

Abstract
Our previous reports showed that the cisplatin exposure induced the ATM-dependent phosphorylation of ΔNp63a, which is subsequently involved in transcriptional regulation of gene promoters encoding mRNAs and microRNAs in squamous cell carcinoma (SCC) cells upon cisplatin-induced cell death. We showed that phosphorylated (p)-ΔNp63a plays a role in upregulation of pro-apoptotic proteins, while non-p-ΔNp63a is implicated in pro-survival signaling. In contrast to non-p-ΔNp63a, p-ΔNp63a modulated expression of specific microRNAs in SCC cells exposed to cisplatin. These microRNAs were shown to attenuate the expression of several proteins involved in cell death/survival, suggesting the critical role for p-ΔNp63a in regulation of tumor cell resistance to cisplatin. Here, we studied the function of ΔNp63a in transcriptional activation and repression of the specific microRNA promoters whose expression is affected by cisplatin treatment of SCC cells. We quantitatively studied chromatin-associated proteins bound to tumor protein (TP) p63-responsive element, we found that p-ΔNp63a along with certain transcription coactivators (e.g., CARM1, KAT2B, TFAP2A, etc.) necessary to induce gene promoters for microRNAs (630 and 885-3p) or with transcription corepressors (e.g., EZH2, CTBP1, HDACs, etc.) needed to repress promoters for microRNAs (181a-5p, 374a-5p and 519a-3p) in SCC cells exposed to cisplatin.
Keywords: p53, p63, cisplatin, squamous cell carcinomas, DNA/protein interactions, microRNAs
Introduction
Acquired or intrinsic resistance of cells to the drug limits the use of cisplatin as an inducer of cell death in cancer chemotherapy.1,2 Cisplatin treatment induces DNA damage stress, oxidative and endoplasmic reticulum stresses.3 Several mechanisms are involved in cisplatin resistance, such as decreased intracellular drug accumulation and mismatch-repair activity and increased levels of cellular thiols and nucleotide excision-repair activity as well as altered expression of regulatory proteins involved in signal transduction pathways that control the cell death/cell survival pathways.3
Many genes are differentially expressed in sensitive and resistant tumor cells and involved in transcriptional regulation of mRNA and microRNA downstream targets implicated in DNA repairs and/or signal transduction, modulation of cell death and cell cycle arrest, and metabolomics.3-5 Among key molecules whose expression is altered in cisplatin-resistant cells as compared with cisplatin-sensitive cells are the tumor proteins (TP) 53, TP63 and TP73, proto-oncoprotein c-Myc, Y-BoX binding protein-1 (YBX1), CCAAT-binding nuclear factor (NF)-Y, activating transcription factor (ATF) 4 and 5, CLOCK, single-stranded recognition protein (SSRP)-1 and some others, which by functioning as transcription factors influence cellular sensitivity to cisplatin exposure.5-24 Loss of normal TP53 function or TP53 modifications confer resistance to cisplatin in various human cancer cells, while TP73 overexpression is associated with cisplatin resistance, and post-translational modifications of TP63 or lack thereof might contribute to tumor cell response to cisplatin exposure.10-20
The role for TP63 in cisplatin resistance is still under much scrutiny, and thereby its role in transcriptional regulation of specific genes implicated in cell survival, metabolomics and autophagy needs to be further examined. We previously reported that the exposure of squamous cell carcinoma (SCC) cells to cisplatin treatment induced the ataxia telangiectasia mutated (ATM) kinase-dependent production of phopshorylated (p)-Δ;Np63α. We further reported that p-Δ;Np63α and non-p-Δ;Np63α (Δ;Np63α-S385G protein, which is not phosphorylated by ATM kinase) differentially regulated transcription of mRNAs and microRNAs (miR) targets implicated in control of cell death and cell survival.20,25-28 Global analysis of cisplatin-modulated gene expression in sensitive SCC cells and resistant SCC cells revealed a number of genes shown to respond to p-Δ;Np63α or non-p-Δ;Np63α, which thereby are likely targets for cisplatin sensitivity or resistance.20 We then showed that both p-Δ;Np63α and non-p-Δ;Np63α are capable of differentially interacting with many proteins involved in signaling pathways of cell death/survival, including other transcription factors.29 Moreover, we found that sensitive SCC cells express a dramatically greater ratio of p-Δ;Np63α over non-p-Δ;Np63α than resistant SCC cells.30
Using the combined DNA pull-down/iTRAQ (isobaric tag for relative and absolute quantitation) approach allowing the global analysis of transcription factors and chromatin accessory proteins bound to specific promoter,31 we defined the critical components necessary to induce or repress Δ;Np63α-dependent gene expression in SCC cells upon cisplatin exposure.
Results
P-Δ;Np63α directly regulates the microRNA promoters in SCC cells upon cisplatin exposure
Our previous reports showed that the cisplatin-induced and ATM-dependent phosphorylation of Δ;Np63α led to a dramatic deregulation of microRNA transcription and processing in SCC cells.17,23 Specifically, a few microRNAs were downregulated [e.g., miR-181a-5p (181a), miR-374a-5p (374a) and miR-519a-3p (519a)], while a couple of microRNAs were upregulated (e.g., miR-630 and miR-885-3p) by p-Δ;Np63α. Our subsequent reports showed that selected microRNAs modulated the expression of proteins implicated in signaling leading to apoptosis, cell cycle arrest, autophagy and DNA damage response.24,25 However, the mechanistic nature of the p-Δ;Np63α -dependent transcriptional regulation leading to activation or repression of microRNA expression in SCC cells upon cisplatin exposure remains unclear.
To examine the effect of p-Δ;Np63α and non-p-Δ;Np63α on the expression of tested microRNAs, we exposed SCC-11 cells and SCC-11M cells to control medium and 10μg/ml cisplatin for 12h. Using quantitative (q)-PCR analysis we showed that cisplatin induced the downregulation of miR-181a-5p and miR-374a-5p and upregulation of miR-630 in SCC-11 cells (Fig. 1A). No such changes were found in SCC-11M cells under these experimental conditions (Fig. 1B). Moreover, the Δ;Np63α-S385G-FL forced expression in SCC-11 cells substantially modulated downregulation of miR-181a-5p and miR-374a-5p, or upregulation of miR-630 in spite of cisplatin treatment (Fig. 1A). However, the Δ;Np63α-FL forced expression in SCC-11M cells exposed to cisplatin mimicked the effect of the drug on SCC-11 cells (Fig. 1B), suggesting the critical role for the Δ;Np63α phosphorylation in regulation of tested microRNA levels. These data essentially confirmed our previous observations.23

Figure 1. P-Δ;Np63α regulated transcription of the specific microRNA promoters in SCC-11 cells upon cisplatin exposure. SCC-11 cells (A) and SCC-11M (B) cells were transfected with an empty vector and with the Δ;Np63α-S385G-FL or Δ;Np63α-wt-FL expression cassettes for 24 h, exposed to control media or 10 μg/ml cisplatin (CIS) for 12 h and then tested for specific microRNA expression using qPCR (A and B) qPCR experiments were performed in triplicate with +SD as indicated (< 0.05). (C) Resulting SCC-11 and SCC-11M cells were used for ChIP analysis to identify the binding of Δ;Np63α to the specific microRNA promoters. The amount of immunoprecipitated-enriched DNA in each sample (ChIP) is represented as signal relative to the total amount of input chromatin DNA (input) using the same primers for the specific promoter region. (D) SCC-11 cells and SCC-11M cells were also transfected with 100 ng of the control promoter-less pLightSwitch_Prom plasmid and the pLightSwitch_Prom plasmids containing promoter sequences of specific microRNAs (as indicated) and luciferase reporter gene as indicated. Cells were exposed to control medium without cisplatin (Con) and medium with 10 μg/ml cisplatin (CIS) for 12 h. RenSP Renilla luciferase reporter activity assays were conducted in triplicate (+SD are indicated, p < 0.05). Data presented as relative to data obtained from the control untreated cells containing the promoter-less reporter plasmid designated as 1.
Using chromatin immunoprecipitation (ChiP) analysis, we further tested the ability of p-Δ;Np63α to bind the selected microRNA promoters in both SCC-11 cells and SCC-11M cells exposed to cisplatin (Fig. 1C). We thus showed that the phosphorylation of Δ;Np63α is necessary for the binding to the specific microRNA promoters, since transfection of SCC-11 cells with the exogenous Δ;Np63α-S385G-FL construct dramatically attenuated this binding, while transfection of SCC-11M cells with the exogenous Δ;Np63α-FL construct substantially increased this binding (Fig. 1C).
