

Abstract
The vomeronasal organ (VNO) is an olfactory structure that detects pheromones and environmental cues. It consists of sensory neurons that express evolutionary unrelated groups of transmembrane chemoreceptors. The predominant V1R and V2R receptor repertoires are believed to detect airborne and water-soluble molecules, respectively. It has been suggested that the shift in habitat of early tetrapods from water to land is reflected by an increase in the ratio of V1R/V2R genes. Snakes, which have a very large VNO associated with a sophisticated tongue delivery system, are missing from this analysis. Here, we use RNA-seq and RNA in situ hybridization to study the diversity, evolution, and expression pattern of the corn snake vomeronasal receptor repertoires. Our analyses indicate that snakes and lizards retain an extremely limited number of V1R genes but exhibit a large number of V2R genes, including multiple lineages of reptile-specific and snake-specific expansions. We finally show that the peculiar bigenic pattern of V2R vomeronasal receptor gene transcription observed in mammals is conserved in squamate reptiles, hinting at an important but unknown functional role played by this expression strategy. Our results do not support the hypothesis that the shift to a vomeronasal receptor repertoire dominated by V1Rs in mammals reflects the evolutionary transition of early tetrapods from water to land. This study sheds light on the evolutionary dynamics of the vomeronasal receptor families in vertebrates and reveals how mammals and squamates differentially adapted the same ancestral vomeronasal repertoire to succeed in a terrestrial environment.
Keywords: vomeronasal organ (VNO), monogenic expression, evolution of sensorial abilities, squamates, snakes, phylogeny
Introduction
The vomeronasal organ (VNO), or Jacobson's organ, contains an olfactory sensory neuroepithelium enclosed in a cartilaginous or bony capsule in contact with the base of the nasal cavity. It plays a major role in interindividual interactions (through the detection of pheromones and kairomones) and environmental recognition (Houck 2009; Su et al. 2009). Chemosensory receptor proteins are present on the microvilli of vomeronasal sensory neuron dendrites and interact with molecules in the VNO cavity. The predominant chemosensory receptors in the vertebrate VNO come in three classes: formyl peptide receptors (FPRs), V1Rs, and V2Rs. These are evolutionarily unrelated families of seven-transmembrane (7TM) G-protein-coupled receptors (GPCRs) (Dulac and Axel 1995; Herrada and Dulac 1997; Matsunami and Buck 1997; Ryba and Tirindelli 1997). Previous analyses of fully sequenced vertebrate genomes (Grus and Zhang 2007) indicated de novo appearance and expansion of V1R and V2R subfamilies in some lineages, as well as the presence of a significant number of pseudogenes in others (Grus et al. 2005; Young et al. 2005; Shi and Zhang 2007; Young and Trask 2007). The remarkable divergence between V1R and V2R gene repertoires is thought to reflect the ecological niche and social habits of a particular species. Indeed, a large expansion of the V1R family accompanied by a sharp decline in the number of V2R genes was observed in mammals in comparison to Xenopus and fish species. Together with the notion that V1Rs and V2Rs might detect volatile and water-soluble molecules, respectively, these results led to the hypothesis that this shift, reflected by the ratio of V1R and V2R functional genes, corresponds to the evolutionary transition of early tetrapods from water to land (Shi and Zhang 2007).
Surprisingly, the vomeronasal chemosensory receptor gene repertoire of snakes, a vertebrate clade that possesses a very large VNO (Dawley 1998), has never been studied, probably because of the under-representation of nonmammalian species among fully sequenced vertebrate genomes (Milinkovitch and Tzika 2007; Milinkovitch et al. 2010). The VNO of snakes is associated with a sophisticated tongue delivery system: The tongue collects chemicals in the environment by means of tongue flicking and transfers them to the vomeronasal openings on the palate. Several neurological and behavioral studies demonstrated that snakes largely depend on their vomeronasal system for prey trailing, capture, and consumption, as well as for courtship and shelter selection (Miller and Gutzke 1999; Zuri and Halpern 2003; Huang et al. 2006). Components of the vomeronasal receptor signal transduction pathway, including several G proteins and the secondary messenger triphosphoinositol (IP3), have been identified in the garter snake VNO (Luo et al. 1994). In the same species, stimulations by prey-derived chemoattractants generate both transient activation of neurons in the vomeronasal sensory epithelium (Cinelli et al. 2002) and an increase in firing rates of individual neurons in the accessory olfactory bulb (AOB), that is, the projection site of vomeronasal sensory neurons in the brain (Jiang et al. 1990).
Recent technological advances allow for de novo sequencing and assembly of transcriptomes (RNA-seq), that is, even in the absence of reference genomes (Gibbons et al. 2009; Schwartz et al. 2010; Surget-Groba and Montoya-Burgos 2010; Tzika et al. 2011). Here, using RNA-seq and RNA in situ hybridization, we report on the diversity and evolution of the corn snake (Elaphe guttata, recently renamed Pantherophis guttatus) vomeronasal receptor repertoire. We found a large V2R repertoire composed of small, but multiple, snake-specific and reptile-specific expansions and only a few V1R receptor genes. We also show punctate expression of V2Rs in the corn snake vomeronasal neuroepithelium, an expression pattern compatible with that observed in mammals (Martini et al. 2001; Silvotti et al. 2007; Ishii and Mombaerts 2011). Our results refute the hypothesis that the distribution of vomeronasal receptors in vertebrates is dictated by the evolutionary transition from water to land (Shi and Zhang 2007) and shed light on the evolutionary dynamics of V1R and V2R families in vertebrates.
Materials and Methods
Animals
Corn snakes (Pantherophis guttatus) were obtained from our in-house breeding colony and maintained according to the Geneva Canton regulations (authorization 1008/3421/1R). All animals were sexually mature, unless indicated otherwise, and 2–5 years old.
Histology
The VNO of one male adult corn snake was dissected, fixed in 4% paraformaldehyde (PFA) overnight at 4 °C, and decalcified in 0.5 M ethylenediaminetetraacetic acid (EDTA) for 3 days at 4 °C. Seven-micrometer paraffin sections were stained with hematoxylin and eosin using standard procedures.
Labeling of VNO Epithelium by Retrograde Neuronal Tracing
The head of a juvenile male corn snake was fixed in 4% PFA for 4 h at 4 °C. The skull was opened and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil) crystals (Invitrogen) were placed on top of the AOB and the main olfactory bulb (MOB). The head was embedded in 4% low-melting agarose and placed in 4% PFA at 37 °C for 6 weeks before it was cut sagittally. The VNO was dissected, embedded in OCT (optimal cutting temperature embedding medium), and frozen on dry ice. Sixteen-micrometer coronal sections were prepared from cryoblocks, counterstained with Hoechst (Invitrogen), and mounted with Vectashield (Vector Labs).
cDNA Preparation and Deep Sequencing
Immediately after sacrifice of the animals, the VNO was dissected and stored in RNA-protective solution (PrepProtect, Miltenyi Biotech). Following tissue disruption with a Polytron device (Kinematica), mRNA was extracted using the μMACS kit (Miltenyi Biotech). After verification of mRNA integrity using a Bionanalyser (Agilent), mRNA was either directly submitted to sequencing, or cDNA was synthesized and DSN normalized (i.e., enriched for less abundant transcripts using Kamchatka crab duplex-specific nuclease) using published procedures (Zhulidov et al. 2004). Normalized cDNA samples were polymerase chain reaction (PCR)-amplified (30 cycles), and the 5′-polyT cDNA synthesis adaptor was removed using restriction enzymes. RNA samples and normalized cDNA samples, pooled or individually (supplementary fig. S1 and table S1, Supplementary Material online), were submitted to Roche/454 (Macrogen) and Illumina single-end sequencing (libraries were prepared according to standard protocols). The samples sequenced using Roche/454 were three males (M1, M2, and M3), pooled and not normalized; three females (F1, F2, and F3), pooled and normalized; and one juvenile female (F4), normalized. The samples sequenced using Illumina were three females (F1, F2, and F3), pooled and normalized, and one male (M4), not normalized.
