 and less so from Lys10, is crucial to the activity of 6DBF7. Although an alanine scan of parent peptide anginex previously also suggested the functional importance of net positive charge (Dings et al., 2003a; Mayo et al., 2003), substitution effects were much less prominent than with 6DBF7, probably because anginex contains several lysine and arginine residues, whereby substitution of any one positively charged residue could be compensated for by the others. From this same study with anginex (Dings et al., 2003a; Mayo et al., 2003), however, it was evident that residues within the hydrophobic face of the folded amphipathic peptide were functionally crucial. Our alanine scan with 6DBF7 had a similar finding, which led us to take these SAR-based conclusions further by demonstrating that the presence of some branched alkyl side chains (Val, Leu, Ile) in fact attenuate activity. This was especially true for Val2, and minimally so for Leu6, Ile8, Val9, and Leu11, where substitution with linear alkyl groups enhanced activity to various extents.
NMR-derived insight into how DB16 interacts with gal-1 further supported the idea that it is the positive charge and hydrophobic character of 6DBF7 that promotes its activity. We found, for example, that there are several aspartate and glutamate groups in the region on gal-1 where DB16 more likely binds that could potentially interact electrostatically with lysine residues in DB16. Accordingly, substitution of the DB16 C-terminal aspartate for alanine, which removes all negative charge from the peptide, also resulted in increased activity (i.e., inhibition of EC proliferation). This is consistent with our working model in which the hydrophobic surface of the amphipathic peptidomimetic directly interfaces with the surface of gal-1, and cationic groups on the hydrophilic surface of the peptidomimetic interact electrostatically with negatively charged residues on the surface of gal-1.
Mechanistically, we hypothesized that DB16 binding to gal-1, even though at a remote site, would alter the strength of glycan binding to the lectin. In fact, HSQC titrations with lactose showed that gal-1 affinity for the disaccharide was reduced in the presence of DB16, supporting the notion that an allosteric effect was operative. Moreover, on the cellular level, our flow cytometry data indicated that binding of FITC-gal-1 to glycans on the surface of leukocytes (CD4+/CD8-/CD31- and CD4-/CD8+/CD31-) and ECs (CD4-/CD8-/CD31+) from gal-1−/−–null mouse splenocytes can be fully inhibited by binding of three DBF-based peptidomimetics (6DBF7 and analogs DB16 and DB21) in a dose-dependent manner. Apparent IC50 values for DB21, 6DBF7, and DB16 are 0.3–1.2, 2–3, and 4–5 µM, respectively. For DB16, this range is generally consistent with the range of apparent Kd values of ∼5 to 100 µM suggested by DB16-induced gal-1 resonance broadening. Overall, DBF-based peptidomimetics are noncompetitive, allosteric inhibitors of gal-1 function. A nonpeptide topomimetic of anginex, calix[4]arene compound 0118, was also shown to bind gal-1 in the same region as DB16 and function as an allosteric inhibitor of gal-1 glycan binding (Dings et al., 2012).
Gal-1, whose levels are elevated on ECs and stroma in several tumors (Gabius et al., 1986; Allen et al., 1990; Rabinovich, 2005), is known to promote EC adhesion and migration, and consequently tumor angiogenesis, on the molecular level by binding to β-galactoside groups of various cell surface glycans. Because allosteric inhibitor DB21, vis-à-vis 6DBF7 and DB16, shows the greatest inhibition of gal-1 binding to splenocyte ECs and the greatest inhibition of tumor angiogenesis, we can conclude that the targeting of gal-1 and antagonizing of its function directly results in attenuation of tumor angiogenesis in our mouse models. Most other known galectin antagonists are β-galactoside analogs and glycomimetics that target the canonical carbohydrate binding site, primarily to antagonize galectins 1, 3, 7, 8, and 9. These include aryl O- and S-galactosides and lactosides (Giguere et al., 2006; Sirois et al., 2006), carbohydrate-based triazoles and isoxazoles (Salameh et al., 2005; Giguere et al., 2006), O-galactosyl aldoximes (Tejler et al., 2005), phenyl thio-β-D-galactopyranoside analogs (Cumpstey et al., 2005a), thioureido N-acetyllactosamine derivatives (Salameh et al., 2006), talosides (Collins et al., 2012), and various multivalent sugar-based compounds (Cumpstey et al., 2005b; Ingrassia et al., 2006; Rabinovich et al., 2006; Tejler et al., 2006). Most of these compounds, unlike 6DBF7 and its analogs, bind galectins rather weakly (Kd values >100 µM). DB21 is currently being developed and holds promise for the management of human cancer in the clinic.