We next tested, whether p-Δ;Np63α affects the luciferase gene expression driven by miR-181a-5p, miR-374a-5p and miR-630 gene promoters. As described in the Materials and Methods section, these custom-made constructs were introduced into both SCC-11 cells and SCC-11M cells subsequently exposed to 10μg/ml cisplatin for 12h (Fig. 1D). As control samples, the SCC-11 cells and SCC-11M cells transfected with an empty pLightSwitch_Prom, were used (Fig. 1D). In some experiments, SCC-11 cells were transfected with the exogenous Δ;Np63α-S385G-FL construct, while SCC-11M cells were transfected with the exogenous Δ;Np63α-FL construct (Fig. 1D). We showed that while the luciferase activities of pLightSwitch_miR-181a-5p-Prom and pLightSwitch_miR-374a-5p-Prom were downregulated, the luciferase activity of pLightSwitch_miR-630-Prom was upregulated in SCC-11 cells, but no changes were observed in SCC-11M cells (Fig. 1D). However, addition of Δ;Np63α-S385G-FL to SCC-11 cells increased the luciferase gene activity under miR-181a-5p and miR-374a-5p promoters and decreased the luciferase activity under miR-630 promoter (Fig. 1D). At the same time, addition of Δ;Np63α-FL to SCC-11M cells decreased the luciferase gene activity under miR-181a-5p and miR-374a-5p promoters and increased the luciferase activity under miR-630 promoter (Fig. 1D).
Interactome analysis of transcription factors bound to TP63-responsive elements in microRNA promoters
To examine what transcription factors and/or chromatin accessory proteins bound to microRNA promoters along with Δ;Np63α near or on TP63-responsive element, we employed the DNA/protein bound iTRAQ-labeled technology coupled with the liquid chromatography (LC)/double mass-spectrometry (MS/MS) analysis.28 SCC-11 cells (three samples) and SCC-11 M cells (two samples) were treated with 10 μg/ml cisplatin for 12 h, and then nuclear lysates were obtained from 5 × 109 cells, which were subsequently incubated with MagnaBind streptavidin beads with the 25nmol bead-bound complementary double-stranded oligonucleotides (50 base pairs) corresponding to the specific regions of the specific promoters for miR-181a-5p, miR-519a-3p, miR-374a-5p, miR-630 and miR-885-3p encompassing TP63-responsive element (Fig. 2; Fig. S1–5), as described in the “Supplemental Methods” section.

Figure 2. Various transcription factors bound to the specific microRNA promoters in SCC upon cisplatin exposure. SCC-11 cells and SCC-11M cells were exposed to 10 μg/ml cisplatin for 12 h. Nuclear lysates were incubated with the 50 bp sequences (locations are indicated in parentheses) derived from the specific microRNA promoters (miR-181a-5p, miR-519-3p, miR-374a-5p, miR-630 and miR-885-3p) containing the predicted TP63 binding site. Proteins bound to the tested sequences were analyzed by iTRAQ (LS/MS/MS) and validated by immunoblotting assays with the indicated antibodies.
We quantitatively compared the TP63-bound protein complexes in SCC-11 cells or SCC-11M cells exposed to cisplatin, as previously described.26 For iTRAQ labeling, 200 μg of eluted protein from large-scale DNA pull-downs were digested with trypsin, and peptides were purified and labeled with the following iTRAQ reagents: products isolated from SCC-11 cells exposed to 10μg/ml cisplatin were labeled with iTRAQ reagents (115, 116 and 117), whereas products isolated from SCC-11M cells exposed to cisplatin were labeled with iTRAQ reagents (113 and 114), respectively. The specific mixes were fractionated by strong cationic exchange high-pressure LC followed by MS/MS analysis, as previously described. Among proteins bound to TP63-responsive element that met the stringent statistical criteria (e.g., iTRAQ ratio > 1.25 and < 0.75), we identified 38 proteins [iTRAQ ratios ranged from 0.219 (depleted) to 8.854 (enriched), Tables S1–5]. As shown, the miR-181a-5p promoter sequence was enriched with 12 proteins, and depleted with three proteins, while the miR-519a-3p and miR-374a-5p promoter sequences were enriched with 12 and 10 proteins, respectively (and depleted with two and five proteins, respectively) in SCC-11 cells compared with SCC-11M cells (Table S1–3). At the same time, the miR-630 and miR-885-3p promoter sequences were enriched with 13 and 12 proteins, respectively (and depleted with five and four proteins, respectively) in SCC-11 cells compared with SCC-11M cells (Table S4 and S5). To confirm data obtained from the quantitative MS analysis, we performed immunoblotting assays with proteins bound to TP63-responsive elements in SCC-11 cells compared with SCC-11M cells exposed to cisplatin treatment. We showed that many proteins identified by quantitative MS are indeed differentially bound to TP63-responsive elements found in the selected microRNA promoters (Fig. 2).
In total ~18 proteins might be potentially involved in transcription repression of the selected microRNA promoters (miR-181a-5p, miR-519-3p and miR-374a-5p; Tables S1–S3), while totally ~16 proteins could play a role in transcription activation of the microRNA promoters (miR-630 and miR-885-3p; Tables S4 and S5) in SCC-11 cells exposed to cisplatin (Fig. 3). At the same time, we found 10 proteins that might function in transcription activation, while eight proteins are likely to act as transcriptional repressors in SCC-11M cells exposed to cisplatin (Fig. 4).
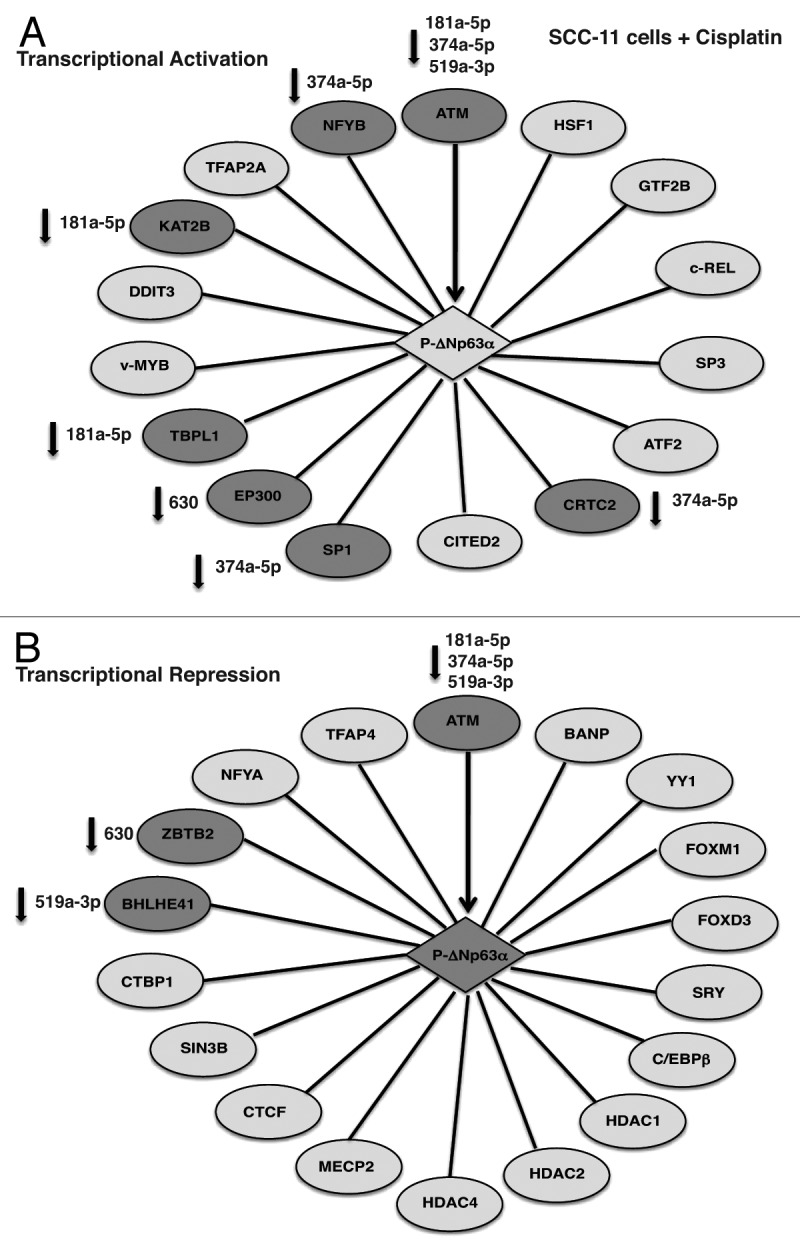
Figure 3. Schematic representation of p-Δ;Np63α-dependent protein interaction network involved in transcriptional activation (A) or repression (B) of the specific microRNA promoters in SCC-11 cells upon cisplatin exposure. Proteins that are potentially affected by specific microRNAs highlighted in darker shades and specific microRNAs indicated.

Figure 4. Schematic representation of Δ;Np63α-dependent protein interaction network involved in transcriptional activation (A) or repression (B) of the specific microRNA promoters in SCC-11M cells upon cisplatin exposure. Proteins that are potentially affected by specific microRNAs highlighted in darker shades and specific microRNAs indicated.