Contig Assembly and Database Searches
The two types of reads (Roche, 200–500 bp, and Illumina, 114 bp) were assembled in two steps (see supplementary fig. S1 and table S1, Supplementary Material online).
Step 1: Preselection Assembly
Adapters removal and quality trimming as well as assembly of all Roche/454 reads (from M1–3, F1–3, and F4) were performed with SeqMan NGen v.2 (DNASTAR). Reads obtained from Illumina were subjected to adapter removal and quality trimming using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit, last accessed February 3, 2013) and assembled using Velvet with the Oases extension (Zerbino and Birney 2008). Optimal k-mer lengths (match length used by Velvet) may differ for transcripts with different abundances (Gibbons et al. 2009; Surget-Groba and Montoya-Burgos 2010). To maximize the chances of identifying VR transcripts, we applied an additive k-method as described in Surget-Groba and Montoya-Burgos (2010). K-mer values of 51, 61, 71, and 81 were used, followed by the removal of redundant contigs with CD-HIT-EST (Li and Godzik 2006). The ShortRead R package (www.r-project.org, last accessed February 3, 2013) was used for analyzing Illumina data (Morgan et al. 2009). Assembled Roche and Illumina contigs and unassembled Roche reads (singletons) with length above 200 bp were annotated using BLASTX, implemented in LANE runner (Tzika et al. 2011), with the following criteria: minimal match length 30 amino acids (aa), minimal sequence identity 50%, maximal E value 0.05, and release 60 of the Ensembl protein database for the following species: Anolis carolinensis (anole lizard), Danio rerio (zebrafish), Monodelphis domestica (opossum), Mus musculus (mouse), Ornithorhynchus anatinus (platypus), Rattus norvegicus (rat), and Xenopus tropicalis (western clawed frog). The anole lizard sequence that we identified as V1R (see below) was included in the search. Assembled Illumina contigs as well as assembled (contigs) and unassembled (singletons >200 bp) Roche/454 sequences are available at http://www.reptilian-transcriptomes.org (last accessed February 3, 2013) (supplementary files S6a and b, Supplementary Material online).
Step 2: Final Assembly
Reads contributing to contigs with a VR hit and singletons with a VR hit (in total 8,048 Roche and 54,191 Illumina reads) were pooled and assembled using SeqMan NGen v.2 (DNASTAR). Assembled contigs were annotated using LANE runner (Tzika et al. 2011) as described earlier. Although stringency of BLASTX search criteria was low, all the V2R contigs were identified with a very high confidence (E <10−9). Read alignments of potential V2R contigs were manually corrected for long homopolymer errors introduced by Roche/454 pyrosequencing. Potential VR transcript fragments were translated into aa sequences and aligned using the MAFFT multiple alignment program (Katoh et al. 2002). After manual editing of the alignment using JalView (Waterhouse et al. 2009), contigs spanning mostly the 3′-UTR or with stop codons in the coding part of the transcript were removed. The remaining set of 196 potentially functional V2R transcripts are listed and their sequences provided, in supplementary file S1, Supplementary Material online. Sequences with stop codons in the coding sequence are listed and their sequences provided, in supplementary file S2, Supplementary Material online. Visualization of contig statistics and properties (fig. 2) was performed using the R software (www.r-project.org, last accessed February 3, 2013).
Fig. 2.—

The V2R repertoire of the corn snake. (A) Length distribution of 196 corn snake V2R contigs. (B) Distribution of the average read depth coverage in relationship to the length of 196 corn snake V2R contigs (those used for phylogenetic analyses are marked in red). (C) Distribution of BLASTX E values in relationship to the length of 196 snake V2R contigs. (D) Identity statistics (based on segments of a multiple alignment of 196 snake V2R contigs) for aa and nt sequence variability analyses and estimation of the number of distinct V2R transcripts; see text for details. (E) Absolute and relative sizes of VR gene repertoires, based on the present (red frame) and published studies (Grus et al. 2007; Shi and Zhang 2007; Young and Trask 2007; Date-Ito et al. 2008; Hashiguchi et al. 2008; Ji et al. 2009; Young et al. 2010; Alfoldi et al. 2011).
Estimation of Transcript Number and Variability
To estimate the number of distinct V2R transcripts and their sequence variability, we used the 350 aa of the alignment, which correspond to the 9-cysteine domain and the 7TM domain. We divided the alignment into four segments (fig. 2D) and, within each segment, we counted the number of unique sequences among those with less than 50% missing data. We aligned the corresponding nucleotide sequences and counted the number of unique nucleotide sequences. To calculate sequence variability, we divided the aa alignment into eight segments and computed pairwise identity among sequences with no missing data.
PCR Validation of Assembled Contigs
PCR primer pairs were designed for each of the 25 selected contigs. cDNA samples from the VNO of one male and one female corn snakes were prepared as described earlier, pooled and used as templates for PCR. Products of the expected sizes were amplified for 24 of the 25 PCR assays and submitted to capillary Sanger sequencing: 23 of the 24 sequences could be aligned to the original corresponding contig. Percentages of nucleotide differences were calculated over the validated sequence length (supplementary fig. S2, Supplementary Material online). Sequences of the 23 primer pairs used for validation (amplification and sequencing) of the predicted sequences are listed in supplementary table S3, Supplementary Material online.
Potential Python V2R Genes
We performed Basic Local Alignment Search Tool (BLAST) analyses using our newly identified corn snake V2R transcripts as input, as well as V2Rs from other vertebrates, to detect V2R genes in the Burmese python (Python molurus bivittatus) draft genome (Castoe et al. 2011). Our TBLASTX analyses revealed the presence of 216 partial sequences of likely V2R genes that were also compared with the nonredundant National Center for Biotechnology Information (NCBI) database to confirm their annotation (supplementary file S3, Supplementary Material online). For each of these shotgun-sequenced fragments of the Python genome, the longest open reading frame (ORF) was identified using the ORF finder of NCBI (supplementary file S4, Supplementary Material online). Among these, 114 ORFs (longer than 140 aa) were selected and aligned with known V2Rs. This alignment indicated that the majority of the Python V2R sequences correspond to the variable N-terminus as only 34 aligned with the 7TM domain.
Phylogenetic Analyses
aa sequences of the 7TM region (265 aa) of 66 V2R corn snake contigs spanning at least 60% of the 7TM region were aligned with 67 representative V2R sequences of zebrafish, frog, anole lizard, mouse, and opossum using MAFFT (Katoh et al. 2002), followed by manual adjustment using JalView (Waterhouse et al. 2009). An alignment with the addition of 15 Python ORFs was also performed. GTR20 was selected as the best aa-substitution model using the Akaike Information Criterion implemented in MetaPIGA-v2.1 (http://www.metapiga.org (last accessed February 3, 2013) [Helaers and Milinkovitch 2010]). Maximum likelihood (ML) trees were computed using the Metapopulation Genetic Algorithm (MetaGA [Lemmon and Milinkovitch 2002]) implemented in MetaPIGA-v2.1 (Helaers and Milinkovitch 2010). We used probability consensus pruning among four populations of four individuals each and estimated posterior probability distribution of possible trees by performing replicated metaGA searches and stopping when the mean relative error values among 10 consecutive consensus trees remained below 2%. ML trees were also inferred using RaxML (v7.2.6) with 500 rapid bootstrap replicates (Stamatakis 2006). Finally, MC3 analyses under a Bayesian framework were performed using MrBayes (v3.2) (Huelsenbeck et al. 2001), and posterior probabilities were estimated after 2 million generations (burn in = 7,000 trees sampled every 100 generations). The RaxML topology is shown in figure 3 (branches with bootstrap support below 50% are collapsed), and RaxML, MetaPIGA, and MrByes branch supports are indicated above the branches of major clades.