Supplementary Material
Abbreviations
- DBF
dibenzofuran
- DMSO
dimethylsulfoxide
- EC
endothelial cell
- FITC
fluorescein isothiocyanate
- Fmoc
fluorenylmethoxycarbonyl
- gal-1
galectin-1
- HPLC
high-performance liquid chromatography
- HSQC
heteronuclear single-quantum coherence
- HUVEC
human umbilical vein endothelial cell
- Kphos
potassium phosphate
- 15N-gal-1
15N-labeled recombinant gal-1
- PBS
phosphate-buffered saline
- PFG
pulse field gradient
- TFE
trifluoroethanol
Authorship Contributions
Participated in research design: Dings, Hoye, Mayo.
Conducted experiments: Dings, Kumar, Miller, Loren.
Contributed new reagents or analytic tools: Rangwala, Hoye.
Performed data analysis: Dings, Kumar, Miller, Hoye, Mayo.
Wrote or contributed to the writing of the manuscript: Dings, Hoye, Mayo.
Footnotes
This work was supported by a research grant from the National Institutes of Health National Cancer Institute [Grant R01 CA-096090] to K.H.M.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Allen HJ, Sucato D, Woynarowska B, Gottstine S, Sharma A, Bernacki RJ. (1990) Role of galaptin in ovarian carcinoma adhesion to extracellular matrix in vitro. J Cell Biochem 43:43–57 [DOI] [PubMed] [Google Scholar]
- Arroyo MM, Mayo KH. (2007) NMR solution structure of the angiostatic peptide anginex. Biochim Biophys Acta 1774:645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K, et al. (1994) Galectins: a family of animal beta-galactoside-binding lectins. Cell 76:597–598 [DOI] [PubMed] [Google Scholar]
- Bekele H, Nesloney CL, McWilliams KW, Zacharias NM, Chitnumsub P, Kelly JW. (1997) Improved Synthesis of the Boc and Fmoc Derivatives of 4-(2′-Aminoethyl)-6-dibenzofuranpropionic Acid: An Unnatural Amino Acid That Nucleates beta-Sheet Folding. J Org Chem 62:2259–2262 [DOI] [PubMed] [Google Scholar]
- Bondos SE, Bicknell A. (2003) Detection and prevention of protein aggregation before, during, and after purification. Anal Biochem 316:223–231 [DOI] [PubMed] [Google Scholar]
- Brandwijk RJ, Dings RP, van der Linden E, Mayo KH, Thijssen VL, Griffioen AW. (2006) Anti-angiogenesis and anti-tumor activity of recombinant anginex. Biochem Biophys Res Commun 349:1073–1078 [DOI] [PubMed] [Google Scholar]
- Carpino LA. (1993) 1-hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J Am Chem Soc 115:4397–4398 [Google Scholar]
- Chae YK, Im H, Zhao Q, Doelling JH, Vierstra RD, Markley JL. (2004) Prevention of aggregation after refolding by balanced stabilization-destabilization: production of the Arabidopsis thaliana protein APG8a (At4g21980) for NMR structure determination. Protein Expr Purif 34:280–283 [DOI] [PubMed] [Google Scholar]
- Collins PM, Oberg CT, Leffler H, Nilsson UJ, Blanchard H. (2012) Taloside inhibitors of galectin-1 and galectin-3. Chem Biol Drug Des 79:339–346 [DOI] [PubMed] [Google Scholar]
- Cumpstey I, Carlsson S, Leffler H, Nilsson UJ. (2005a) Synthesis of a phenyl thio-beta-D-galactopyranoside library from 1,5-difluoro-2,4-dinitrobenzene: discovery of efficient and selective monosaccharide inhibitors of galectin-7. Org Biomol Chem 3:1922–1932 [DOI] [PubMed] [Google Scholar]
- Cumpstey I, Sundin A, Leffler H, Nilsson UJ. (2005b) C2-symmetrical thiodigalactoside bis-benzamido derivatives as high-affinity inhibitors of galectin-3: efficient lectin inhibition through double arginine-arene interactions. Angew Chem Int Ed Engl 44:5110–5112 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293 [DOI] [PubMed] [Google Scholar]