Notably, in addition to p-Δ;Np63α (known to be phosphorylated by ATM kinase), which essentially recognized TP63-responsive element in the tested microRNA promoters, the following proteins were a part of the complex that support activation of the miR-630 and miR-885-3p promoters: heat shock factor 1 (HSF1), general transcription factor 2B (GTF2B), proto-oncogene c-REL, specificity factors SP1 and 3, activating transcription factor 2 (ATF2), CREB-regulated transcription coactivator 2 (CRTC2), Cbp/p300-interacting transactivator 2 (CITED2), specificity factor (SP1 and 3), E1A-binding protein, p300 (EP300), TATA-binding protein-like 1 (TBPL1), proto-oncogene c-MYB, DNA damage-inducible transcript 3 (DDIT3), K(lysine) acetyltransferase 2B (KAT2B, PCAF), transcription factor activation protein 2A (TFAP2A) and nuclear factor YB (NFYB) as shown in Figure 3A. At the same time, the following proteins took a part in the repression complex for the miR-181a-5p, miR-519a-3p and miR-374a-5p promoters: BTG3-associated nuclear protein (BANP, SMAR1), Yin and Yang 1 (YY1), Forkhead box M1 FOXM1, FOXD3, sex-determining region Y SRY, CCAAT/enhancer binding protein β (C/EBPβ), histone deacetylase (HDAC1, HDAC2, HDAC4), methyl CpG binding protein 2 (MECP2), CCCTC-binding factor (CTCF), SIN3 transcription regulator homolog B (SIN3B), C-terminal binding protein 1 (CTBP1), basic helix-loop-helix family member E41 (BHLHE41), zinc finger and BTB domain containing 2 ZBTB2, nuclear factor YA (NFYA) and transcription factor activation protein 4 (TFAP4) as shown in Figure 3B. On the other hand, non-p-Δ;Np63α was shown to bind to the following proteins likely involved in gene activation: GTF2B, CRTC2, CITED2, SP1, TBPL1, TFAP2A, KAT2B, K(lysine) acetyltransferase 5 (KAT5) and coactivator-associated arginine methyltransferase 1 (CARM1) shown in Figure 4A, while transcription repressors bound to non-p-Δ;Np63α include enhancer of zeste homolog 2 (EZH2), HDAC1, HDAC2, CTCF, SIN3B, suppressor of zeste 12 homolog (SUZ12) and C-terminal binding protein 1 (CTBP1) as shown in Figure 4B.
MicroRNAs modulate the protein levels of TP63 and other transcription factors in SCC cells upon cisplatin exposure
Using miRDB-MicroRNA target prediction and functional study database, we found that the microRNAs previously shown to be induced or repressed by p-Δ;Np63α in SCC-11 cells upon cisplatin exposure, in fact, showed a homology to the respective “seed” sequences at the Δ;Np63α mRNA 3′-untranslated region (UTR, target prediction scores ranging from 53 to 71 as shown in Fig. 5A). These observations suggested that the specific microRNAs (e.g., miR-181a-5p, miR-374a-5p, miR-519a-3p, miR-630 and miR-885-3p) are likely to modulate the expression of Δ;Np63α protein in SCC-11 and/or SCC-11M cells.

Figure 5. Specific microRNA mimics and inhibitors modulated the expression of Δ;Np63α via its 3′-UTR sequences upon cisplatin exposure. (A) Predicted “seed” sequences for specific miRs in the TP63 3′-UTR with the target prediction scores in parentheses. SCC-11 cells (B) and SCC-11M cells (C) were transfected with the LightSwitch_3UTR vector for the TP63 3′-UTR along with the scrambled microRNA, or mimics and inhibitors for miR-181a-5p, miR-374a-5p, miR-519-3p, miR-630 and miR-885-3p for 36 h. Cells were treated with control medium without cisplatin (Con) or medium with 10 μg/ml cisplatin (CIS) for additional 12 h and then tested for the RenSP Renilla luciferase reporter activity. Measurements (in triplicate) for the luciferase activity presented as relative units (RU). Values obtained from cells transfected with the scrambled RNA and treated with control medium were designated as 1. (D) Total lysates from the resulting SCC-11M cells from (C) treated with control medium were subsequently analyzed by immunoblotting with antibodies to Δ;Np63α and β-actin. Relative levels of Δ;Np63α normalized for β-actin levels were quantified and shown above immunoblot images. Levels of Δ;Np63α in cells with the scrambled microRNA were designated as 1.
First, we showed that when SCC-11 cells and SCC-11M cells were transfected with the scrambled RNA, the cisplatin treatment caused the downregulation of Δ;Np63α level in SCC-11 cells, but failed to do so in SCC-11M cells suggesting that p-Δ;Np63α -dependent expression of microRNA species might play a role in this process (Fig. 5B). We further examined the direct effect of mimics and inhibitors for miR-181a-5p, miR-374a-5p, miR-519a-3p, miR-630 and miR-885-3p on the luciferase activity controlled by TP63 3′-UTR using the pLightSwitch_3UTR-TP63 plasmid. We thus found that mimics for miR-181a-5p, miR-374a-5p, miR-519a-3p, miR-630 and miR-885-3p repressed the pLightSwitch_3UTR-TP63 luciferase activity, while inhibitors for these microRNAs induced the luciferase activity in SCC-11 cells and SCC-11M cells grown in control medium (Fig. 5B and C). We further found that the Δ;Np63α protein levels were differentially attenuated by selected microRNAs tested on SCC-11 cells (ranging from 0.11 to 0.82 from scrambled control designated as 1 and normalized for the β-actin levels in total lysates), as shown in Figure 5D.
We next examined whether selected microRNAs might affect the expression of transcription factors and chromatin accessory proteins shown to bind the TP63-responsive element in the microRNA promoters. Again, using miRDB-microRNA target prediction and functional study database, we found that the 3′-UTR for several mRNAs of interest contained the respective “seed” sequences (target prediction scores ranging from 65 to 91, as shown in Fig. 6A). We then tested whether, in fact, specific microRNA species could affect the luciferase activity regulated by 3′-UTR sequences derived from ATM, CARM1, EP300, NFYB, BHLHE41, KAT2B, EZH2 and TBPL1. SCC-11 cells were transfected with the control scrambled RNA along with the empty pLightSwitch_3UTR vector (Fig. 6B). SCC-11 cells were also transfected with the pLightSwitch_3UTR-ATM, pLightSwitch_3UTR-BHLHE41, pLightSwitch_3UTR-CARM1, pLightSwitch_3UTR-NFYB, pLightSwitch_3UTR-KAT2B, pLightSwitch_3UTR-EP300, pLightSwitch_3UTR-EZH2 and pLightSwitch_3UTR-TBPL1 (Fig. 6B). We observed that while individual microRNAs had a slight inhibitory effect on the ATM-luciferase activity, the combination of all three microRNAs (miR-181a-5p, miR-374a-5p and miR-519a-3p) dramatically reduced the ATM-luciferase activity (Fig. 6B). MiR-519a-3p miR-885-3p, miR-374a-5p, miR-181a-5p and miR-630 were shown to markedly reduce the luciferase activities of the BHLHE41, CARM1, NFYB, KAT2B, EP300, EZH2 and TBPL1 vectors (Fig. 6B).

Figure 6. Specific microRNA mimics modulated the expression of the Δ;Np63α protein interacting targets via their 3′-UTR sequences. (A) Predicted “seed” sequences for specific microRNAs in the protein target 3′-UTRs with the target prediction scores in parentheses. (B) SCC-11 cells were transfected with the LightSwitch_3UTR plasmids for the indicated protein 3′-UTRs along with the scrambled microRNA and mimics for miR-181a-5p, miR-374a-5p, miR-519-3p, miR-630 and miR-885-3p for 36 h. Cells were then tested for the RenSP Renilla luciferase reporter activity. Measurements (in triplicate) for the luciferase activity presented as relative units (RU). Values from cells transfected with the scrambled RNA were designated as 1.
We next tested the effect of cisplatin treatment, as well as tested microRNAs on levels of the selected target proteins (e.g., ATM, BHLHE41, EZH2 and CARM1) in total lysates of SCC-11 cells and SCC-11M cells (Fig. 7). We also used siRNA directed against ATM, BHLHE41, EZH2 and CARM1 to modulate the expression of tested proteins in SCC-11 cells and SCC-11M cells (Fig. 7). We found that cisplatin induced the expression of ATM and BHLHE41 in SCC-11 cells transfected with the scrambled siRNA. However, miR-181a-5p together with miR-374a-5p and miR-519-3p or miR-519a-3p offseted the cisplatin effect on these proteins, respectively (Fig. 7A and C). Similarly, siRNAs against ATM and BHLHE41 attenuated the expression of both tested proteins (Fig. 7A and C). We further observed that cisplatin induced the expression of EZH2 and CARM1 in SCC-11M cells transfected with the scrambled siRNA. However, miR-630 or miR-885-3p counteracted the cisplatin effect on these proteins, respectively (Fig. 7B and D). Similarly, siRNAs against EZH2 and CARM1 decreased the expression of both tested proteins (Fig. 7B and D). Although, the cisplatin exposure of SCC-11 cells caused a dramatic decrease in cell viability, siRNAs against both ATM and BHLHE41 partially reversed this cisplatin effect on sensitive SCC-11 cells, rendering them to become more resistant to cisplatin treatment (Fig. 7E). On the other hand, the cisplatin exposure of SCC-11M cells had a more moderate effect on the cell viability, while siRNAs against both CARM1 and EZH2 rendered the resistant SCC-11M cells to be more sensitive to cisplatin-induced cell death (Fig. 7F).

