

Abstract
Amide carbonyl groups in proteins can engage in C=O···C=O and C–X···C=O interactions, where X is a halogen. The putative involvement of four poles suggests that these interactions are primarily dipolar. Our survey of crystal structures with a C–X···C=O contact that is short (i.e., within the sum of the X and C van der Waals radii) revealed no preferred C–X···C=O dihedral angle. Moreover, we found that structures with a short X−···C=O contact display the signatures of an n→π* interaction. We conclude that intimate interactions with carbonyl groups do not require a dipole.
INTRODUCTION
The amide carbonyl groups in the main chain of proteins are the foci for many noncovalent interactions. For example, this main-chain carbonyl group engages in a C=O···C=O interaction with another main-chain carbonyl group in common secondary structures.1 Many protein·ligand complexes are stabilized by analogous C–X···C=O interactions, where X is a halogen located on the ligand.2 Two well-known examples include a serine protease·inhibitor complex, which is stabilized by a C–F···C=O interaction,3 and the histamine N-methyltransferase·quinacrine complex, which is stabilized by a C–Cl···C=O interaction.4 The possible involvement of at least four poles in these interacting pairs has led to the proposal that such interactions are primarily dipolar.2 According to this argument, the functional group interacting favorably with the carbonyl group must have a dipole.
We were skeptical of this proposal for several reasons. In many interacting pairs, the van der Waals surface of the negative pole and that of the carbonyl carbon interpenetrate, forming a short contact. The ensuing orbital overlap places such interactions in the realm of quantum mechanics rather than classical electrostatics. In addition, a short contact is observed even when the interacting dipoles are not oriented favorably. For example, the two adjacent carbonyl dipoles are in a repulsive orientation in an α-helix.5 Yet, a short contact is observed between these carbonyl groups.1
An intimate C=O···C=O interaction can be modeled as n→π* interaction. This n→π* interaction involves delocalization of a lone pair (n) of the donor carbonyl group into the antibonding orbital (π*) of the acceptor carbonyl group. Evidence for n→π* interactions has been detected in small molecules,6 peptides, 7 peptoids, 8 proteins, 1,9 and nucleic acids, 10 they have been postulated to stabilize transition states.11 These interactions have three signatures. First, a short contact exists between the donor atom and the acceptor carbonyl carbon, allowing for orbital overlap. Secondly, the donor atom approaches the carbonyl group along the Bürgi–Dunitz trajectory,12 maximizing that overlap. Finally, this interaction pyramidalizes the acceptor carbonyl group.
We sought a means to determine whether or not a dipole was truly necessary for an intimate interaction with a carbonyl group. We reasoned that focusing on the interaction of a monopole with a carbonyl group could provide insight. Accordingly, we analyzed X−···C=O short contacts in detail (where X− is a halide ion), as well as C–X···C=O short contacts. The latter serve as surrogates for intimate C=O···C=O interactions, as both contain two interacting dipoles and have the potential for an n→π* interaction. Herein, we report on the results of these analyses.
RESULTS AND DISCUSSION
We began our analysis with a survey of C–X···C=O short contacts in crystal structures of the Cambridge Structural Database (CSD). The n→π* interaction, unlike the dipolar interaction, restricts the orientation of the negative but not the positive pole of the dipole that interacts with the carbonyl group. Typically, the negative pole of the dipole interacting with the carbonyl group is located along the Bürgi–Dunitz trajectory.12–13 Our analysis did not, however, reveal a preferred C–X···C=O dihedral angle (Figure 1). In accord with this finding, we have observed an orientational preference in O···C=O angle, but not C=O···C=O dihedral angle, in protein secondary structures with C=O···C=O interactions.7l Because there is no orientational restriction on the positive pole of the dipole that interacts with the carbonyl group, this result is contrary to the expectations from a meaningful dipole–dipole interaction.
Figure 1.