Fig. 3.—

Evolutionary history of snake and other vertebrate V2Rs. ML tree, based on a multiple alignment of 7TM-domain aa sequences from 66 corn snake V2Rs and 67 representative V2R sequences from five vertebrate species. Branch support values (under RaxML/MetaPIGA/MrBayes) are indicated for major clades. Branches with <50% support values are collapsed. Pink boxes highlight five of the snake-specific expansions. Arrows indicate sequences used for in situ hybridization (figs. 4 and 5). The tree is rooted with taste receptors (TAS1Rs) as an outgroup. Scale bar: Mean number of aa substitutions per site.
Identification of Lizard and Snake V1R Sequences
Representative V1R nucleotide sequences of zebrafish, opossum, mouse, platypus, rat, and frog were used as input of TBLASTX queries against the anole genome (Ensembl release 64). This led to the identification (by “best-reciprocal hit”) of a single Anolis sequence (ENSACAG00000025768) as a V1R homolog. The presence of the corresponding sequence in the genomic DNA of the anole lizard was confirmed by PCR, followed by Sanger sequencing. Known V1R nucleotide sequences of various vertebrates along with the newly identified Anolis V1R gene were also compared (TBLASTX using LANE runner) with the fully sequenced genome of the Burmese python (Python molurus bivittatus), as well as the multiorgan transcriptome of the garter snake (Thamnophis sirtalis). Although no significant hit was retrieved from the queries against the garter snake database, two potential Python V1R sequences were identified as two independent shotgun-sequenced fragments of the species genome (Python V1r1—GenBank: AEQU010364851.1 and Python V1rb—GenBank: AEQU010376814.1; from the whole-genome shotgun sequencing project AEQU000000000.1). Searching our corn snake VNO transcriptome with TBLASTX did not reveal any V1R homologs. On the other hand, two slightly different copies of each Python V1ra1 and V1r2 were successfully amplified from Pantherophis guttatus genomic DNA using primers based on the Python sequences (supplementary file S7, Supplementary Material online). The distances of the two copies to each other and to several vertebrate V1Rs were computed using MetaPIGA-v2.1 (Helaers and Milinkovitch 2010), both for the nucleotide (with Jukes–Cantor correction) and the aa sequences (with Poisson correction). The lizard and snake V1Rs thus identified were translated and aligned with their mouse, fish, and frog homologs. The poorly aligned regions of the 438-aa MAFFT alignment were trimmed with the “strict” criterion of trimal (Capella-Gutierrez et al. 2009) implemented into MetaPIGA-v2.1 (Helaers and Milinkovitch 2010), and a final alignment of 230 aa per sequence was phylogenetically analyzed as described earlier for the V2R alignment.
mRNA In Situ Hybridization
Templates for probes were amplified (primers listed in supplementary table S3, Supplementary Material online) from a mixed sample of VNO cDNA from male and female corn snakes. Digoxigenin (DIG) and fluorescein (FLUO) probes were synthesized according to the DIG and FLUO RNA labeling kits protocols (Roche). Sixteen-micrometer cryosections were prepared from freshly frozen VNO of male and female corn snakes. Sections were fixed in 4% PFA for 20 min at room temperature. Single- and double-staining in situ hybridizations were performed using published protocols (Riviere et al. 2009). Hybridizations were carried out at 62 °C for 14 h. Anti-DIG antibody coupled to alkaline phosphatase (AP) and anti-FLUO antibody coupled to horseradish peroxidase (POD) were used (Roche). FastRed (Sigma) and biotinyl-tyramide (PerkinElmer) were used as substrates for AP and POD, respectively. Biotinyl-tyramide was detected by streptavidin-conjugated Alexa Fluor 488 (Invitrogen). Sections were counterstained with Hoechst (Invitrogen) and mounted with Vectashield (Vector Labs). Low-magnification images were taken with a standard fluorescent microscope (Zeiss). High-magnification images were taken with a confocal microscope (Leica). The number of cells expressing receptors and the total number of cells in the sensory epithelium were manually counted and used to calculate the percentage of expressing neurons in supplementary figure S5, Supplementary Material online.
Data Availability
All developed tools and supplementary files, Supplementary Material online, are available at http://www.reptilian-transcriptomes.org (last accessed February 3, 2013).
Results and Discussion
The Corn Snake VNO
The corn snake, suggested as one of the most convenient reptilian model species (Milinkovitch and Tzika 2007), was selected for the investigation of the vomeronasal chemoreceptor repertoire of snakes (fig. 1A and B). First, to identify the sensory part of the vomeronasal epithelium in the corn snake VNO, we retrogradely labeled sensory neurons by applying a lipophilic neuronal tracer (Godement et al. 1987) on the AOB of a fixed brain. Figure 1C–E indicates that the majority of cells present in the vomeronasal epithelium are neurons whose axons project to the AOB and whose dendrites protrude into the lumen of the VNO cavity. The corn snake VNO neuroepithelium is organized in columns (fig. 1B and E), like in other snake species (Wang and Halpern 1980; Taniguchi et al. 2000). The unlabeled cells in the basal zone of the epithelium likely represent neuronal stem cells. At the edges of the epithelium, we observe groups of labeled cells that possibly represent a pool of immature neurons, as their dendritic projections into the lumen are not labeled (arrowheads, fig. 1D).
Fig. 1.—

The VNO of the corn snake. (A) Corn snake. (B) Coronal section of a corn snake VNO stained with hematoxylin and eosin (HE). Full organ (left), scale bar 200 μm; close-ups of the columnar sensory epithelium (top right) and the zone in contact with the outside world (bottom right), scale bars 20 μm; SE, sensory epithelium; NE, nonsensory epithelium; L, lumen. (C) Schematic representation of a head hemi-section with a picture of the fluorescence detected in the VNO and the main olfactory epithelium (MOE) after application of a retrograde tracing dye onto the AOB and main olfactory bulb (MOB). (D,E) Coronal section of the VNO with fluorescence (red) detected after application of the tracing dye onto the AOB. Arrowheads and arrows in (D) indicate groups of potentially immature neurons at the edges and base, respectively, of the neuroepithelium; arrows and dotted frame in (E) indicate dendritic projections to the lumen. DNA is stained with Hoechst (blue). Scale bar in (D), 200 μm and in (E), 50 μm.