Figure 7. Specific microRNA mimics modulated expression of the Δ;Np63α protein interacting targets in SCC cells exposed to cisplatin and affected cell viability. Immunoblotting assay. SCC-11 cells (A and C) and SCC-11M cells (B and D) were transfected with the scrambled microRNA and indicated microRNA mimics for 36 h. Cells were then treated with control medium without cisplatin (Con) or medium with 10 μg/ml cisplatin (CIS) for additional 12 h and nuclear lysates were tested for indicated endogenous proteins. Loading levels were tested using a TBP antibody. Relative protein levels normalized for the TBP levels were quantified and shown above immunoblot images. Protein levels in cells with the scrambled miR were designated as 1. Cell viability assay. (E) SCC-11 cells were transfected with the scrambled siRNA (Scr) and siRNAs against ATM (si-ATM) or BHLHE41 (si-BHLHE41). (F) SCC-11M cells were transfected with the scrambled siRNA (Scr) and siRNAs against CARM1 (si-CARM1) or EZH2 (si-EZH2). Resulting cells were cultured in the presence (CIS) or absence (Con) of the 10 μg/ml cisplatin for indicated times. 104 cells/well in 96-well plates were then incubated in serum-free medium with 5 μg/ml of the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide in the dark for 4 h at 37°C. Cells were lysed and incubated for 2 h at 37°C, and the measurements were obtained on a Spectra Max-250 plate reader. Each assay was repeated at three times in triplicate. The bars are the mean ± SD of triplicate; p < 0.05, t-test.
Discussion
Defects in the DNA damage response signaling (e.g., ATM signaling) often lead to an increased susceptibility to cancer and thereby represent novel therapeutic targets.10,13,32-35 Exposure of tumor cells to cisplatin chemotherapy often induces DNA damage associated with expression of mRNAs whose encoded proteins implicated in cell cycle arrest and apoptosis.16,18,32-35 Moreover, numerous microRNAs involved in regulation of transcription and protein stability are also deregulated in cancer cells exposed to cisplatin treatment.4,5,26,36-40
We previously reported that SCC cells enabling the ATM-dependent phosphorylation of Δ;Np63α upon cisplatin exposure showed altered expression of the specific mRNAs or microRNAs, displaying reduction of the miR-181a-5p, miR-519-3p and miR-374a-5p levels and induction of the miR-630 and miR-885-3p levels.26 We further found that the p-Δ;Np63α protein, in association with NFY proteins, binds to the specific mRNA and microRNA promoters in SCC-11 cells, while it failed to bind these promoters in SCC-11M cells.25,26 We found here that the cisplatin-induced p-Δ;Np63α decreased the reporter activity driven by the miR-181a-5p and miR-374a-5p promoters, while increased the activity of reporter luciferase gene fused to the miR-630 and miR-885-3p promoters.
In this study, we focused on the cisplatin-induced TP63/microRNA functional relationship in SCC cells attempting to address the question why p-Δ;Np63α reduced transcription of some microRNAs and induced others. Since, expression of microRNAs is maintained by RNA polymerase II and III transcription machinery, we suggested that the regulatory role of p-Δ;Np63α is intimately intertwined with other transcription factors and other chromatin accessory proteins.
Since TP63-responsive elements in the tested microRNA promoters are surrounded by other transcription factor cognate binding sites (Figs. S1–5), we analyzed the protein complexes formed between ~50 oligos derived from the specific regions of the microRNA promoters in question (miR-181a-5p, miR-519-3p, miR-374a-5p, miR-630 and miR-885-3p, Fig. 2), as described elsewhere.31 We used a pull-down of proteins (from SCC-11 cells and SCC-11M cells exposed to cisplatin) bound to 50bp oligos followed by tryptic digestion. Resulting peptides were labeled with iTRAQ reagents and subsequently subjected to LC/MS/MS fractionation and characterization, as was previously described for TP53 and Δ;Np63α-interacting proteins. Using the combination of DNA binding and quantitative mass spectrometry identification of proteins bound to specific promoter DNAs, we defined the critical components necessary to induce or repress p-Δ;Np63α-dependent microRNA gene expression in SCC cells upon cisplatin exposure, including transcription factors, regulators and chromatin accessory proteins bound to the specific microRNA promoters. We thus found that the following proteins might be involved in transcriptional induction of the miR-630 and miR-885-3p promoters in SCC-11 cells upon cisplatin exposure: HSF1, GTF2B, c-REL, SP1 and 3, ATF2, CRTC2, CITED2, EP300, TBPL1, v-MYB, DDIT3, KAT2B, TFAP2A and NFYB. We further found that the following proteins could take a part in the repression complex for the miR-181a-5p, miR-519a-3p and miR-374a-5p promoters in SCC-11 cells treated with cisplatin: BANP, YY1, FOXM1, FOXD3, SRY, C/EBPβ, HDAC1, HDAC2, HDAC4, MECP2, CTCF, SIN3B, CTBP1, BHLHE41, ZBTB2, NFYA and TFAP4. On the other hand, the following proteins are likely to be involved in gene activation of the specific promoters in SCC-11M cells treated with cisplatin: GTF2B, CRTC2, CITED2, SP1, TBPL1, TFAP2A, KAT2B, KAT5 and CARM1, while transcription repressors include EZH2, HDAC1, HDAC2, CTCF, SIN3B, SUZ12 and CTBP1.
We further observed that the selected microRNAs (miR-181a-5p, miR-519-3p, miR-374a-5p, miR-630 and miR-885-3p) modulated the stability of specific transcription factors (ATM, CARM1, EP300, NFYB, BHLHE41, KAT2B, EZH2 and TBPL1), including TP63 shown by the prediction target analysis of “seed” sequences, luciferase reporter and immunoblotting assays in SCC cells. Finally, we showed that siRNA knockdown of selected targets (modulated by microRNAs whose transcription is regulated by p-Δ;Np63α), notably ATM and BHLHE41 might render sensitive SCC-11 cells to become more resistant to cisplatin-induced cell death. However siRNA silencing of EZH2 and CARM1 would render more resistant SCC-11M cells to be more sensitive to cisplatin-induced cell death.
Taken together, we demonstrated that the p-Δ;Np63α protein can transcriptionally regulate the microRNA gene promoters by forming protein complexes with other transcriptional and chromatin-associated factors, while total Δ;Np63α levels, and p-Δ;Np63α levels (via downregulation of ATM) are maintained through a microRNA-mediated post-transcriptional/ translational machinery, thereby providing a regulatory feedback for selected microRNAs and their respective promoters.