- Dings RP, Arroyo MM, Lockwood NA, van Eijk LI, Haseman JR, Griffioen AW, Mayo KH. (2003a) Beta-sheet is the bioactive conformation of the anti-angiogenic anginex peptide. Biochem J 373:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings RP, Chen X, Hellebrekers DM, van Eijk LI, Zhang Y, Hoye TR, Griffioen AW, Mayo KH. (2006) Design of nonpeptidic topomimetics of antiangiogenic proteins with antitumor activities. J Natl Cancer Inst 98:932–936 [DOI] [PubMed] [Google Scholar]
- Dings RP, Loren M, Heun H, McNiel E, Griffioen AW, Mayo KH, Griffin RJ. (2007) Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clin Cancer Res 13:3395–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings RP, Mayo KH. (2007) A journey in structure-based drug discovery: from designed peptides to protein surface topomimetics as antibiotic and antiangiogenic agents. Acc Chem Res 40:1057–1065 [DOI] [PubMed] [Google Scholar]
- Dings RP, Miller MC, Nesmelova I, Astorgues-Xerri L, Kumar N, Serova M, Chen X, Raymond E, Hoye TR, Mayo KH. (2012) Antitumor agent calixarene 0118 targets human galectin-1 as an allosteric inhibitor of carbohydrate binding. J Med Chem 55:5121–5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings RP, van der Schaft DW, Hargittai B, Haseman J, Griffioen AW, Mayo KH. (2003b) Anti-tumor activity of the novel angiogenesis inhibitor anginex. Cancer Lett 194:55–66 [DOI] [PubMed] [Google Scholar]
- Dings RP, Van Laar ES, Loren M, Webber J, Zhang Y, Waters SJ, Macdonald JR, Mayo KH. (2010) Inhibiting tumor growth by targeting tumor vasculature with galectin-1 antagonist anginex conjugated to the cytotoxic acylfulvene, 6-hydroxylpropylacylfulvene. Bioconjug Chem 21:20–27 [DOI] [PubMed] [Google Scholar]
- Dings RP, Van Laar ES, Webber J, Zhang Y, Griffin RJ, Waters SJ, MacDonald JR, Mayo KH. (2008) Ovarian tumor growth regression using a combination of vascular targeting agents anginex or topomimetic 0118 and the chemotherapeutic irofulven. Cancer Lett 265:270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings RP, Vang KB, Castermans K, Popescu F, Zhang Y, Oude Egbrink MG, Mescher MF, Farrar MA, Griffioen AW, Mayo KH. (2011) Enhancement of T-cell-mediated antitumor response: angiostatic adjuvant to immunotherapy against cancer. Clin Cancer Res 17:3134–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings RP, Williams BW, Song CW, Griffioen AW, Mayo KH, Griffin RJ. (2005) Anginex synergizes with radiation therapy to inhibit tumor growth by radiosensitizing endothelial cells. Int J Cancer 115:312–319 [DOI] [PubMed] [Google Scholar]
- Dings RP, Yokoyama Y, Ramakrishnan S, Griffioen AW, Mayo KH. (2003c) The designed angiostatic peptide anginex synergistically improves chemotherapy and antiangiogenesis therapy with angiostatin. Cancer Res 63:382–385 [PubMed] [Google Scholar]
- Fischer C, Sanchez-Ruderisch H, Welzel M, Wiedenmann B, Sakai T, André S, Gabius HJ, Khachigian L, Detjen KM, Rosewicz S. (2005) Galectin-1 interacts with the alpha5beta1 fibronectin receptor to restrict carcinoma cell growth via induction of p21 and p27. J Biol Chem 280:37266–37277 [DOI] [PubMed] [Google Scholar]
- Gabius HJ, Brehler R, Schauer A, Cramer F. (1986) Localization of endogenous lectins in normal human breast, benign breast lesions and mammary carcinomas. Virchows Arch B Cell Pathol Incl Mol Pathol 52:107–115 [DOI] [PubMed] [Google Scholar]