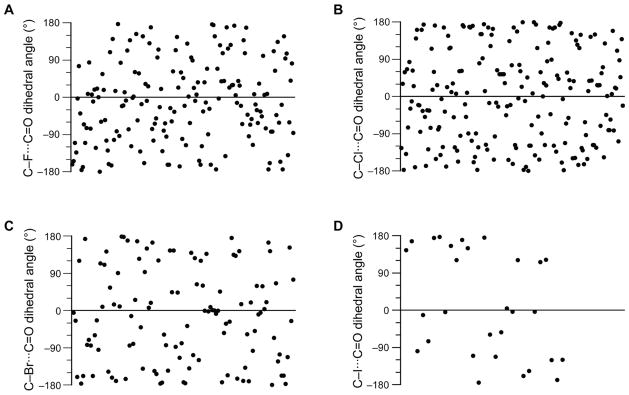
Values of C–X···C=O dihedral angles. The abscissa is used to distribute the values randomly. (A) X = F. (B) X = Cl. (C) X = Br. (D) X = I.
Next, we examined X−···C=O short contacts. Our premise was that the absence or presence of intimate interactions between halide ions (which are monopoles) and carbonyl groups would reveal whether or not a dipole is required for a favorable interaction with a carbonyl group. In addition, we reasoned that an examination of orientational restriction on the halide ions with respect to the carbonyl group in these interacting pairs would illuminate the nature of an X− ···C=O interaction. Towards this end, we sought small-molecule crystal structures containing short halide–carbonyl group contacts.
For fluoride, we found 4 structures that met these search criteria, containing 5 fluoride–carbonyl short contacts. Likewise, there were 110 structures for chloride with 130 short contacts, 22 structures for bromide with 27 short contacts, and 6 structures for iodide with 7 short contacts. For each of these short contacts, we determined the distance (d) and the angle (θ) of approach of the halide to the carbonyl (Figure 2).
Figure 2.

Definition of distance d, and angles θ and Θ.
The X− ···C=O angles between the halide donors and carbonyl acceptors fall mainly within the Bürgi–Dunitz trajectory (θ ~107°; Figure 3). Steric effects could contribute to an orientational preference. Accordingly, we re-examined the crystal structures for carbonyl pyramidalization, which can be measured by the parameter Θ (Figure 2). A positive Θ value indicates that the acceptor carbonyl oxygen is displaced away from the donor halide, while a negative value indicates that the displacement is toward the halide donor. In general, we found that Θ > 0 (Figure 4), consistent with the halide donating electron density into the carbonyl π* orbital in an n→π* interaction and thus pulling the carbonyl carbon out of its canonical planar geometry and towards the halide donor. To confirm that these trends in the θ and Θ values are indeed meaningful, we examined “control” structures with chloride ions located at 0.90–1.00 Å beyond the sum of the van der Waals radii of carbon and chlorine. We measured the distance, d, and the angles θ and Θ for these long Cl− ···C=O interactions. The values of d and θ indicate that the chloride ions are not necessarily located along the Bürgi–Dunitz trajectory (Figure 5A); moreover, the values of Θ are not indicative of carbonyl group pyramidalization towards the chloride ions (Figure 5B). We note that the location and effect of the halide could be altered in some structures by the accompanying cation (which maintains electrical neutrality).Moreover, crystal packing forces could have some influence on our observed geometrical preferences.14 We did not observe a strong correlation between d and Θ. The degree of pyramidalization, Θ, is a function of many variables besides X−···C=O distance. For example, the degree of pyramidalization depends on the Bürgi–Dunitz angle (θ) and the elasticity of the acceptor carbonyl group. For the same X−···C=O distance, the degree of pyramidalization for an amide carbonyl will be different from that of a ketone. The structures examined in our search vary not only in the nature of the carbonyl group, but also in the θ angle. Hence, we should not expect a strong correlation between the degree of pyramidalization and the X−···C=O distance.
Figure 3.

Plots of X−···C=O distance (d) and angle (θ). (A) X = F. (B) X = Cl. (C) X = Br. (D) X = I.
Figure 4.

Values of carbonyl group pyramidalization induced by a sub-van der Waals X− ···C=O interaction. The abscissa is used to distribute the values randomly. (A) X = F. (B) X = Cl. (C) X = Br. (D) X = I.
Figure 5.