Control of gene expression is exerted at a number of different levels, one of which is the accessibility of gene promoters to the transcriptional machinery.41-52 Intriguingly, among many proteins forming complexes with p-Δ;Np63α bound to the TP63-responsive element in the tested microRNA promoters, one can find specific transcription coactivators (CARM1, CITED2, CTRC2, DDIT3, c-REL, SP1, SSRP1, TFAP2A and YAP) or corepressors (BHLHE41, CTBP1, EZH2, YY1 and ZBTB20), histone acetyltransferases (EP300, KAT2B) and histone deacetylases (HDAC1, 2 and 4) are also likely to be associated with drug-induced cell death and especially with cisplatin resistance of tumor cells.46,47,53-59
Among transcription activators, the CARM1-mediated arginine methylation is shown to play a role in regulation of histone acetylation and transcription: facilitating transcription by discharging corepressors from chromatin and thereby has been linked to transcriptional regulation, cell cycle regulation and DNA repair.60-62 CARM1 is a novel transcriptional coactivator of nuclear factor kappa B (NFκB) and functions as a promoter-specific regulator of NFκB recruitment to chromatin. CARM1 forms a complex with EP300 and NFκB in vivo and interacts directly with the NFκB subunit p65 in vitro. Moreover, CARM1 synergistically coactivates NFκB-mediated transactivation, in concert with the transcriptional coactivators, EP300 and /CREB-binding protein (CREBBP), as previously described.60
Second, Yes-associated Protein (YAP) transcriptional coactivator has been implicated in tumorigenesis by regulating cell proliferation and apoptosis.63,64 YAP is phosphorylated in response to genotoxic stress induced by cisplatin treatment.63 Physical association of YAP and Δ;Np63α was markedly enhanced by cisplatin. YAP coactivator activity correlated with its state of phosphorylation and sensitivity to cisplatin-induced apoptosis.64
Third, short-hairpin RNA against Cited2 transcriptional modulator sensitized cancer cells to cisplatin, suggesting that acquired cisplatin resistance of cancer cells could be reversed by Cited2 silencing.65 Fourth, histone acetyltransferase genes, Kat2b (Pcaf), Clock and Tip60, are overexpressed, subsequently inducing the expression of DNA repair genes in cisplatin-resistant cells.54 Finally, Tfap2a was identified as a strong independent predictive marker for a good response and survival after cisplatin-containing chemotherapy.22 siRNA-mediated knockdown of Tfap2a increased the cell proliferation and rendered the cancer cells to become more resistant to cisplatin.22
Among transcription repressors, EZH2, a specific histone-3 lysine-27 (H3K27) methyltransferase, plays a critical role in tumorigenesis and cancer progression through epigenetic gene silencing and chromatin remodeling.53,66-69 EZH2 was overexpressed in cisplatin-resistant ovarian cancer cells compared with cisplatin-sensitive cells.53 Knockdown of EZH2 by siRNA resensitized drug-resistant ovarian cancer cells to cisplatin and decreased the level of H3K27 trimethylation.53 Loss of EZH2 also enhanced sensibility of tumor xenografts to cisplatin and inhibited tumor growth in vivo.53 EZH2 is one of the potent regulators of the accessibility of gene promoters and a part of the Polycomb repressive complex 2 (PRC2) along with SUZ12 and EED) catalyzing trimethylation of H3K27 and subsequently recruiting other DNA methyltransferases and histone deacetylases, resulting in transcriptional repression.68,69 EZH2 overexpression promotes the proliferation and invasion of epithelial ovarian and prostate cancer cells, contributing to cell resistance to cisplatin exposure and suggesting that EZH2 is a potential target for developing cancer therapeutics.66,67
Second, FOXM1, an oncogenic transcription factor, promotes tumorigenesis by regulating genes associated with cell cycle progression and cell proliferation, and its inhibition has been shown to sensitize cancer cells to apoptosis.55,56 The anti-FoxM1 siRNA can be functional when administered into tumors in vivo and holds potential as part of a therapy for cancer treatment.70
Third, BHLHE41 (DEC2/SHARP1) is a basic helix-loop-helix transcription repressor involved in the regulation of apoptosis, cell proliferation and cisplatin resistance.71-73 The expression of BHLHE41 was upregulated by cisplatin, while its forced expression inhibited pro-apoptotic facilitator BCL2-interacting protein (BIM), thereby blocking apoptosis. Interestingly, CTBP1 corepressor was shown to be a component of the RBP-Jκ/SHARP-co-repressor complex, which augmented the SHARP-mediated transcription repression.75,76
Fourth, ZBTB2, a POK family transcription factor, repressed transcription of the ADP-ribosylation factor (ARF), TP53 and cyclin-dependent kinase inhibitor 1A (CDKN1A) genes.77 ZBTB2 was shown to interact with SP1 and TP53, thereby inhibiting SP1-induced and TP53-dependent transcription activation. ZBTB2 also interacted with the complex of HDAC3, BCL6 corepressor (BCOR) and nuclear receptor corepressors (NCOR1 and 2), leading to an additional transcription repression.77 Although the forced ZBTB2 expression stimulated cell proliferation, its knockdown decreased cell proliferation.77
Finally, recent reports showed strong evidence that HDACs are implicated in transcription repression and cisplatin resistance.78-82 Following DNA damage, HDAC4 becomes recruited on NFY-dependent repressed G2/M gene promoters through a TP53-dependent mechanism.78 Platinum therapy induced a significantly enhanced apoptosis in resistant ovarian cancer cells transfected with HDAC4 siRNA, suggesting that HDAC4 is likely to be a beneficial target to counter platinum resistance in ovarian cancer.78
Δ;Np63α was specifically shown to associate with HDAC1 and HDAC2 to form an active transcriptional repressor complex that can be targeted to therapeutic advantage.82 Cisplatin chemotherapy as well as HDAC inhibitors promoted dissociation of Δ;Np63α and HDAC from the pro-apoptotic gene Puma promoter, in turn leading to increased histone acetylation, Puma expression and apoptosis.82
Our study established a new functional link between p-Δ;Np63α and the deregulated microRNA promoters in SCC cells exposed to cisplatin, suggesting that a complex transcriptional machinery involving p-Δ;Np63α could potentially act as a regulator of death or survival of SCC cells during chemotherapy. Thus, therapeutic compounds deactivating Δ;Np63α phosphorylation and/or its downstream microRNA targets could be used in combination with cisplatin to induce optimal tumor regression of human cancers that overexpress p-Δ;Np63α.
Transcriptional regulation of both mRNA and microRNA genes is maintained by multiple layers of molecular control including binding of transcription factors to promoter sequences and RNA polymerase initiation complex, modifications (acetylation/deacetylation, phosphorylation/ dephoshorylation, methylation/demethylation) of DNA and histones, gene accessibility via nucleosome and chromatin remodeling, and transcriptional cycling.41,42,44,47,52 Each of these regulatory layers plays a critical role in activation/repression of target gene promoters and future investigations needed to clarify their contributions to the mRNA and microRNA regulatory network under chemotherapeutic treatments.
Materials and Methods
Antibodies
We used a rabbit polyclonal antibody Ab-1 directed against human Δ;Np63 (EMD Chemicals), and monoclonal antibodies against human β-actin (Sigma) and TATA-binding protein (TBP, 1TBP18, ab818, Abcam). Mouse monoclonal antibodies to p63 (4A4, sc-8431), to SIN3B (H-4, sc-1314), to C/EBPβ (47A1, sc-56637), to TFAP2A (H-79, sc-8975), to c-MYB (3H2746, sc-73247), to TBPL1 (C-16, sc-10105) and to ATM (ATM 11G12, sc-53173) were obtained from Santa Cruz Biotechnology). We also used rabbit polyclonal antibodies against human NFYA (NBP1-19146), HDAC2 (NB100-2232, Novus), CtBP1 (NBP1-44886), FOXD3 (NB100-78525), TFAP4 (NBP1-46201), CARM1 (NB100-920) and a monoclonal antibody against BHLHE41 (SHARP1, 4H6, H00079365-M01), all purchased from Novus Biologicals. Antibodies to NFYB (PAB0659), to (PAB12512), to HDAC1 (PAB0647), to SRY (clone SRY.G12, MAB8814) were all obtained from Abnova. We then used the following rabbit polyclonal antibodies from Bethyl Laboratories: anti-FOXM1 (A301-532A), anti-YY1 (A302-778A), anti-PCAF (KAT2B, A301-666A), anti-SP1 (A300-133A), anti-HSF1 (A303-174A), anti-TORC2 (CRTC2, A300-637A), anti-ZBTB2 (A303-262A), anti-SMAR1/BANP (A300-278A) and anti-c-REL (A301-825A) and antibodies against EP300 (554215) and EZH2 (612666) from BD Transduction Laboratories. Custom rabbit polyclonal antibody against phosphorylated peptide encompassing the Δ;Np63α protein sequence (ATM motif, NKLPSV-pS-QLINPQQ, residues 379-392) was purified against the phosphorylated peptide vs. non-phosphorylated peptide.20
Cells and reagents
The cell line SCC-11 (expressing wt-TP53, wt-TP63 is amplified and Δ;Np63α is overexpressed) was characterized, tested and authenticated by a short tandem repeat profiling analysis using the AmpFISTR Identifiler PCR Amplification Lit (Applied Biosystems) at the JHMI Fragment Analysis Facility.20,25-29 The stable SCC cell lines expressing wild type Δ;Np63α (SCC-11) or Δ;Np63α-S385G (SCC-11M) were generated using Flp-In technology.20 Cells were maintained in RPMI medium 1640 and 10% fetal bovine serum and incubated with control medium without cis-diamminedichloro-platinum-dichloride (cisplatin, CIS, Sigma, P4394) or medium with10 μg/ml cisplatin (Sigma) for the indicated time periods. Cells were lysed with 50 mM Tris, pH7.5, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% Brij-50, 1 mM PMSF, 0.5 mM NaF, 0.1 mM Na3VO4, 2× complete protease inhibitor cocktail, sonicated for 10 sec intervals, and spun for 30 min at 15,000× g. Total and nuclear supernatants were analyzed by immunoblotting, and the levels of tested proteins were normalized against β-actin or TBP levels, respectively. Blots were scanned and quantified by the Image Quant software version 3.3 (Molecular Dynamics). Values were expressed as percentage of a control sample (defined as 1).