The Snake VNO Chemoreceptor Transcriptome
We isolated vomeronasal mRNA from four male and four female individuals, of which all but one female were sexually mature. Some of the produced cDNA samples were normalized to enrich for less abundant transcripts. We submitted two normalized and two non-normalized samples to Roche/454 and Illumina sequencing (see supplementary table S1, Supplementary Material online, for sequencing and assembly statistics). In total, we obtained 343,062 reads of 400 bp mean length (Roche/454) and 54,394,908 reads of 114 bp (Illumina). The high number of Illumina reads allowed for greater depth coverage, whereas the longer Roche/454 reads reduced the chances of chimeric assemblies. Following a two-step approach (supplementary fig. S1, Supplementary Material online), we first generated contigs separately from the two types of reads and identified (using BLASTX against all protein-coding sequences of seven vertebrate species) the snake contigs likely to encode for vomeronasal receptors. Second, the reads contributing to these contigs were pooled into a single set of 62,239 sequences that were assembled into 467 mixed-read contigs. Out of these, 196 contigs were identified as fragments of likely functional snake V2R transcripts based on high-confidence BLASTX similarity hits (E < 10−9) against V2R transcripts from other species (fig. 2C) and the absence of stop codons in the protein-coding part of the sequence (the 7TM and cysteine domains) (supplementary file S1, Supplementary Material online). We also identified 39 contigs representing putative pseudogenes as they included stop codons leading to a translation termination before the end of the seventh transmembrane domain (supplementary file S2, Supplementary Material online). We did not observe any large insertions or deletions in any of these putative V2Rs or pseudogenes. To validate our results, we selected 25 contigs for PCR amplification and sequencing. Among these, 92% (23/25) could be amplified, and their sequences were identical or nearly so with the corresponding target sequence (average percentage of nucleotide differences =0.012; supplementary fig. S2, Supplementary Material online). These few differences between target and amplified sequences were attributed to transcriptome sequencing errors, amplification of closely related gene family members, or polymorphisms between individuals used for sequencing and validation. Taken as a whole, our validation demonstrates that the majority of assembled contigs represent genuine transcripts.
Both the mean and median lengths of snake V2R contigs were approximately 500 bp (fig. 2A). Hence, most of them represent a fragment of a full transcript (the approximate mean length of a V2R coding sequence in vertebrates is 2,100 bp). V2R sequences corresponding to the N-terminus are under-represented in our data set for at least two reasons: 1) The low evolutionary conservation of the V2R N-terminal region makes homology assignment difficult using BLASTX searches against the genomes of other vertebrates and 2) cDNA libraries tend to be biased against 5′ sequences. To estimate the minimum number of distinct transcripts present in our assembly, we first translated the 196 putative V2R snake transcripts into aa sequences. Next, we generated multiple alignments of nucleotide (nt) and aa sequences with MAFFT (Katoh et al. 2002). We divided the alignments into four segments (of 228–279 nt or 76–93 aa). In each of these segments, we counted the number of unique sequences (fig. 2D); note the N-terminus high variability of V2R sequences. The third and fourth segments of the alignment, located in the more conserved 7TM domain, indicated that our 196 snake V2R sequences expressed in the corn snake VNO correspond to a minimum of 116 distinct transcripts, encoding 109 different proteins.
The suggestion of a large and variable V2R repertoire in snakes is also supported by our TBLASTX analyses (supplementary file S3, Supplementary Material online) against 1) the Burmese python (Python molurus bivittatus) draft genome (Castoe et al. 2011), in which we identified 216 partial sequences of potential V2R genes and 2) a garter snake (Thamnophis elegans) multiorgan transcriptome (from brain, gonad, heart, kidney, liver, spleen, and blood tissues but not olfactory tissue [Schwartz et al. 2010]) in which we identified 38 potential V2R genes.
Reptile-Specific Expansion of V2Rs
To infer the evolutionary history of snake V2Rs, we performed phylogenetic analyses of snake V2R aa sequences with homologs from various vertebrate species. A multiple alignment was built among a selection of 67 sequences from Anolis carolinensis, Mus musculus, Monodelphis domestica, Xenopus tropicalis, and Danio rerio, as well as 66 of our corn snake sequences (only sequences that spanned at least 60% of the 7TM domain were used; supplementary fig. S3, Supplementary Material online). Note that the majority of these corn snake sequences have an average depth coverage above 10 (fig. 2B). The consensus topology shown in figure 3 is supported by three different heuristics for large phylogeny inference: the metapopulation genetic algorithm (Lemmon and Milinkovitch 2002) implemented in metaPIGA-v2 (Helaers and Milinkovitch 2010), the rapid bootstrap analysis carried out with RaxML (Stamatakis 2006), and MC3 Bayesian estimation performed with MrBayes (Huelsenbeck et al. 2001). The gene-family tree includes the three well-described mammalian V2R subfamilies A, B, and D, as well as the more distantly related subfamily C (Yang et al. 2005; Grus et al. 2007; Young and Trask 2007). Our analyses indicate that family C is also present in nonmammalian vertebrates but is represented by a single member both in the anole lizard and the corn snake. The V2R gene tree also includes a large reptile-specific clade whose topology indicates that 1) a significant portion of the snake V2R repertoire arose before the split of this lineage from other squamates (i.e., several monophyletic groups include both corn snake and anole sequences) and 2) snakes experienced further evolution of their V2R repertoire but in the form of multiple small expansions (of which five are indicated in fig. 3). Given the short size of the V2R alignment (265 aa/sequence), increasing the number of sequences in the alignment will tend to reduce the robustness of branches during phylogeny inference. We anyway built a second alignment with the addition of 15 Python putative V2R fragments spanning at least 60% of the 7TM domain. Phylogenetic analyses of this larger data set (supplementary fig. S4, Supplementary Material online) yield a topology highly similar to that shown in figure 3, with Python sequences clustering with corn snake sequences (including one Python sequence in family C; supplementary fig. S4, Supplementary Material online).
It is most likely that the clear separation between mammalian, frog, and squamate non-C clades reflects phylogenetic signal rather than long-branch attraction or homogenization through gene conversion (Mallon et al. 2004; Ezawa et al. 2006). The latter hypothesis could be investigated through analysis of synteny conservation and patterns of sequence divergence among closely related paralogs (Sawyer 1999).
Expression Patterns of V2R Subfamilies Are Conserved among Vertebrates
The expression of snake V2R genes was investigated with in situ hybridizations on corn snake VNO sections (fig. 4) using probes corresponding to putatively functional V2R transcripts (indicated with arrows in fig. 3: V2R-EG001, EG107, EG108, and EG154). We also analyzed the expression pattern of one identified pseudogene (V2R-EG204). At least three snakes were analyzed for each receptor gene, and similar patterns of expression were observed among individuals (supplementary fig. S5, Supplementary Material online). All tested transcripts were detected in both males and females. For each of the five V2R genes, we detected expression in the vomeronasal sensory neuroepithelium (fig. 4) and not in the MOE. Similar to what has been reported for mice (Herrada and Dulac 1997; Matsunami and Buck 1997; Ryba and Tirindelli 1997), we observed punctate signals (e.g., V2R-EG107 and EG154) corresponding to single neuron expression, with a cytoplasmic localization of V2R mRNA. As observed in other vertebrate species, the probability of expression of a given V2R from non-C subfamilies is gene dependent. This probability is high for V2R-EG108 and lower for V2R-EG107/154/204 (figs. 4 and 5). We note that we cannot exclude that distinct, but very closely related, receptor transcripts were codetected by some probes.