Parameters for structures with a long Cl−···C=O interaction. (A) Plot of Cl−···C=O distance (d) and angle (θ). (B) Values of carbonyl group pyramidalization. The abscissa is used to distribute the values randomly.
Finally, we note that an n→π* interaction should not be confused15 with an n→π* electronic transition, which is distinct electronically and energetically (Figure 6). An n→π* electronic transition within a carbonyl group refers to the excitation of its non-bonded electron (n) to its π* orbital. An n→π* interaction, on the other hand, refers to electron delocalization between a donor atom and an acceptor carbonyl group. Moreover, an n→π* electronic transition requires the addition of energy to the system, whereas an n→π* interaction releases energy from the system.
Figure 6.

Orbital energy diagrams for two distinct electronic phenomena of carbonyl groups. (A) n→π* electronic transition of a single carbonyl group. The inset depicts the Zimmerman “circle-dot-y” notation for the ground state and excited state in which “o” represents an s-rich electron, “●” represents a π electron, and “y” represents a p-rich electron.16( B) n→π* interaction between two carbonyl groups.
CONCLUSION
We have argued previously that intimate interactions between carbonyl groups with interpenetrating van der Waals surfaces involve significant delocalization of an electron pair of the oxygen of the donor carbonyl group into the antibonding orbital (π*) of the acceptor carbonyl group. Our current findings support earlier ones,1,6–11 and suggest that these intimate interactions involving carbonyl groups do not require a dipole.
EXPERIMENTAL SECTION
ConQuest and Mercury software were used to search CSD version 5.31 (updated November 2009) for structures with a sub-van der Waals contact between a halide ion and a carbonyl group.13c Specifically, we searched for structures where the distance (d in Figure 2) between the halide and the carbonyl carbon was ≤3.17 Å, ≤3.45 Å, ≤3.55 Å, and ≤3.68 Å for fluoride, chloride, bromide, and iodide, respectively. All of these structures have R ≤ 5%, and none have any errors or disorder. In addition, none were polymeric, and none were organometallic or powder structures.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of our colleague, Howard E. Zimmerman (1926–2012). We thank G. J. Bartlett, C. N. Bradford, B. R. Caes, G. R. Krow, M. D. Shoulders, B. VanVeller, and D. N. Woolfson for assistance and contributive discussions. This work was supported by Grants R01 AR044276 (NIH) and CHE-1124944 (NSF). K.J.K. was supported by a Mary Shine Peterson Award from the Department of Biochemistry.
Footnotes
Data used to construct Figures 1 and 3–5. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bartlett GA, Choudhary A, Raines RT, Woolfson DN. Nat Chem Biol. 2010;6:615–620. doi: 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Olsen JA, Banner DW, Seiler P, Obst Sander U, D’Arcy A, Stihle M, Müller K, Diederich F. Angew Chem Int Ed. 2003;42:2507–2511. doi: 10.1002/anie.200351268. [DOI] [PubMed] [Google Scholar]; (b) Olsen JA, Banner DW, Seiler P, Wagner B, Tschopp T, Obst-Sander U, Kansy M, Müller K, Diederich F. ChemBioChem. 2004;5:666–675. doi: 10.1002/cbic.200300907. [DOI] [PubMed] [Google Scholar]; (c) Hof F, Scofield DM, Schweizer WB, Diederich F. Angew Chem Int Ed. 2004;43:5056–5059. doi: 10.1002/anie.200460781. [DOI] [PubMed] [Google Scholar]; (d) Paulini R, Müller K, Diederich F. Angew Chem Int Ed. 2005;44:1788–1805. doi: 10.1002/anie.200462213. [DOI] [PubMed] [Google Scholar]; (e) Fischer FR, Schweizer WB, Diederich F. Angew Chem Int Ed. 2007;46:8270–8273. doi: 10.1002/anie.200702497. [DOI] [PubMed] [Google Scholar]; (f) Bissantz C, Kuhn B, Stahl M. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adler M, Davey DD, Phillips GB, Kim SH, Jancarik J, Rumennik G, Light DR, Whitlow M. Biochemistry. 2000;39:12534–12542. doi: 10.1021/bi001477q. [DOI] [PubMed] [Google Scholar]