Isolation of nuclear fractions
1–2 × 106 cells were resuspended in hypotonic lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA) added with protease inhibitors (Sigma). After resuspension, 0.6% Triton X-100 (final concentration) was added and the nuclei were pelleted by centrifugation at 2,500–3,000xg for 10 min at 4°C. Nuclear pellets were resuspended in the extract buffer (20 mM HEPES pH 7.9, 25% glycerol, 0.4 M NaCl, 0.1mM EDTA, 0.1 mM EGTA), rocked for 15 min at 4°C and nuclear lysate (supernatant) was recovered by centrifugation at 10,000 × g for 5 min at 4°C as described elsewhere.29
qPCR of microRNAs
For validation of differential expression of microRNAs, we isolated total small RNAs using mirVana miRNA Isolation kit (#AM1560, Applied Biosystems). We then used the High Capacity cDNA Reverse Transcription kit (#4374966, Applied Biosystems) to produce single-stranded cDNA probes. Next, we applied a quantitative two-step qRT-PCR using the TaqMan MicroRNA Assay Kit TaqMan® U47 (#4380911) and TaqMan® Gene Expression Master Mix, 1-Pack (#4369016) both obtained from Applied Biosystems. For precursor pri-microRNAs, we used the following individual kits: pri-hsa-mir-181a-5p (Hs03302966_pri), pri-hsa-mir-519a-3p (Hs03302632_pri), pri-hsa-mir-374a-5p (Hs03304235_pri), pri-hsa-mir-630 (Hs03304713_pri) and pri-hsa-mir-885-3p (Hs03305150_pri). The reaction conditions were 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min with a sample volume of 20 μl. Expression was normalized to the U47 expression (gene ID 26802) and expression levels were determined as the average Ct of the U47 control. This averaged value was used to normalize the sample’s Ct. The averaged normalized value for each sample was then entered into this formula = 10 (NL avg/-3.5) and the average microRNA expression was determined using the Mann-Whitney's U test.26
MicroRNA mimics and inhibitors and transfection
The following individual microRNA mimics (precursors): hsa-miR-181a-5p (PM10381), hsa-miR-519a-3p (PM12949), hsa-miR-374a-5p (PM12702), hsa-miR-630 (PM11552) and hsa-miR-885-3p (PM12458), and inhibitors: hsa-miR-181a (AM10381), hsa-miR-519a (AM12949), hsa-miR-374a (AM12702), hsa-miR-630 (AM11552) and hsa-miR-885-3p (AM12458) were purchased from Ambion/Applied Biosystems.26 Cells were transfected for 24 h in a 6-well plate with 100 pmol of the mimic, inhibitor or control in 500 μl serum-free media with 5 μl of Lipofectamine-2000 reagent (Invitrogen). Each experiment was performed independently at least three times and in triplicate. Cells were also transfected with scrambled siRNA (SR30004), siRNA against ATM (si-ATM, SR300330) and siRNA against BHLHE41 (SR312407), all from Origene, CARM1 siRNA (sc-44875, Santa Cruz Biotechnology) and SignalSilence Ezh2 siRNA (6509S, Cell Signaling Technology).
Chromatin immunoprecipitation (ChIP)
Five × 106 cell equivalents of chromatin (2–2.5 kbp in size) were immunoprecipitated (IP) with 10 μg of anti-p-Δ;Np63α antibody, as described elsewhere.26 After reversal of formaldehyde cross-linking, RNA-ase A and proteinase K treatments, IP-enriched DNAs were used for PCR amplification.26 PCR was performed for 40 cycles (30s at 94°C, 30s at 60°C and 30s at 72°C) using Taq DNA polymerase (Invitrogen). Although the tested promoters contain multiple potential TP63 binding sites, the regions for PCR were selected based on the efficiency of amplification, choosing the highest PCR outcome. The specific regions (containing tested binding sites defined by the web browser: www.cbrc.jp/research/db/TFSEARCH) and non-specific regions (containing no tested binding sites) of selected gene promoters were amplified for ChIP-PCR assay (primers are underlined in Figs. S1–S5) and yielding the 250 bp or 150 bp PCR products, respectively. To quantify the binding of p-Δ;Np63α to the selected gene promoter sequences (enrichment), we used qPCR. ChIP-PCR values were obtained from the ChIP and Input samples and were normalized to GAPDH PCR values. For each transcription factor, values obtained from the input samples were designated as 1. ChIP/input ratio was plotted from all biological experiments using the Microsoft Excel software. Experiments were performed in triplicate.
Cloning of the reporter plasmids for microRNA promoters
Promoter sequences for miR-181a-5p, miR-374a-5p and miR-630 were amplified using the following primers: for miR-181a-5p, sense - 5′-TCCATCAAAACAAAACGAAACAACTCGAAATAATTTAGAATAT-3′and antisense - 5′-TGTGGAGGTTTGCCAAACTCAGTCGAGCACGTTCATCTGCTT-3′ yielding the 1,895bp PCR product; for miR-374a-5p, sense - 5′-TCCATCAAAACAAAACGA AACAATTATCGAAGAGACTTCTAGA-3′and antisense - 5′-CTTTTCTAACTTATTCCTAC AGTCGAGCACGTTCATCTGCTT-3′ yielding the 1,745 bp PCR product; for miR-630, sense - 5′-TCCATCAAAACAAAACGAAACAA GTTTGAGTGTCATAAATCCA-3′ and antisense - 5′-TACTCTTATTTGGATCTGT AAGTCGAGCACGTTCATCTGCTT-3′ yielding the 1,545bp PCR product. The PCR fragments were subcloned into the promoter-less pLightSwitch_Prom vector upstream the luciferase reporter gene (S790005, SwitchGear Genomics) and subsequently used for transfections and reporter assays.
Luciferase reporter assay
We used the 3′-UTR luciferase reporter plasmids for ATM (SC221017, Origene), TP63 (S811809), CRTC2 (S803503), CARM1 (S807909), BHLHE41 (S705709), EP300 (S808354), KAT2B (S810567), EZH2 (S811982), NFYB (S811604) and TBPL1 (S804783) were obtained from SwitchGear Genomics. For the promoter-mediated luciferase activity assay, and 3′-UTR-mediated luciferase activity assay, a total of 5 × 104 cells/ well in a 24-well plate were transfected with the control (empty) pLightSwitch_Prom vector (S790005) or with the empty pLightSwitch_3UTR vector (S890005), respectively, using Fugene HD reagent (Roche) as recommended by the manufacturer. Both vectors represent a fully optimized reporter system that includes an improved luminescent reporter gene (RenSP). In addition, cells were transfected with the selected 3′-UTR plasmids as listed above. The LightSwitch Luciferase Assay Reagent (SwitchGear Genomics) enables to monitor luciferase reporter signal according to the manufacturer’s protocol. For the 3′-UTR assays, cells were also transfected with 100 ng of the mimics or inhibitors of tested microRNAs. At 36 h, cells were also treated with 10 μg/ml cisplatin or control medium without cisplatin for an additional 12 h. The RenSP Renilla luciferase activity was measured at 480 nm using a luminometer.
Cell viability assay
104 cells/well in 96-well plates were incubated in serum-free medium with 5 μg/ml of the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT assay, American Tissue Culture Collection) in the dark for 4 h at 37°C as described elsewhere.21 Cells were lysed and incubated for 2 h at 37°C, and the measurements (A570 nm to A650 nm) were obtained on a Spectra Max 250 plate reader (Molecular Devices) as described. Each assay was repeated three times in triplicate.
Statistical analysis
Results were expressed as means ± SD from three independent experiments in triplicate. Differences in variables between experimental and control group were assessed by using the Student’s t-test. Statistically significant difference was accepted at p < 0.05.
Bioinformatics
Putative responsive elements were deifned using the TFSEARCH (www.cbrc.jp/research/db/TFSEARCH.html) and TRANSFAC (version 7.4, www.generegulation.com/pub/databases.html) databases. For prediction of the microRNA-specific “seed” sequences in target 3′-UTRs, we used the following databases: microRNA.org-Targets and Expression (August 2010, Computational Biology Center, Memorial Sloan-Kettering Cancer Center, www.microrna.org), TargetScan: Prediction of microRNA targets, version 5.2 (June 2011, www.targetscan.org) and miRDB-MicroRNA Target Prediction And Functional Study Database, v3.0 (April 2009, www.mirdb.org). All the targets were selected based on mirSVR scores (from -0.1479 to -0.4518), PhastCons scores (0.4775–0.6217) and target prediction scores (from 55 to 91) assigned by the computational target prediction algorithm.
Concluding Remarks
Drug resistance acquired by tumor cells limits the successful use of cisplatin chemotherapy. Many genes encoding mRNAs and microRNAs are differentially expressed in sensitive and resistant tumor cells, suggesting that the transcriptional regulation of genes for mRNA and microRNAs involved in mechanisms underlying chemoresistance. We demonstrated that the p-Δ;Np63α transcriptionally regulates the microRNA gene promoters by forming protein complexes with other transcriptional and chromatin-associated factors, while total Δ;Np63α levels are maintained through a microRNA-mediated post-transcriptional/ translational machinery, thereby providing a regulatory feedback for selected microRNAs and their respective promoters. Our study established a new functional link between p-Δ;Np63α and the deregulated microRNA promoters in SCC cells exposed to cisplatin, suggesting that a complex transcriptional machinery involving p-Δ;Np63α could potentially act as a regulator of death or survival of SCC cells during chemotherapy. Thus, therapeutic compounds deactivating Δ;Np63α phosphorylation and/or its downstream microRNA targets could be used in combination with cisplatin to induce optimal tumor regression of human cancers that overexpress p-Δ;Np63α.
Supplementary Material
Acknowledgments
This study was supported in part by the Flight Attendant Research Institutions grant (#082469 to E.A.R.), and by National Cancer Institute grants K01-CA164092 and U01-CA84986 (R.G-P.).