Fig. 4.—
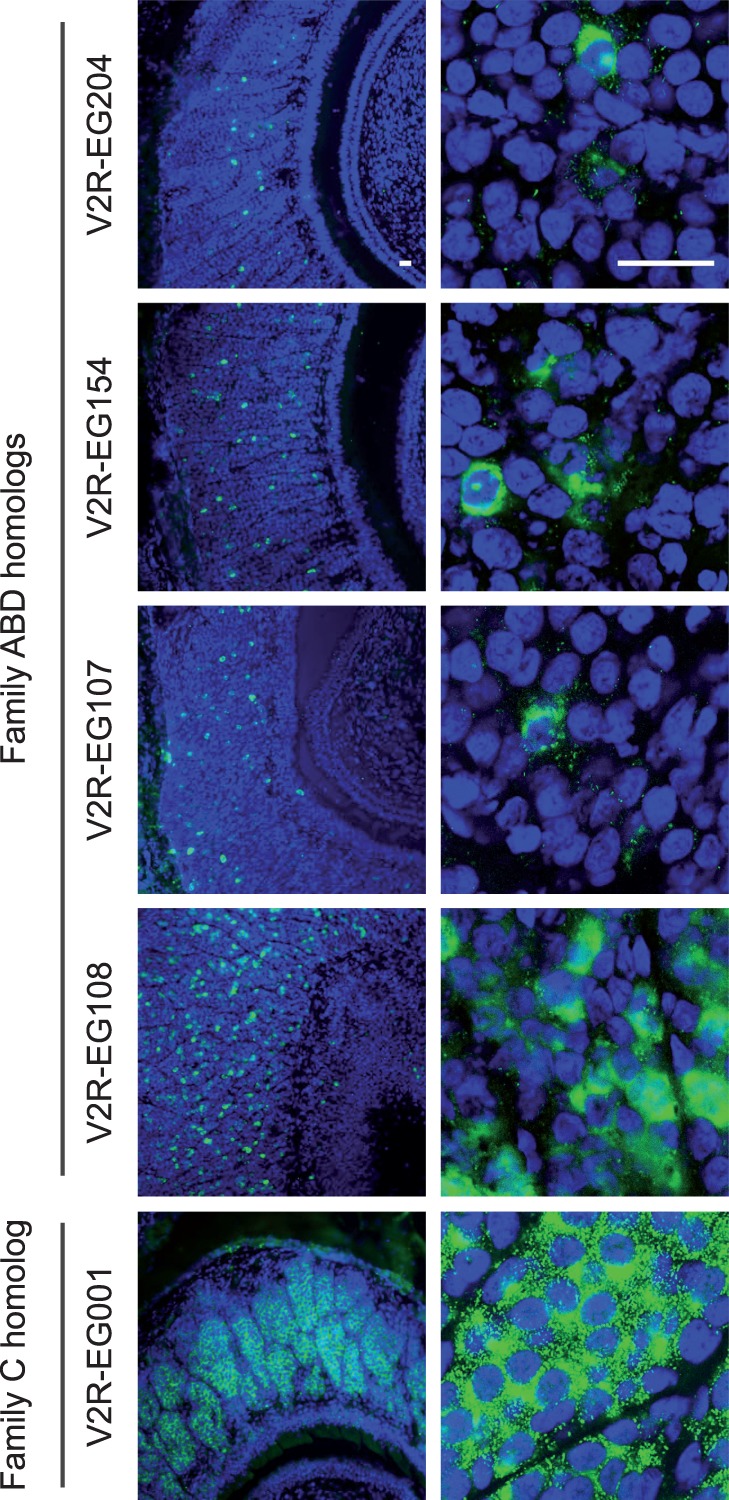
Expression of V2R transcripts in the snake vomeronasal sensory epithelium. In situ hybridizations (green) of coronal sections of a female corn snake VNO, with antisense RNA probes for one pseudogene (V2R-EG204) and four likely functional V2R transcripts. DNA is stained with Hoechst (blue). Scale bar, 20 μm.
Fig. 5.—
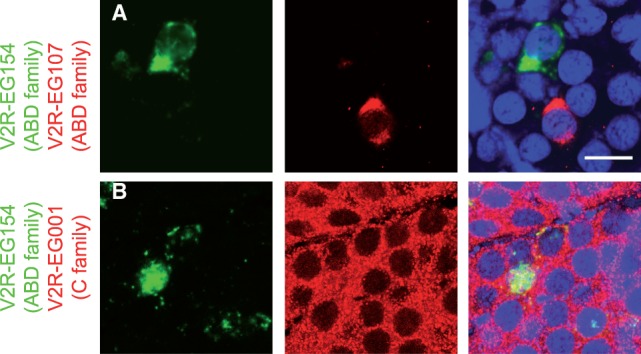
Monogenic versus nonmonogenic expression of V2Rs. In situ hybridizations with antisense RNA probes for (A) two ABD-subfamily members V2R-EG154 (green) and V2R-EG107 (red), (B) one C (V2R-EG001; red), and one ABD (V2R-EG154; green) subfamily members; coexpression of V2Rs is only observed between family C and family A/B/D members. Left and middle panels show single channels in green and red, and right panel shows the merge plus Hoechst staining of DNA in blue. Scale bar, 10 μm.
One striking feature of V2Rs in the mouse VNO is that subfamily-C members are broadly expressed and are coexpressed in the same cells with non-C (ABD) V2Rs (Martini et al. 2001; Silvotti et al. 2007; Ishii and Mombaerts 2011). The functional relevance of this peculiar pan- and coexpression pattern, if any, is unknown but could reflect that these receptors work as heterodimers with one monomer widely expressed and the other specifically expressed. Interestingly, we find that the corn snake subfamily-C gene (V2R-EG001) is expressed in the majority of the vomeronasal sensory neurons (fig. 4 and supplementary fig. S5, Supplementary Material online). To test for monogenic expression of non-C and C V2Rs, we performed double-staining in situ hybridizations. Figure 5A indicates that V2R-EG154 and V2R-EG107, that is, two close members of the non-C family of snake V2Rs (fig. 3), show mutually exclusive transcription. In contrast, the snake gene belonging to subfamily C (V2R-EG001) is coexpressed with members of non-C subfamilies, such as V2R-EG154 (fig. 5B).
V1R versus V2R Receptor Repertoires
No V1R sequences were found in our snake vomeronasal transcriptome, even under relaxed criteria of BLASTX similarity searches. To further investigate the potential lack of V1R genes in corn snakes and other Sauropsida reptiles, we performed TBLASTX searches against the fully sequenced genomes of the anole lizard (Ensembl release 64) and the Burmese python (Castoe et al. 2011), as well as the multiorgan transcriptome of the garter snake (Schwartz et al. 2010). Although no lizard V1R gene is predicted in the Ensembl database (whereas 39 genes are annotated as V2Rs), our analyses recognized as a V1R a single anole previously unidentified protein-coding sequence. We confirmed the presence of this sequence in the anole lizard genome by PCR amplification followed by sequencing. Our in silico analyses also identified two potential V1Rs (called here Python V1r1 and V1r2) in the Python draft genome, and their presence was confirmed by PCR amplification from Python genomic DNA. On the other hand, no significant match was obtained by searching the multiorgan garter snake transcriptome. We then designed PCR primers based on the Python V1R sequences that were used to probe the corn snake genomic DNA. This approach led to the identification of four V1R genes. Based on the distances among corn snake and Python sequences (supplementary table S2, Supplementary Material online), it is likely that these four corn snake sequences (called Pantherophis V1r1–4) correspond to two alleles or close paralogs for each of the two V1Rs.
We also performed phylogenetic analyses of a 230-aa data set including representatives of the 12 V1R mouse subfamilies (Rodriguez et al. 2002), the 19 known frog V1Rs (Date-Ito et al. 2008), V1Rs from teleost fish (Pfister et al. 2007), the detected anole lizard V1R along with additional fish and frog homologs (as provided by Ensembl release 66), and the Python and corn snake V1Rs identified above. The resulting ML phylogenetic tree indicates that the snake V1Rs (along with their orthologs from lower vertebrates) belong to two distant lineages of paralogs (fig. 6), one of which includes the anole V1r. Hence, the reptilian V1Rs are not reptilian acquisitions. In situ hybridizations of corn snake VNO sections revealed vomeronasal transcripts of Pantherophis V1r3 and 4 that were absent or present depending on the individual examined, whereas no Pantherophis V1r1 or 2 transcription was detected. The reason for this variability is unknown but may be correlated with seasonal expression because positive staining was observed during the mating period but not during hibernation (supplementary fig. S6, Supplementary Material online). Additional experiments are required to statistically determine whether this is true.
