

Abstract
Vagal sensory nerves innervate the majority of visceral organs (e.g. heart, lungs, GI tract, etc) and their activation is critical for defensive and regulatory reflexes. Intracellular Ca2+ is a key regulator of neuronal excitability and is largely controlled by the Ca2+ stores of the endoplasmic reticulum. In other cell types store-operated channels (SOC) have been shown to contribute to the homeostatic control of intracellular Ca2+. Here, using Ca2+ imaging, we have shown that ER depletion in vagal sensory neurons (using thapsigargin or caffeine) in the absence of extracellular Ca2+ evoked Ca2+ influx upon reintroduction of Ca2+ into the extracellular buffer. This store-operated Ca2+ entry (SOCE) was observed in approximately 25–40% of vagal neurons, equally distributed among nociceptive and non-nociceptive sensory subtypes. SOCE was blocked by Gd3+ but not by the Orai channel blocker SKF96365. We found Orai channel mRNA in extracts from whole vagal ganglia, but when using single cell RT-PCR analysis we found only 3 out of 34 neurons expressed Orai channel mRNA, indicating that Orai channel expression in the vagal ganglia was likely derived from non-neuronal cell types. Confocal microscopy of vagal neurons in 3 day cultures demonstrated rich ER tracker fluorescence throughout axonal and neurite structures and ER store depletion (thapsigargin) evoked Ca2+ transients from these structures. However, no SOCE could be detected in the axonal/neurite structures of vagal neurons. We conclude that SOCE occurs in vagal sensory neuronal cell bodies through non-Orai mechanisms but is absent at nerve terminals.
Keywords: Vagal sensory nerves, Endoplasmic reticulum, Calcium, Nerve terminal, Store-operated calcium entry, Orai channel
1. Introduction
The mammalian viscera, including the airways, GI tract and heart, are densely innervated by sensory fibers projected by bilateral vagal nerves. These afferent fibers serve to sense the local environment and relay such information to brainstem areas involved in homeostatic control and defensive strategies. The large majority of vagal sensory afferent nerves are slowly conducting fibers (sparse to no myelination) that express proteins involved in the transduction of noxious or potentially noxious stimuli (Taylor-Clark and Undem, 2006). These fibers are called ‘nociceptors’ and their activation initiates defensive strategies, for example in the airways: cough, reflex bronchospasm, mucus secretion and neurogenic inflammation (Carr and Undem, 2003). The sensitivity or excitability of afferent nerves is critical to their function in health and disease.
Intracellular Ca2+ ions have significant effects on nerve excitability. Studies of dissociated sensory neurons from vagal, trigeminal and dorsal root ganglia have yielded important details of Ca2+ regulation and function (Cordoba-Rodriguez et al., 1999; Gover et al., 2007; Solovyova et al., 2002; Taylor-Clark et al., 2005). In the cell body, there is substantial endoplasmic reticulum (ER) that acts as a controllable sink of free cytosolic Ca2+. Ca2+ release from the ER occurs downstream of either Gq protein activation and IP3 generation or following influx of Ca2+ through voltage-gated Ca2+ channels (Ca2+-induced Ca2+ release) (Cohen et al., 1997; Nicolson et al., 2002). Ca2+ then modulates excitability, particularly through the modulation of K+ and Cl− ion conductances (Gover et al., 2009). For example Ca2+ can activate BK and SK channels, thus causing K+ efflux and hyperpolarization (Bahia et al., 2005; Li et al., 2007). Alternatively, Ca2+ can induce the inhibition of KCNQ channels, thus causing a decrease in K+ leak and depolarization (Liu et al., 2010). In addition, Ca2+ activates CLCA channels, thus causing Cl− efflux (due to depolarized reversal potential set by NKCC1 co-transporter expression) and depolarization (Lee et al., 2005; Liu et al., 2010).