Glossary
Abbreviations:
- ARF
ADP-ribosylation factor
- ATF
activation transcription factor
- ATM
ataxia telangiectasia mutated
- BANP
BTG3 associated nuclear protein
- BCOR
BCL6 corepressor
- BHLH
basic helix-loop-helix
- CDKN
cyclin-dependent kinase inhibitor
- C/EBP
CCAAT/enhancer binding protein
- CCTF
CCCTC-binding factor
- ChIP
chromatin immunoprecipitation
- CIS
cisplatin
- Con
control
- CITED
CBP/p300-interacting transactivator
- CRTC
CREB-regulated transcription coactivator
- CTBP
c-terminal binding protein
- DDIT
DNA-damage-inducible transcript
- EP
E1A-binding protein
- EZH
enhancer of zeste homolog
- FL
FLAG
- HDAC
histone deacetylase
- GTF
general transcription factor
- HSF
heat shock factor
- IP
immunoprecipitation
- iTRAQ
isobaric tags for relative and absolute quantification
- JHMI
Johns Hopkins Medical Institutions
- KAT
K(lysine) acetyltransferase
- LC
liquid chromatography
- MECP
methyl CpG binding protein
- miR
microRNA
- MS
mass spectrometry
- NF
nuclear factor
- p
phosphorylated
- qPCR
quantitative PCR
- RU
relative units
- SCC
squamous cell carcinoma
- SCX
strong cationic exchange
- SP
specificity factor
- SRY
sex determining region Y
- TBPL
TATA-binding protein-Like
- TFAP
transcription factor activation protein
- TP
tumor protein
- YAP
Yes-associated protein
- ZBTB
zinc finger and BTB domain
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/23598
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23598
References
- 1.Helmbach H, Kern MA, Rossmann E, Renz K, Kissel C, Gschwendt B, et al. Drug resistance towards etoposide and cisplatin in human melanoma cells is associated with drug-dependent apoptosis deficiency. J Invest Dermatol. 2002;118:923–32. doi: 10.1046/j.1523-1747.2002.01786.x. [DOI] [PubMed] [Google Scholar]
- 2.Zangen R, Ratovitski EA, Sidransky D. DeltaNp63α levels correlate with clinical tumor response to cisplatin. Cell Cycle. 2005;4:1313–5. doi: 10.4161/cc.4.10.2066. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70:1793–803. doi: 10.1158/0008-5472.CAN-09-3112. [DOI] [PubMed] [Google Scholar]
- 5.Ory B, Ellisen LW. A microRNA-dependent circuit controlling p63/p73 homeostasis: p53 family cross-talk meets therapeutic opportunity. Oncotarget. 2011;2:259–64. doi: 10.18632/oncotarget.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torigoe T, Izumi H, Ishiguchi H, Yoshida Y, Tanabe M, Yoshida T, et al. Cisplatin resistance and transcription factors. Curr Med Chem Anticancer Agents. 2005;5:15–27. doi: 10.2174/1568011053352587. [DOI] [PubMed] [Google Scholar]
- 7.Igarashi T, Izumi H, Uchiumi T, Nishio K, Arao T, Tanabe M, et al. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene. 2007;26:4749–60. doi: 10.1038/sj.onc.1210289. [DOI] [PubMed] [Google Scholar]
- 8.Sheng Z, Evans SK, Green MR. An activating transcription factor 5-mediated survival pathway as a target for cancer therapy? Oncotarget. 2010;1:457–60. doi: 10.18632/oncotarget.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng SX, Dai MS, Keller DM, Lu H. SSRP1 functions as a co-activator of the transcriptional activator p63. EMBO J. 2002;21:5487–97. doi: 10.1093/emboj/cdf540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. P63 and p73 are required for TP53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 11.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 12.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–51. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–39. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Miyamoto N, Izumi H, Noguchi T, Nakajima Y, Ohmiya Y, Shiota M, et al. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J Biol Chem. 2008;283:18218–26. doi: 10.1074/jbc.M802332200. [DOI] [PubMed] [Google Scholar]
- 15.Maclaine NJ, Hupp TR. The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging (Albany NY) 2009;1:490–502. doi: 10.18632/aging.100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin AG, Trama J, Crighton D, Ryan KM, Fearnhead HO. Activation of p73 and induction of Noxa by DNA damage requires NF-κ B. Aging (Albany NY) 2009;1:335–49. doi: 10.18632/aging.100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakasugi T, Izumi H, Uchiumi T, Suzuki H, Arao T, Nishio K, et al. ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene. 2007;26:5194–203. doi: 10.1038/sj.onc.1210326. [DOI] [PubMed] [Google Scholar]
- 18.Vousden KH. Partners in death: a role for p73 and NFκB in promoting apoptosis. Aging (Albany, NY Online) 2009;1:275–7. doi: 10.18632/aging.100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muppani N, Nyman U, Joseph B. TAp73α protects small cell lung carcinoma cells from caspase-2 induced mitochondrial mediated apoptotic cell death. Oncotarget. 2011;2:1145–54. doi: 10.18632/oncotarget.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, et al. ATM kinase is a master switch for the Δ; Np63 α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2008;7:2846–55. doi: 10.4161/cc.7.18.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen T, Sen N, Brait M, Begum S, Chatterjee A, Hoque MO, et al. Δ;Np63α confers tumor cell resistance to cisplatin treatment through the transcriptional regulation of AKT. Cancer Res. 2011;71:1167–76. doi: 10.1158/0008-5472.CAN-10-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nordentoft I, Dyrskjøt L, Bødker JS, Wild PJ, Hartmann A, Bertz S, et al. Increased expression of transcription factor TFAP2α correlates with chemosensitivity in advanced bladder cancer. BMC Cancer. 2011;11:135. doi: 10.1186/1471-2407-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garand C, Guay D, Sereduk C, Chow D, Tsofack SP, Langlois M, et al. An integrative approach to identify YB-1-interacting proteins required for cisplatin resistance in MCF7 and MDA-MB-231 breast cancer cells. Cancer Sci. 2011;102:1410–7. doi: 10.1111/j.1349-7006.2011.01948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiota M, Yokomizo A, Tada Y, Uchiumi T, Inokuchi J, Tatsugami K, et al. P300/CBP-associated factor regulates Y-box binding protein-1 expression and promotes cancer cell growth, cancer invasion and drug resistance. Cancer Sci. 2010;101:1797–806. doi: 10.1111/j.1349-7006.2010.01598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Chuang AY, Romano RA, Liegeois NJ, Sinha S, Trink B, et al. Phospho-Δ;Np63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle. 2010;9:332–42. doi: 10.4161/cc.9.2.10432. [DOI] [PubMed] [Google Scholar]
- 26.Huang YP, Chuang A, Hao H, Talbot CC, Trink B, Sidransky D, et al. Phospho-Δ;Np63α is a key regulator of cisplatin-induced microRNAome in head and neck squamous cell carcinoma cells. Cell Death Differ. 2011;18:1220–30. doi: 10.1038/cdd.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-Δ;Np63α-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle. 2012;11:1247–59. doi: 10.4161/cc.11.6.19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Chuang AY, Ratovitski EA. Phospho-Δ;Np63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle. 2011;10:3938–47. doi: 10.4161/cc.10.22.18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y, Jeong JS, Okamura J, Sook-Kim M, Zhu H, Guerrero-Preston R, et al. Global tumor protein p53/p63 interactome: making a case for cisplatin chemoresistance. Cell Cycle. 2012;11:2367–79. doi: 10.4161/cc.20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Ratovitski EA. Phospho-Δ;Np63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging (Albany, NY Online) 2010;2:959–68. doi: 10.18632/aging.100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ku WC, Chiu SK, Chen YJ, Huang HH, Wu WG, Chen YJ. Complementary quantitative proteomics reveals that transcription factor AP-4 mediates E-box-dependent complex formation for transcriptional repression of HDM2. Mol Cell Proteomics. 2009;8:2034–50. doi: 10.1074/mcp.M900013-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida K, Ozaki T, Furuya K, Nakanishi M, Kikuchi H, Yamamoto H, et al. ATM-dependent nuclear accumulation of IKK-α plays an important role in the regulation of p73-mediated apoptosis in response to cisplatin. Oncogene. 2008;27:1183–8. doi: 10.1038/sj.onc.1210722. [DOI] [PubMed] [Google Scholar]
- 33.Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010;2:535–7. doi: 10.18632/aging.100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seviour EG, Lin SY. The DNA damage response: Balancing the scale between cancer and ageing. Aging (Albany NY) 2010;2:900–7. doi: 10.18632/aging.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–15. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le MT, Shyh-Chang N, Khaw SL, Chin L, Teh C, Tay J, et al. Conserved regulation of p53 network dosage by microRNA-125b occurs through evolving miRNA-target gene pairs. PLoS Genet. 2011;7:e1002242. doi: 10.1371/journal.pgen.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20:187–99. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Au SL, Wong CC, Lee JM, Fan DN, Tsang FH, Ng IO, et al. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology. 2012;56:622–31. doi: 10.1002/hep.25679. [DOI] [PubMed] [Google Scholar]