Fig. 6.—

Evolutionary history of snake and other vertebrate V1Rs. ML tree, based on a 230-aa multiple alignment of reptilian, mouse, frog, and fish V1Rs. The topology of the RaxML analyses is shown, and branch support values (under RaxML/MetaPIGA/MrBayes) are indicated for major clades. Branches with <50% support values are collapsed. The tree is unrooted and has been arbitrarily oriented for display purposes (there is no outgroup). The dashed arrow indicates the Anolis single V1R. Scale bar: Mean number of aa substitutions per site.
Furthermore, we searched our snake VNO transcriptome data sets for other GPCR classes. We did not find any sequences with significant similarity to trace amine-associated receptors (TAARs) (Liberles and Buck 2006) or FPRs (Riviere et al. 2009) both of which are expressed in the murine olfactory system. We, however, found several olfactory receptor (ORs) transcripts (data not shown) that may have originated from a minor contamination of our samples by snake MOE. To test this possibility, we used a Pantherophis OR probe (Locus_18943—supplementary file S5, Supplementary Material online), corresponding to a transcript that was identified in our sequence data set, and found punctate transcription in the MOE and no transcription in the VNO (data not shown), consistent with the idea of MOE contamination.
Taken together, our results strongly suggest that squamate reptiles (snakes and lizards) have a very limited repertoire of functional V1Rs. This makes the ratio of squamate V1R versus V2R sequences very similar to the ones observed in amphibians and teleosts, in contrast to mammals whose genomes encode more V1Rs than V2Rs (fig. 2E).
Conclusions
We report on the identification of the vomeronasal receptor repertoire in snakes (i.e., the lineage with the most developed VNO among all vertebrates), as well as the analysis of its expression pattern and evolutionary history. Our analyses demonstrate that 1) snakes exhibit a large V2R repertoire but a very limited number of V1R genes and 2) the peculiar V2R expression pattern observed in the VNO of mice (monogenic expression for V2R ABD-subfamily members and broad expression of C-subfamily members) is conserved in snakes.
Our deep-sequencing analyses suggest that the corn snake genome contains more than 116 V2R genes. Although we cannot exclude that some reconstructed transcripts combine polymorphisms among individuals, this number is likely to reflect a minimal estimation of the snake V2R repertoire because 1) we identified 196 high-quality partial V2R sequences that potentially represent distinct transcripts (thus making the snake V2R gene repertoire the second largest known after Xenopus), 2) we identified 216 potential V2R genes in the Python draft genome, 3) our transcriptome approach, contrary to genomic surveys, might have missed poorly transcribed and temporally regulated VR genes, and 4) some VR genes may be expressed in nonolfactory structures and be absent from olfactory neurons. Our phylogenetic analyses indicate the presence of reptile-specific and also of snake-specific clades of V2R genes (fig. 3) that may reflect specialization of specific snake receptors to particular ligands. This specialization is, however, unlikely to be a rule because electrophysiological recordings in garter snakes showed that individual neurons of the vomeronasal sensory epithelium respond to multiple classes of stimuli, including different peptides and aa (Inouchi et al. 1993). The snake vomeronasal neuroepithelium is elaborate and highly developed, and almost all VNO sensory neurons express V2Rs. One might have expected a correspondingly large chemoreceptor gene repertoire. In fact, we find that the snake repertoire is quite sizable but not significantly larger than the one found in rodents. This shows that the large size of the snake VNO is not the result of a need to host a larger chemoreceptor repertoire but suggests that this organ is under another selective pressure.
The snake large V2R repertoire is accompanied by a surprisingly small number (2–4) of V1R genes. Note that expression of a G-protein subunit specifically associated with V1R receptors in mammals (Gi2alpha) has previously been detected in vomeronasal epithelial cells of garter snakes (Luo et al. 1994). Our own analyses of the snake V2R expression patterns also support the existence of only a few snake vomeronasal sensory neurons devoted to the expression of V1R receptors because 1) in mice, V1Rs and V2Rs expressing neurons are distributed in distinct zones of the VNO and are never coexpressed in sensory neurons, 2) the expression patterns of V2Rs observed in rodents are largely conserved in snakes (figs. 4 and 5), and 3) a V2R C-subfamily homolog is expressed in nearly all cells of the snake VNO sensory epithelium (fig. 4, supplementary fig. S5, Supplementary Material online). The very limited number of V1R genes in the snake genome could be the result of two different evolutionary histories. The first one would be characterized by a small repertoire of V1Rs in the tetrapod ancestor followed by an expansion of V2Rs in squamates and an expansion of V1Rs in mammals, both after the split between the two lineages. A second potential history could involve a pre-existing large V1R repertoire that would have contracted in squamates and resulted in a few remnants today. This latter possibility should have left traces, that is, V1R pseudogenes in squamates. Our analysis of the Anolis genome, which did not reveal V1R pseudogenes, supports a history that lacked V1R expansions.
Our analyses of the snake VNO transcriptome elucidates how the repertoire of vomeronasal receptors evolved in different lineages of terrestrial vertebrates. Both fully aquatic and semiaquatic amphibians possess a well-developed VNO (Scalia et al. 1991; Eisthen 2000). Similar to mammals, they have large vomeronasal receptor repertoires, such as the frog Xenopus tropicalis, whose genome encodes 330 V2R and 21 V1R genes (Shi and Zhang 2007). Therefore, the common ancestor of extant amphibians and amniotes possessed a VNO and had both V1R and V2R receptors (Swaney and Keverne 2009). We detected four V1R-like sequences in the corn snake genome, two in the Python genome and a single V1R gene in the anole lizard genome. Therefore, contrary to the situation observed in mammals, the V1R repertoire did not expand in squamate reptiles. This is inconsistent with the hypothesis that the expansion of the V1R family arose in response to the transition of early tetrapods from detecting water-soluble to identifying air-borne ligands (Shi and Zhang 2007). Hence, mammals and squamate reptiles developed different strategies for the detection of ligands via their VNOs: In mammals, the number and diversity of the V1R family members are high (accompanied by a V2R repertoire of variable size), whereas squamate reptiles rely almost entirely on their V2R repertoire. Although the ability of mammals to detect airborne ligands (i.e., without contact with the source of the ligand) with their VNO remains controversial (Luo et al. 2003), we suggest that snake V2Rs are very likely able to detect airborne molecules because 1) our data show that the snake repertoire of vomeronasal receptors is largely dominated by V2Rs and 2) an increase in tongue flicking frequencies (delivering molecules to the VNO) has been observed in response to airborne chemical stimuli (Zuri and Halpern 2003). Therefore, we argue here that adaptation and expansion of the V2R repertoire in squamate reptiles for the detection of ligands was facilitated by the development, especially in snakes and allies, of a sophisticated tongue delivery system allowing for both volatile and nonvolatile molecules to efficiently reach the VNO.
Our phylogenetic analyses of V2R sequences, as well as our in situ hybridization experiments in corn snakes, indicate that the dichotomy between C-subfamily and ABD-subfamily members exists in all vertebrates investigated so far. Indeed, similar to what was previously reported in fishes, amphibians, and mammals (Yang et al. 2005), at least one family-C member is present in squamate reptiles as well. Our analyses further indicate that the expression pattern of the C-subfamily of V2Rs (expression in most VNO sensory neurons and coexpression with subfamily-ABD members) is conserved in squamate reptiles, hinting at an important functional role played by this coexpression strategy. This role is still elusive but could reflect a necessary heterodimerization for receptor function as is observed for other GPCRs.