ER Ca2+ stores can become depleted following repeated Ca2+ mobilization signals. In many cell types store-operated Ca2+ entry (SOCE) facilitates the rapid refilling of the ER by extracellular Ca2+. SOCE has principally been described in non-neuronal cells and requires the gating of plasma membrane store-operated channels (SOC) (Parekh and Putney, 2005). Evidence suggests that plasma membrane Ca2+-permeable Orai channels are the major SOC in many cell types, which are activated by the ER protein Stim1 upon loss of Ca2+ from the store (Varnai et al., 2009). Evidence supporting SOCE in central and peripheral neurons has been published (Gemes et al., 2011; Gruszczynska-Biegala et al., 2011; Steinbeck et al., 2011; Usachev and Thayer, 1999), but at present there are no published reports of putative SOCE in vagal neurons. It is likely that the presence of SOC would significantly modulate Ca2+-mediated regulation of vagal neuronal excitability (Gover et al., 2009; Parekh and Putney, 2005). In this article we present data demonstrating SOCE in vagal sensory neuronal cell bodies. However, SOCE in vagal neurons is not mediated by Orai channels and SOCE is absent at nerve terminals.
2. Results
2.1 Ca2+ store in dissociated vagal sensory neurons
We first investigated the presence of functional Ca2+ stores in the soma of dissociated vagal sensory neurons using thapsigargin, an inhibitor of the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pump. Thapsigargin (10μM) evoked a Ca2+ transient in vagal neurons (Fig 1), indicating the presence of ER stores in these cells: 14.4±2.2% of ionomycin (n=111). Previous studies have suggested that the thapsigargin-induced increase in [Ca2+]cyt can lead to the activation of the sensory nerve plasmalemmal non-selective ion channel TRPA1 (Jordt et al., 2004). As such, the thapsigargin-induced Ca2+ transient observed in these neurons may be secondary to TRPA1 activation. Nevertheless, thapsigargin (10μM) evoked Ca2+ transients in vagal neurons derived from TRPA1−/ − mice (13.8±2.2% of ionomycin (n=74)), suggesting the vast majority of the Ca2+ transient was directly as a result of Ca2+ store mobilization.
Fig 1.

Mean +/− SEM Ca2+ responses of WT (n=111, red) and TRPA1−/ − (n=74, black) vagal neurons to thapsigargin (10μM). Blocked line denotes the 60-s application of agonist. Data are presented as mean change in 340/380 ratio as a % of ionomycin peak. All neurons responded to KCl (75mM) applied immediately before ionomycin (not shown).
2.2 Store-operated Ca2+ entry in vagal sensory neurons
As part of intracellular Ca2+ homeostatic machinery in many cells, SOC allow for the refilling of Ca2+ stores (e.g. ER) following physiological mobilization or pathophysiological depletion. SOC activation is responsible for SOCE. To investigate SOCE, we studied the effect of removal and re-introduction of extracellular Ca2+ on the [Ca2+]cyt of dissociated vagal neurons. The removal of Ca2+ for 3 minutes produced a small decrease in [Ca2+]cyt in 75 of 88 neurons (−4.9±0.5% of ionomycin). Following the re-introduction of 2.2mM Ca2+, the [Ca2+]cyt returned to baseline. In 4 of 88 neurons, the re-introduction of Ca2+ evoked a significant Ca2+ influx above the baseline (Fig 2). Thus, Ca2+ removal alone is unable to elicit a classic SOCE or ‘Ca2+ addback response’ in vagal neurons.
Fig 2.
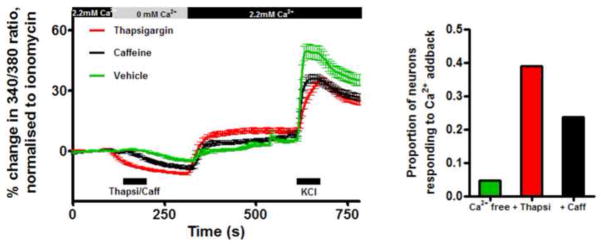
Left, mean +/− SEM Ca2+ responses of vagal neuron soma to thapsigargin (Thapsi, 10 μM, red, n=310), caffeine (Caff, 10mM, black, n=233) and vehicle (green, n=88) during changes in [Ca2+]extracellular. Response to KCl (75mM) is included. Blocked line denotes the 60-s application of drugs. Right, proportion of vagal neurons responding to the Ca2+ addback.