- 40.Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, et al. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany NY) 2010;2:333–43. doi: 10.18632/aging.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashraf SI, Ip YT. Transcriptional control: repression by local chromatin modification. Curr Biol. 1998;8:R683–6. doi: 10.1016/S0960-9822(98)70435-X. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen TT, Cho K, Stratton SA, Barton MC. Transcription factor interactions and chromatin modifications associated with p53-mediated, developmental repression of the α-fetoprotein gene. Mol Cell Biol. 2005;25:2147–57. doi: 10.1128/MCB.25.6.2147-2157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–42. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 44.Shalgi R, Brosh R, Oren M, Pilpel Y, Rotter V. Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging (Albany NY) 2009;1:762–70. doi: 10.18632/aging.100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsumura N, Huang Z, Baba T, Lee PS, Barnett JC, Mori S, et al. Yin yang 1 modulates taxane response in epithelial ovarian cancer. Mol Cancer Res. 2009;7:210–20. doi: 10.1158/1541-7786.MCR-08-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suenaga Y, Ozaki T, Tanaka Y, Bu Y, Kamijo T, Tokuhisa T, et al. TATA-binding Protein (TBP)-like Protein Is Engaged in Etoposide-induced Apoptosis through Transcriptional Activation of Human TAp63 Gene. J Biol Chem. 2009;284:35433–40. doi: 10.1074/jbc.M109.050047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B, Xiao Z, Ko HL, Ren EC. The p53 response element and transcriptional repression. Cell Cycle. 2010;9:870–9. doi: 10.4161/cc.9.5.10825. [DOI] [PubMed] [Google Scholar]
- 48.Mallette FA, Calabrese V, Ilangumaran S, Ferbeyre G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging (Albany NY) 2010;2:445–52. doi: 10.18632/aging.100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill R, Madureira PA, Waisman DM, Lee PW. DNA-PKCS binding to p53 on the p21WAF1/CIP1 promoter blocks transcription resulting in cell death. Oncotarget. 2011;2:1094–108. doi: 10.18632/oncotarget.378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Bedford DC, Brindle PK. Is histone acetylation the most important physiological function for CBP and p300? Aging (Albany NY) 2012;4:247–55. doi: 10.18632/aging.100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imbriano C, Gnesutta N, Mantovani R. The NF-Y/p53 liaison: Well beyond repression. Biochim Biophys Acta. 2012;1825:131–9. doi: 10.1016/j.bbcan.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther. 2010;10:788–95. doi: 10.4161/cbt.10.8.12913. [DOI] [PubMed] [Google Scholar]
- 54.Hirano G, Izumi H, Kidani A, Yasuniwa Y, Han B, Kusaba H, et al. Enhanced expression of PCAF endows apoptosis resistance in cisplatin-resistant cells. Mol Cancer Res. 2010;8:864–72. doi: 10.1158/1541-7786.MCR-09-0458. [DOI] [PubMed] [Google Scholar]
- 55.Gomes AR, Brosens JJ, Lam EW. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle. 2008;7:3133–6. doi: 10.4161/cc.7.20.6920. [DOI] [PubMed] [Google Scholar]
- 56.Kwok JM, Peck B, Monteiro LJ, Schwenen HD, Millour J, Coombes RC, et al. FOXM1 confers acquired cisplatin resistance in breast cancer cells. Mol Cancer Res. 2010;8:24–34. doi: 10.1158/1541-7786.MCR-09-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stronach EA, Alfraidi A, Rama N, Datler C, Studd JB, Agarwal R, et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011;71:4412–22. doi: 10.1158/0008-5472.CAN-10-4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davies AH, Dunn SE. YB-1 drives preneoplastic progression: Insight into opportunities for cancer prevention. Oncotarget. 2011;2:401–6. doi: 10.18632/oncotarget.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sen T, Chang X, Sidransky D, Chatterjee A. Regulation of Δ;Np63α by NFκΒ. Cell Cycle. 2010;9:4841–7. doi: 10.4161/cc.9.24.14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, et al. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 2005;24:85–96. doi: 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu J, Cui N, Wang R, Li J, Wong J. A role for CARM1-mediated histone H3 arginine methylation in protecting histone acetylation by releasing corepressors from chromatin. PLoS One. 2012;7:e34692. doi: 10.1371/journal.pone.0034692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee YH, Stallcup MR. Roles of protein arginine methylation in DNA damage signaling pathways is CARM1 a life-or-death decision point? Cell Cycle. 2011;10:1343–4. doi: 10.4161/cc.10.9.15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chatterjee A, Sen T, Chang X, Sidransky D. Yes-associated protein 1 regulates the stability of DeltaNp63α. Cell Cycle. 2010;9:162–7. doi: 10.4161/cc.9.1.10321. [DOI] [PubMed] [Google Scholar]
- 64.Lee KK, Yonehara S. Identification of mechanism that couples multisite phosphorylation of Yes-associated protein (YAP) with transcriptional coactivation and regulation of apoptosis. J Biol Chem. 2012;287:9568–78. doi: 10.1074/jbc.M111.296954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu ZZ, Sun NK, Chao CC. Knockdown of CITED2 using short-hairpin RNA sensitizes cancer cells to cisplatin through stabilization of p53 and enhancement of p53-dependent apoptosis. J Cell Physiol. 2011;226:2415–28. doi: 10.1002/jcp.22589. [DOI] [PubMed] [Google Scholar]
- 66.Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget. 2011;2:669–83. doi: 10.18632/oncotarget.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012;72:3091–104. doi: 10.1158/0008-5472.CAN-11-3546. [DOI] [PubMed] [Google Scholar]
- 68.Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18:1771–9. doi: 10.1038/cdd.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tong ZT, Cai MY, Wang XG, Kong LL, Mai SJ, Liu YH, et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene. 2012;31:583–94. doi: 10.1038/onc.2011.254. [DOI] [PubMed] [Google Scholar]
- 70.Wang M, Gartel AL. The suppression of FOXM1 and its targets in breast cancer xenograft tumors by siRNA. Oncotarget. 2011;2:1218–26. doi: 10.18632/oncotarget.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garriga-Canut M, Roopra A, Buckley NJ. The basic helix-loop-helix protein, sharp-1, represses transcription by a histone deacetylase-dependent and histone deacetylase-independent mechanism. J Biol Chem. 2001;276:14821–8. doi: 10.1074/jbc.M011619200. [DOI] [PubMed] [Google Scholar]
- 72.Liu JJ, Chung TK, Li J, Taneja R. Sharp-1 modulates the cellular response to DNA damage. FEBS Lett. 2010;584:619–24. doi: 10.1016/j.febslet.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Falvella FS, Colombo F, Spinola M, Campiglio M, Pastorino U, Dragani TA. BHLHB3: a candidate tumor suppressor in lung cancer. Oncogene. 2008;27:3761–4. doi: 10.1038/sj.onc.1211038. [DOI] [PubMed] [Google Scholar]
- 74.Wu Y, Sato F, Bhawal UK, Kawamoto T, Fujimoto K, Noshiro M, et al. BHLH transcription factor DEC2 regulates pro-apoptotic factor Bim in human oral cancer HSC-3 cells. Biomed Res. 2012;33:75–82. doi: 10.2220/biomedres.33.75. [DOI] [PubMed] [Google Scholar]
- 75.Oswald F, Winkler M, Cao Y, Astrahantseff K, Bourteele S, Knöchel W, et al. RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol Cell Biol. 2005;25:10379–90. doi: 10.1128/MCB.25.23.10379-10390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madison DL, Lundblad JR. C-terminal binding protein and poly(ADP)ribose polymerase 1 contribute to repression of the p21(waf1/cip1) promoter. Oncogene. 2010;29:6027–39. doi: 10.1038/onc.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jeon BN, Choi WI, Yu MY, Yoon AR, Kim MH, Yun CO, et al. ZBTB2, a novel master regulator of the p53 pathway. J Biol Chem. 2009;284:17935–46. doi: 10.1074/jbc.M809559200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Basile V, Mantovani R, Imbriano C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J Biol Chem. 2006;281:2347–57. doi: 10.1074/jbc.M507712200. [DOI] [PubMed] [Google Scholar]
- 79.Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67:3145–52. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 80.Bansal N, Kadamb R, Mittal S, Vig L, Sharma R, Dwarakanath BS, et al. Tumor suppressor protein p53 recruits human Sin3B/HDAC1 complex for down-regulation of its target promoters in response to genotoxic stress. PLoS One. 2011;6:e26156. doi: 10.1371/journal.pone.0026156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jurkin J, Zupkovitz G, Lagger S, Grausenburger R, Hagelkruys A, Kenner L, et al. Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle. 2011;10:406–12. doi: 10.4161/cc.10.3.14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramsey MR, He L, Forster N, Ory B, Ellisen LW. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011;71:4373–9. doi: 10.1158/0008-5472.CAN-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.