- Gibbs SJ, Johnson CS. (1991) APFG NMR experiment for accurate diffusion and flow studies in the presence of eddy currents. J Magn Reson 93:395–402 [Google Scholar]
- Giguère D, Sato S, St-Pierre C, Sirois S, Roy R. (2006) Aryl O- and S-galactosides and lactosides as specific inhibitors of human galectins-1 and -3: role of electrostatic potential at O-3. Bioorg Med Chem Lett 16:1668–1672 [DOI] [PubMed] [Google Scholar]
- Griffioen AW, Damen CA, Mayo KH, Barendsz-Janson AF, Martinotti S, Blijham GH, Groenewegen G. (1999) Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int. J. Cancer 80:315–319 [DOI] [PubMed] [Google Scholar]
- Hermanson GT. (2008) Bioconjugate Techniques, Academic Press, Massachusetts [Google Scholar]
- Hungerford G, Benesch J, Mano JF, Reis RL. (2007) Effect of the labelling ratio on the photophysics of fluorescein isothiocyanate (FITC) conjugated to bovine serum albumin. Photochem Photobiol Sci 6:152–158 [DOI] [PubMed] [Google Scholar]
- Ilyina E, Roongta V, Mayo KH. (1997) NMR structure of a de novo designed, peptide 33mer with two distinct, compact beta-sheet folds. Biochemistry 36:5245–5250 [DOI] [PubMed] [Google Scholar]
- Ingle JD, Crouch SR. (1988) Spectrochemical Analysis, Prentice Hall, New Jersey [Google Scholar]
- Ingrassia L, Nshimyumukiza P, Dewelle J, Lefranc F, Wlodarczak L, Thomas S, Dielie G, Chiron C, Zedde C, Tisnès P, et al. (2006) A lactosylated steroid contributes in vivo therapeutic benefits in experimental models of mouse lymphoma and human glioblastoma. J Med Chem 49:1800–1807 [DOI] [PubMed] [Google Scholar]
- Johnson BA. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol 278:313–352 [DOI] [PubMed] [Google Scholar]
- Keeler J. (2005) Understanding NMR spectroscopy, John Wiley & Sons, West Sussex [Google Scholar]
- King DS, Fields CG, Fields GB. (1990) A cleavage method which minimizes side reactions following Fmoc solid phase peptide synthesis. Int J Pept Protein Res 36:255–266 [DOI] [PubMed] [Google Scholar]
- Lefranc F, Mathieu V, Kiss R. (2011) Galectin-1-mediated biochemical controls of melanoma and glioma aggressive behavior. World J Biol Chem 2:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FT, Patterson RJ, Wang JL. (2002) Intracellular functions of galectins. Biochim Biophys Acta 1572:263–273 [DOI] [PubMed] [Google Scholar]
- Liu FT, Rabinovich GA. (2005) Galectins as modulators of tumour progression. Nat Rev Cancer 5:29–41 [DOI] [PubMed] [Google Scholar]
- Mayo KH, Dings RP, Flader C, Nesmelova I, Hargittai B, van der Schaft DW, van Eijk LI, Walek D, Haseman J, Hoye TR, et al. (2003) Design of a partial peptide mimetic of anginex with antiangiogenic and anticancer activity. J Biol Chem 278:45746–45752 [DOI] [PubMed] [Google Scholar]
- Mayo KH, Ilyina E, Park H. (1996) A recipe for designing water-soluble, beta-sheet-forming peptides. Protein Sci 5:1301–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MC, Klyosov A, Mayo KH. (2009a) The alpha-galactomannan Davanat binds galectin-1 at a site different from the conventional galectin carbohydrate binding domain. Glycobiology 19:1034–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MC, Nesmelova IV, Platt D, Klyosov A, Mayo KH. (2009b) The carbohydrate-binding domain on galectin-1 is more extensive for a complex glycan than for simple saccharides: implications for galectin-glycan interactions at the cell surface. Biochem J 421:211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R. (1973) Self diffusion in normal and heavy water in the range 1-45°. J Phys Chem 77:685–688 [Google Scholar]