Ca2+ stores can be depleted using thapsigargin (inhibits SERCA pump) or caffeine (activates ryanodine receptors). We treated vagal neurons with the store depletors in the absence of extracellular Ca2+, then after 3 minutes, Ca2+ was re-introduced. Under these conditions, caffeine (10mM) caused a greater decrease in [Ca2+]cyt than Ca2+ removal alone (−8.6±0.9% of ionomycin) and upon re-introduction of Ca2+ evoked a small increase above baseline (4.6±0.9% of ionomycin)(n=233). Thapsigargin (10μM) also caused a decrease in [Ca2+]cyt (−11.4±0.5% of ionomycin) and upon re-introduction of Ca2+ evoked a small increase above baseline (9.6±1.2% of ionomycin)(n=310) (Fig 2). Thapsigargin (1μM) caused similar responses (n=108, data not shown). Vagal neurons are a heterogeneous population of sensory nerve subtypes, thus we analyzed these data to determine if significant Ca2+ addback responses were only evoked in some cells. We found that compared to the 4.5% of neurons that evoked SOCE following Ca2+ reintroduction without treatment with store depletors, SOCE were evoked in 23.6% following caffeine, 29.6% following 1μM thapsigargin and 39.0% following 10μM thapsigargin (Figs 2 and 3). These neurons were further characterized by either capsaicin (TRPV1 agonist, 1μM) or allyl isothiocyanate (AITC, TRPA1 agonist, 100μM), which selectively activate nociceptor subtypes (Patapoutian et al., 2009). Ca2+ addback responses following caffeine and thapsigargin were equally distributed in nociceptive and non-nociceptive subtypes (Table 1 and data not shown).
Fig 3.

(A) Left, mean +/− SEM Ca2+ responses of vagal neurons that responded to the Ca2+ addback following thapsigargin (1 μM, green, n=32 out of 108) or following thapsigargin (10 μM) in the absence (black, n=121 out of 310) or presence of SKF96365 (SKF, 10 μM, red, n=35 out of 102); right, proportion of vagal neurons responding to the Ca2+ addback. (B) Left, mean +/− SEM Ca2+ responses of vagal neurons that responded to the Ca2+ addback following caffeine (10 mM) under control conditions (black, n=55 out of 233) or in the presence of SKF96365 (10 μM, red, n=13 out of 33) or Gd3+ (10 μM, green, n=5 out of 117). Blocked line denotes the 60-s application of drugs. All neurons responded to KCl (75mM) applied immediately before ionomycin.
Table 1.
SOCE responses are evoked in both nociceptive and non-nociceptive vagal neurons
| Control | Caffeine (10mM) | Thapsigargin (10μM) | Thapsigargin (10μM) | |||||
|---|---|---|---|---|---|---|---|---|
| Capsaicin-sensitive | Capsaicin-insensitive | Capsaicin-sensitive | Capsaicin-insensitive | Capsaicin-sensitive | Capsaicin-insensitive | AITC-sensitive | AITC-insensitive | |
| Responds to Ca2+ addback | 4/60 | 0/28 | 37/110 | 18/123 | 18/45 | 6/16 | 49/141 | 48/108 |
SOCE in non-neuronal cells occurs through the gating of Orai channels, which are low conductance Ca2+-selective channels. Orai channels (1, 2 and 3) are inhibited by SKF96365 and by trivalent cations such as La3+ or Gd3+ (Shuttleworth, 2012; Varnai et al., 2009). We tested the hypothesis that Orai channels were responsible for the SOCE evoked in vagal neurons. In the presence of 10μM SKF-96365, re-introduction of Ca2+ following 10μM thapsigargin-induced store depletion evoked significant SOCE in 35 of 102 neurons (Fig 3A). The peak mean response of these ‘addback-sensitive’ neurons was 31.2±3.6% of ionomycin compared to 28.0±2.0% of ionomycin under control conditions. Similarly, in the presence of 10μM SKF-96365, re-introduction of Ca2+ following caffeine-induced store depletion evoked significant SOCE in 13 of 33 neurons (peak mean response was 29.0±8.0% of ionomycin compared to 26.6±2.7% of ionomycin under control conditions)(Fig 3B). Therefore SKF96365 failed to inhibit SOCE. However, in the presence of 10μM Gd3+, re-introduction of Ca2+ following caffeine-induced store depletion failed to evoke significant SOCE: only 5 of 117 neurons responded (peak mean response was −2.8±0.8% of ionomycin). The data indicate that SOCE in vagal neurons is inhibited by Gd3+ but not SKF96365, which suggests that SOCE is not mediated by Orai channels.