- 4.Horton JR, Sawada K, Nishibori M, Zhang X, Cheng X. Structure. 2001;9:837–849. doi: 10.1016/s0969-2126(01)00643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Perczel A, Angyan JG, Kajtar M, Viviani W, Rivail JL, Marcoccia JF, Csizmadia IG. J Am Chem Soc. 1991;113:6256–6265. [Google Scholar]; (b) Yang AS, Honig B. J Mol Biol. 1995;252:351–365. doi: 10.1006/jmbi.1995.0502. [DOI] [PubMed] [Google Scholar]; (c) Cantor CR, Schimmel PR. Biophysical Chemistry, Part III: The Behavior of Biological Macromolecules. Freeman and Company; New York, NY: 2004. [Google Scholar]
- 6.(a) Lesarri A, Cocinero EJ, López JC, Alonso JL. J Am Chem Soc. 2005;127:2572–2579. doi: 10.1021/ja045955m. [DOI] [PubMed] [Google Scholar]; (b) Blanco S, López JC, Mata S, Alonso JL. Angew Chem Int Ed. 2010;49:9187–9192. doi: 10.1002/anie.201002535. [DOI] [PubMed] [Google Scholar]; (c) Choudhary A, Kamer KJ, Raines RT. J Org Chem. 2011;76:7933–7937. doi: 10.1021/jo201389d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cabezas C, Alonso JL, López JC, Mata S. Angew Chem Int Ed. 2012;51:1375–1378. doi: 10.1002/anie.201106621. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bretscher LE, Jenkins CL, Taylor KM, DeRider ML, Raines RT. J Am Chem Soc. 2001;123:777–778. doi: 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]; (b) DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL. J Am Chem Soc. 2002;124:2497–2505. doi: 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]; (c) Hinderaker MP, Raines RT. Protein Sci. 2003;12:1188–1194. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jenkins CL, Lin G, Duo J, Rapolu D, Guzei IA, Raines RT, Krow GR. J Org Chem. 2004;69:8565–8573. doi: 10.1021/jo049242y. [DOI] [PubMed] [Google Scholar]; (e) Horng JC, Raines RT. Protein Sci. 2006;15:74–83. doi: 10.1110/ps.051779806. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hodges JA, Raines RT. Org Lett. 2006;8:4695–4697. doi: 10.1021/ol061569t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Sonntag LS, Schweizer S, Ochsenfeld C, Wennemers H. J Am Chem Soc. 2006;128:14697–14703. doi: 10.1021/ja0654938. [DOI] [PubMed] [Google Scholar]; (h) Kümin M, Sonntag LS, Wennemers H. J Am Chem Soc. 2007;129:466–467. doi: 10.1021/ja067148o. [DOI] [PubMed] [Google Scholar]; (i) Shoulders MD, Guzei IA, Raines RT. Biopolymers. 2008;89:443–454. doi: 10.1002/bip.20864. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Dai N, Wang XJ, Etzkorn FA. J Am Chem Soc. 2008;130:5396–5397. doi: 10.1021/ja711021m. [DOI] [PubMed] [Google Scholar]; (k) Chiang YC, Lin YJ, Horng JC. Protein Sci. 2009;18:1967–1977. doi: 10.1002/pro.208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Choudhary A, Gandla D, Krow GR, Raines RT. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Dai N, Etzkorn FA. J Am Chem Soc. 2009;131:13728–13732. doi: 10.1021/ja904177k. [DOI] [PubMed] [Google Scholar]; (n) Kuemin M, Schweizer S, Ochsenfeld C, Wennemers H. J Am Chem Soc. 2009;131:15474–15482. doi: 10.1021/ja906466q. [DOI] [PubMed] [Google Scholar]; (o) Choudhary A, Fry CG, Raines RT. ARKIVOC. 