- Neri D, Bicknell R. (2005) Tumour vascular targeting. Nat Rev Cancer 5:436–446 [DOI] [PubMed] [Google Scholar]
- Nesmelova IV, Dings RPM, Mayo KH. (2008a) Understanding galectin structure-function relationships to design effective antagonists, in Galectins (Klyosov AA, Witczak ZJ, Platt D. eds) pp 33–69, John Wiley & Sons, Hoboken [Google Scholar]
- Nesmelova IV, Ermakova E, Daragan VA, Pang M, Menéndez M, Lagartera L, Solís D, Baum LG, Mayo KH. (2010) Lactose binding to galectin-1 modulates structural dynamics, increases conformational entropy, and occurs with apparent negative cooperativity. J Mol Biol 397:1209–1230 [DOI] [PubMed] [Google Scholar]
- Nesmelova IV, Pang M, Baum LG, Mayo KH. (2008b) 1H, 13C, and 15N backbone and side-chain chemical shift assignments for the 29 kDa human galectin-1 protein dimer. Biomol NMR Assign 2:203–205 [DOI] [PubMed] [Google Scholar]
- Rabinovich GA. (2005) Galectin-1 as a potential cancer target. Br J Cancer 92:1188–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Cumashi A, Bianco GA, Ciavardelli D, Iurisci I, D’Egidio M, Piccolo E, Tinari N, Nifantiev N, Iacobelli S. (2006) Synthetic lactulose amines: novel class of anticancer agents that induce tumor-cell apoptosis and inhibit galectin-mediated homotypic cell aggregation and endothelial cell morphogenesis. Glycobiology 16:210–220 [DOI] [PubMed] [Google Scholar]
- Salameh BA, Leffler H, Nilsson UJ. (2005) 3-(1,2,3-Triazol-1-yl)-1-thio-galactosides as small, efficient, and hydrolytically stable inhibitors of galectin-3. Bioorg Med Chem Lett 15:3344–3346 [DOI] [PubMed] [Google Scholar]
- Salameh BA, Sundin A, Leffler H, Nilsson UJ. (2006) Thioureido N-acetyllactosamine derivatives as potent galectin-7 and 9N inhibitors. Bioorg Med Chem 14:1215–1220 [DOI] [PubMed] [Google Scholar]
- Schumacher JJ, Dings RP, Cosin J, Subramanian IV, Auersperg N, Ramakrishnan S. (2007) Modulation of angiogenic phenotype alters tumorigenicity in rat ovarian epithelial cells. Cancer Res 67:3683–3690 [DOI] [PubMed] [Google Scholar]
- Sirois S, Giguère D, Roy R. (2006) A first QSAR model for galectin-3 glycomimetic inhibitors based on 3D docked structures. Med Chem 2:481–489 [DOI] [PubMed] [Google Scholar]
- Stejskal EO, Tanner JE. (1965) Spin diffusion measurements: spin echoes in the presence of a time-dependent field gradient. J Chem Phys 42:288–292 [Google Scholar]
- Takenaka Y, Fukumori T, Raz A. (2004) Galectin-3 and metastasis. Glycoconj J 19:543–549 [DOI] [PubMed] [Google Scholar]
- Tejler J, Leffler H, Nilsson UJ. (2005) Synthesis of O-galactosyl aldoximes as potent LacNAc-mimetic galectin-3 inhibitors. Bioorg Med Chem Lett 15:2343–2345 [DOI] [PubMed] [Google Scholar]
- Tejler J, Tullberg E, Frejd T, Leffler H, Nilsson UJ. (2006) Synthesis of multivalent lactose derivatives by 1,3-dipolar cycloadditions: selective galectin-1 inhibition. Carbohydr Res 341:1353–1362 [DOI] [PubMed] [Google Scholar]
- Thijssen VL, Postel R, Brandwijk RJ, Dings RP, Nesmelova I, Satijn S, Verhofstad N, Nakabeppu Y, Baum LG, Bakkers J, et al. (2006) Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci USA 103:15975–15980 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.