In recent years, evolutionary dynamics of vomeronasal receptor repertoires have been extensively studied using fully sequenced mammalian genomes. Shifts in vomeronasal receptor repertoires have been associated with adaptations to different habitats and/or life histories, as well as to the development of social structures (Grus et al. 2005, 2007; Young et al. 2005; Young and Trask 2007; Wang et al. 2010). Our study opens new perspectives for the exploration of vomeronasal receptor repertoires in Sauropsida reptiles, a group for which an increasing number of new model species are being developed (Milinkovitch and Tzika 2007; Tzika and Milinkovitch 2007) and genomic/transcriptomic data are emerging (Schwartz et al. 2010; Alfoldi et al. 2011; Castoe et al. 2011; Tzika et al. 2011; St John et al. 2012). This vertebrate lineage includes Testudines (turtles), Lepidosauria (the tuatara and squamates), and Archosauria (crocodiles and birds). It comprises (after exclusion of the 10,000 species of birds) twice as many species as mammals, and it exhibits adaptations to very diverse habitats ranging from dry deserts to almost exclusive riverine or marine waters. Moreover, morphological, behavioral, and electrophysiological studies point to a broad usage of the vomeronasal system not only in snakes but also in lizards and turtles (Mason and Parker 2010). Evolutionary and functional analyses of vomeronasal receptor repertoires in multiple Sauropsidia reptiles will uncover how reptiles and mammals differentially adapted the same ancestral chemoreceptor toolkit to exploit the terrestrial environment.
Supplementary Material
Supplementary files S1–S7, tables S1–S3, and figures S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank the “NCCR Frontiers in Genetics” genomic platform for Illumina sequencing. They are grateful to Tom Bozza and Ueli Schibler for comments on the manuscript and to Adrien Debry for technical assistance. This work was supported by grants from the University of Geneva (Switzerland), the Swiss National Science Foundation (FNSNF, grant 31003A_125060), the Georges & Antoine Claraz Foundation, and the Ernst & Lucie Schmidheiny Foundation.
Literature Cited
- Alfoldi J, et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477:587–591. doi: 10.1038/nature10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoe TA, et al. Sequencing the genome of the Burmese python (Python molurus bivittatus) as a model for studying extreme adaptations in snakes. Genome Biol. 2011;12:406. doi: 10.1186/gb-2011-12-7-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinelli AR, Wang D, Chen P, Liu W, Halpern M. Calcium transients in the garter snake vomeronasal organ. J Neurophysiol. 2002;87:1449–1472. doi: 10.1152/jn.00651.2001. [DOI] [PubMed] [Google Scholar]
- Date-Ito A, Ohara H, Ichikawa M, Mori Y, Hagino-Yamagishi K. Xenopus V1R vomeronasal receptor family is expressed in the main olfactory system. Chem Senses. 2008;33:339–346. doi: 10.1093/chemse/bjm090. [DOI] [PubMed] [Google Scholar]
- Dawley EM. Species, sex, and seasonal differences in VNO size. Microsc Res Tech. 1998;41:506–518. doi: 10.1002/(SICI)1097-0029(19980615)41:6<506::AID-JEMT6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Dulac C, Axel R. A novel family of genes encoding putative pheromone receptors in mammals. Cell. 1995;83:195–206. doi: 10.1016/0092-8674(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Eisthen HL. Presence of the vomeronasal system in aquatic salamanders. Philos Trans R Soc Lond B Biol Sci. 2000;355:1209–1213. doi: 10.1098/rstb.2000.0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezawa K, Oota S, Saitou N. Proceedings of the SMBE Tri-National Young Investigators' Workshop 2005. Genome-wide search of gene conversions in duplicated genes of mouse and rat. Mol Biol Evol. 2006;23:927–940. doi: 10.1093/molbev/msj093. [DOI] [PubMed] [Google Scholar]
- Gibbons JG, et al. Benchmarking next-generation transcriptome sequencing for functional and evolutionary genomics. Mol Biol Evol. 2009;26:2731–2744. doi: 10.1093/molbev/msp188. [DOI] [PubMed] [Google Scholar]
- Godement P, Vanselow J, Thanos S, Bonhoeffer F. A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development. 1987;101:697–713. doi: 10.1242/dev.101.4.697. [DOI] [PubMed] [Google Scholar]
- Grus WE, Shi P, Zhang J. Largest vertebrate vomeronasal type 1 receptor gene repertoire in the semiaquatic platypus. Mol Biol Evol. 2007;24:2153–2157. doi: 10.1093/molbev/msm157. [DOI] [PubMed] [Google Scholar]
- Grus WE, Shi P, Zhang Y, Zhang J. Dramatic variation of the vomeronasal pheromone receptor gene repertoire among five orders of placental and marsupial mammals. Proc Natl Acad Sci U S A. 2005;102:5767–5772. doi: 10.1073/pnas.0501589102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grus WE, Zhang J. Origin and evolution of the vertebrate vomeronasal system viewed through system-specific genes. BioEssays. 2007;28:709–718. doi: 10.1002/bies.20432. [DOI] [PubMed] [Google Scholar]
- Hashiguchi Y, Furuta Y, Nishida M. Evolutionary patterns and selective pressures of odorant/pheromone receptor gene families in teleost fishes. PLoS One. 2008;3:e4083. doi: 10.1371/journal.pone.0004083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helaers R, Milinkovitch MC. MetaPIGA v2.0: maximum likelihood large phylogeny estimation using the metapopulation genetic algorithm and other stochastic heuristics. BMC Bioinformatics. 2010;11:379. doi: 10.1186/1471-2105-11-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrada G, Dulac C. A novel family of putative pheromone receptors in mammals with a topographically organized and sexually dimorphic distribution. Cell. 1997;90:763–773. doi: 10.1016/s0092-8674(00)80536-x. [DOI] [PubMed] [Google Scholar]
- Houck LD. Pheromone communication in amphibians and reptiles. Annu Rev Physiol. 2009;71:161–176. doi: 10.1146/annurev.physiol.010908.163134. [DOI] [PubMed] [Google Scholar]
- Huang G, Zhang J, Wang D, Mason RT, Halpern M. Female snake sex pheromone induces membrane responses in vomeronasal sensory neurons of male snakes. Chem Senses. 2006;31:521–529. doi: 10.1093/chemse/bjj056. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Inouchi J, Wang D, Jiang XC, Kubie J, Halpern M. Electrophysiological analysis of the nasal chemical senses in garter snakes. Brain Behav Evol. 1993;41:171–182. doi: 10.1159/000113835. [DOI] [PubMed] [Google Scholar]
- Ishii T, Mombaerts P. Coordinated coexpression of two vomeronasal receptor V2R genes per neuron in the mouse. Mol Cell Neurosci. 2011;46:397–408. doi: 10.1016/j.mcn.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Ji Y, Zhang Z, Hu Y. The repertoire of G-protein-coupled receptors in Xenopus tropicalis. BMC Genomics. 2009;10:263. doi: 10.1186/1471-2164-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XC, Inouchi J, Wang D, Halpern M. Purification and characterization of a chemoattractant from electric shock-induced earthworm secretion, its receptor binding, and signal transduction through the vomeronasal system of garter snakes. J Biol Chem. 1990;265:8736–8744. [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon AR, Milinkovitch MC. The metapopulation genetic algorithm: an efficient solution for the problem of large phylogeny estimation. Proc Natl Acad Sci U S A. 2002;99:10516–10521. doi: 10.1073/pnas.162224399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Liberles SD, Buck LB. A second class of chemosensory receptors in the olfactory epithelium. Nature. 2006;442:645–650. doi: 10.1038/nature05066. [DOI] [PubMed] [Google Scholar]