2010;8:251–262. [PMC free article] [PubMed] [Google Scholar]; (p) Kuemin M, Nagel YA, Schweizer S, Monnard FW, Ochsenfeld C, Wennemers H. Angew Chem Int Ed. 2010;49:6324–6327. doi: 10.1002/anie.201001851. [DOI] [PubMed] [Google Scholar]; (q) Jakobsche CE, Choudhary A, Miller SJ, Raines RT. J Am Chem Soc. 2010;132:6651–6653. doi: 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Choudhary A, Pua KH, Raines RT. Amino Acids. 2011;41:181–186. doi: 10.1007/s00726-010-0504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Choudhary A, Raines RT. Protein Sci. 2011;20:1077–1081. doi: 10.1002/pro.627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Pollock SB, Kent SBH. Chem Commun. 2011;47:2342–2344. doi: 10.1039/c0cc04120c. [DOI] [PubMed] [Google Scholar]; (u) Erdmann RS, Wennemers H. J Am Chem Soc. 2012;134:17117–17124. doi: 10.1021/ja3066418. [DOI] [PubMed] [Google Scholar]
- 8.(a) Gorske BC, Bastian BL, Geske GD, Blackwell HE. J Am Chem Soc. 2007;129:8928–8929. doi: 10.1021/ja071310l. [DOI] [PubMed] [Google Scholar]; (b) Gorske BC, Stringer JR, Bastian BL, Fowler SA, Blackwell HE. J Am Chem Soc. 2009;131:16555–16567. doi: 10.1021/ja907184g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Caumes C, Roy O, Faure S, Taillefumier C. J Am Chem Soc. 2012;134:9553–9556. doi: 10.1021/ja302342h. [DOI] [PubMed] [Google Scholar]
- 9.(a) Gao J, Kelly JW. Protein Sci. 2008;17:1096–1101. doi: 10.1110/ps.083439708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fufezan C. Proteins. 2010;78:2831–2838. doi: 10.1002/prot.22800. [DOI] [PubMed] [Google Scholar]; (c) Choudhary A, Kamer KJ, Raines RT. Protein Sci. 2012;21:171–177. doi: 10.1002/pro.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhary A, Kamer KJ, Powner MW, Sutherland JD, Raines RT. ACS Chem Biol. 2010;5:655–657. doi: 10.1021/cb100093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Liu P, Yang X, Birman VB, Houk KN. Org Lett. 2012;14:3288–3291. doi: 10.1021/ol301243f. [DOI] [PubMed] [Google Scholar]; (b) Wang H, Kohler P, Overman LE, Houk KN. J Am Chem Soc. 2012;134:16054–16058. doi: 10.1021/ja3075538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bürgi HB, Dunitz JD, Shefter E. Acta Cryst. 1974;B30:1517–1527. [Google Scholar]
- 13.(a) Allen FH. Acta Cryst. 2002;B58:380–388. [Google Scholar]; (b) Bruno IJ, Cole JC, Edgington PR, Kessler M, Macrae CF, McCabe P, Pearson J, Taylor R. Acta Cryst. 2002;B58:389–397. doi: 10.1107/s0108768102003324. [DOI] [PubMed] [Google Scholar]; (c) Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. J Appl Cryst. 2006;39:453–457. [Google Scholar]
- 14.(a) Dauber P, Hagler AT. Acc Chem Res. 1980;13:105–112. [Google Scholar]; (b) Dickerson RE, Goodsell DS, Neidle S. Proc Natl Acad Sci USA. 1994;91:3579–3583. doi: 10.1073/pnas.91.9.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Steiner T. Angew Chem Int Ed. 2002:48–76. [Google Scholar]
- 15.Worley B, Richard G, Harbison GS, Powers R. PLoSONE. 2012;7:e42075. doi: 10.1371/journal.pone.0042075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Zimmerman HE. 17th National Organic Symposium of the American Chemical Society, Abstracts; Bloomington, IN. 1961. pp. 31–41. [Google Scholar]; (b) Zimmerman HE. In: Advances in Photochemistry. Noyes WA, Hammond GS, Pitts JN, editors. Vol. 1. John Wiley; New York, NY: 1963. pp. 183–208. [Google Scholar]; (c) Zimmerman HE. Pure Appl Chem. 2006;78:2193–2203. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.