- Luo M, Fee MS, Katz LC. Encoding pheromonal signals in the accessory olfactory bulb of behaving mice. Science. 2003;299:1196–1201. doi: 10.1126/science.1082133. [DOI] [PubMed] [Google Scholar]
- Luo Y, Lu S, Chen P, Wang D, Halpern M. Identification of chemoattractant receptors and G-proteins in the vomeronasal system of garter snakes. J Biol Chem. 1994;269:16867–16877. [PubMed] [Google Scholar]
- Mallon AM, et al. Organization and evolution of a gene-rich region of the mouse genome: a 12.7-Mb region deleted in the Del(13)Svea36H mouse. Genome Res. 2004;14:1888–1901. doi: 10.1101/gr.2478604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini S, Silvotti L, Shirazi A, Ryba NJ, Tirindelli R. Co-expression of putative pheromone receptors in the sensory neurons of the vomeronasal organ. J Neurosci. 2001;21:843–848. doi: 10.1523/JNEUROSCI.21-03-00843.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RT, Parker MR. Social behavior and pheromonal communication in reptiles. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2010;196:729–749. doi: 10.1007/s00359-010-0551-3. [DOI] [PubMed] [Google Scholar]
- Matsunami H, Buck LB. A multigene family encoding a diverse array of putative pheromone receptors in mammals. Cell. 1997;90:775–784. doi: 10.1016/s0092-8674(00)80537-1. [DOI] [PubMed] [Google Scholar]
- Milinkovitch MC, Helaers R, Depiereux E, Tzika AC, Gabaldon T. 2x genomes—depth does matter. Genome Biol. 2010;11:R16. doi: 10.1186/gb-2010-11-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milinkovitch MC, Tzika AC. Escaping the mouse trap: the selection of new evo-devo model species. J Exp Zool B Mol Dev Evol. 2007;308B:337–346. doi: 10.1002/jez.b.21180. [DOI] [PubMed] [Google Scholar]
- Miller LR, Gutzke WHN. The role of the vomeronasal organ of crotalines (Reptilia: Serpentes: Viperidae) in predator detection. Anim Behav. 1999;58:53–57. doi: 10.1006/anbe.1999.1126. [DOI] [PubMed] [Google Scholar]
- Morgan M, et al. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–2608. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister P, Randall J, Montoya-Burgos JI, Rodriguez I. Divergent evolution among teleost V1r receptor genes. PLoS One. 2007;2:e379. doi: 10.1371/journal.pone.0000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere S, Challet L, Fluegge D, Spehr M, Rodriguez I. Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature. 2009;459:574–577. doi: 10.1038/nature08029. [DOI] [PubMed] [Google Scholar]
- Rodriguez I, Del Punta K, Rothman A, Ishii T, Mombaerts P. Multiple new and isolated families within the mouse superfamily of V1r vomeronasal receptors. Nat Neurosci. 2002;5:134–140. doi: 10.1038/nn795. [DOI] [PubMed] [Google Scholar]
- Ryba NJ, Tirindelli R. A new multigene family of putative pheromone receptors. Neuron. 1997;19:371–379. doi: 10.1016/s0896-6273(00)80946-0. [DOI] [PubMed] [Google Scholar]
- Sawyer SA. Department of Mathematics, Washington University in St. Louis. 1999. GENECONV: a computer package for the statistical detection of gene conversion. Available from: http://www.math.wustl.edu/∼sawyer (last accessed February 3, 2013) [Google Scholar]
- Scalia F, Gallousis G, Roca S. Differential projections of the main and accessory olfactory bulb in the frog. J Comp Neurol. 1991;305:443–461. doi: 10.1002/cne.903050308. [DOI] [PubMed] [Google Scholar]
- Schwartz TS, et al. A garter snake transcriptome: pyrosequencing, de novo assembly, and sex-specific differences. BMC Genomics. 2010;11:694. doi: 10.1186/1471-2164-11-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Zhang J. Comparative genomic analysis identifies an evolutionary shift of vomeronasal receptor gene repertoires in the vertebrate transition from water to land. Genome Res. 2007;17:166–174. doi: 10.1101/gr.6040007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvotti L, Moiani A, Gatti R, Tirindelli R. Combinatorial co-expression of pheromone receptors, V2Rs. J Neurochem. 2007;103:1753–1763. doi: 10.1111/j.1471-4159.2007.04877.x. [DOI] [PubMed] [Google Scholar]
- St John JA, et al. Sequencing three crocodilian genomes to illuminate the evolution of archosaurs and amniotes. Genome Biol. 2012;13:415. doi: 10.1186/gb-2012-13-1-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Su CY, Menuz K, Carlson JR. Olfactory perception: receptors, cells, and circuits. Cell. 2009;139:45–59. doi: 10.1016/j.cell.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20:1432–1440. doi: 10.1101/gr.103846.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney WT, Keverne EB. The evolution of pheromonal communication. Behav Brain Res. 2009;200:239–247. doi: 10.1016/j.bbr.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Wang D, Halpern M. Chemosensitive conductance and inositol 1,4,5-trisphosphate-induced conductance in snake vomeronasal receptor neurons. Chem Senses. 2000;25:67–76. doi: 10.1093/chemse/25.1.67. [DOI] [PubMed] [Google Scholar]
- Tzika A, Milinkovitch MC. A pragmatic approach for selecting evo-devo model species in amniotes. In: Minelli A, Fusco G, editors. Evolving pathways; key themes in evolutionary developmental biology. Cambridge (United Kingdom): Cambridge University Press; 2008. pp. 119–140. [Google Scholar]
- Tzika AC, Helaers R, Schramm G, Milinkovitch MC. Reptilian-transcriptome v1.0, a glimpse in the brain transcriptome of five divergent Sauropsida lineages and the phylogenetic position of turtles. Evodevo. 2011;2:19. doi: 10.1186/2041-9139-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Shi P, Zhu Z, Zhang YP. More functional V1R genes occur in nest-living and nocturnal terricolous mammals. Genome Biol Evol. 2010;2:277–283. doi: 10.1093/gbe/evq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RT, Halpern M. Scanning electron microscopic studies of the surface morphology of the vomeronasal epithelium and olfactory epithelium of garter snakes. Am J Anat. 1980;157:399–428. doi: 10.1002/aja.1001570408. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Shi P, Zhang YP, Zhang J. Composition and evolution of the V2r vomeronasal receptor gene repertoire in mice and rats. Genomics. 2005;86:306–315. doi: 10.1016/j.ygeno.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Young JM, Kambere M, Trask BJ, Lane RP. Divergent V1R repertoires in five species: amplification in rodents, decimation in primates, and a surprisingly small repertoire in dogs. Genome Res. 2005;15:231–240. doi: 10.1101/gr.3339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Massa HF, Hsu L, Trask BJ. Extreme variability among mammalian V1R gene families. Genome Res. 2010;20:10–18. doi: 10.1101/gr.098913.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Trask BJ. V2R gene families degenerated in primates, dog and cow, but expanded in opossum. Trends Genet. 2007;23:212–215. doi: 10.1016/j.tig.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhulidov PA, et al. Simple cDNA normalization using Kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32:e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuri I, Halpern M. Differential effects of lesions of the vomeronasal and olfactory nerves of garter snake (Thamnophis sirtalis) responses to airborne chemical stimuli. Behav Neurosci. 2003;117:169–183. doi: 10.1037//0735-7044.117.1.169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All developed tools and supplementary files, Supplementary Material online, are available at http://www.reptilian-transcriptomes.org (last accessed February 3, 2013).