We further addressed this hypothesis by detecting Orai mRNA expression using single cell RT-PCR. For these studies vagal neurons were dissociated and, after 2 hours in culture, individual neurons (>10μm) were pipette-picked for single cell analysis of multiple transcripts using intron-spanning primers (Fig 4). Single cells were only considered for analysis if they demonstrated a positive signal for the housekeeping gene β-actin. Neurons were excluded if either the bath sample or the negative reverse transcription samples yielded bands. 30% of neurons expressed mRNA for the nociceptor marker TRPV1. Out of 34 neurons, 31 neurons failed to express mRNA for any Orai channel, one neuron expressed mRNA for both Orai 1 and 3, one neuron expressed Orai 1 and one neuron expressed Orai 3. None of the neurons expressed Orai 2. As expected, RT-PCR of the whole vagal ganglia showed expression of all three Orai channels, likely derived from non-neuronal sources (mast cells, fibroblasts, glial cells, blood vessels, etc). Based on the pharmacological data and the single cell RT-PCR we conclude that the SOCE evoked in vagal neurons is not mediated by Orai channels.
Fig 4.

Representative data of single neuron RT-PCR detection of Orai channels. (A) Expression of housekeeping gene β-actin in individual neurons (1–4) and bath control (5B); top, with reverse transcription (RT+), and bottom, without reverse transcription (RT−). (B) Left, expression of TRPV1, Orai 1–3 in the same single neuron samples as (A). All samples were pooled for RT− for each gene. Right, expression of TRPV1, Orai 1–3 in whole nodose ganglia (NG).
2.3 Ca2+ stores in cultured vagal sensory neurites
Using confocal microscopy and the ER-specific fluorescent dye ER-Tracker, we investigated the presence of ER in an in vitro culture of dissociated vagal sensory neurons. Within 24 hours of dissociation, vagal neurons began to grow processes. By 3 days these processes developed into simple axonal structures (100 to 300μm long) terminating in growth cone/neurite structures. As expected, the soma of dissociated vagal neurons contained substantial ER within non-nuclear somal regions (Fig 5). We also observed significant ER-tracker staining throughout the neuronal processes, including the growth cone/neurite structures (Fig 5). In axonal/neurite structures, we found that thapsigargin (10μM) evoked Ca2+ transients in Ca2+ imaging studies of vagal sensory neurons following 3 day culture (Fig 6): 15.6±4.8% of ionomycin (n=7). Consistent with the somal studies, the nerve terminal Ca2+ transients were unaffected by genetic deletion of TRPA1 (15.6±4.8% of ionomycin (n=5)). Identical Ca2+ transients occurred in axonal and terminal regions (data not shown). These data demonstrate the presence of functional Ca2+ stores in vagal sensory nerve growth cone/neurites in culture.
Fig 5.

Confocal images of an in vitro 3 day culture of dissociated vagal neuron soma (top) and terminus (bottom) labeled with ER-tracker (200 nM, green) and Mito-tracker (100 nM, red).
Fig 6.

Mean +/− SEM Ca2+ responses of WT (n=7, red) and TRPA1−/ − (n=5, black) vagal neurites to thapsigargin (Thapsi, 10μM). Blocked line denotes the 60-s application of agonist. All neurite structures responded to KCl (75mM) applied immediately before ionomycin. (Insert) Mean +/− SEM Ca2+ responses of vagal neurites to the TRPA1 agonist AITC (100 μM).
2.4 Lack of store-operated Ca2+ entry in vagal sensory neurites
We next investigated whether the mechanism for SOCE is also present in the terminal neurites. Using the 3 day culture of vagal sensory neurons, we detected changes in [Ca2+]cyt in growth cone/neurite structures. The removal of extracellular Ca2+ for 5 minutes produced a substantial decrease in [Ca2+]cyt: −37.7±11% of ionomycin. Following the re-introduction of 2.2mM Ca2+, the [Ca2+]cyt slowly increased to 11.7±6.7% of ionomycin (n=7). In the presence of thapsigargin (10μM), the removal of extracellular Ca2+ evoked similar decreases in [Ca2+]cyt (−36.6±14% of ionomycin), and the reintroduction of 2.2mM Ca2+ again caused a minor increase of [Ca2+]cyt beyond the baseline (19.7±6.1% of ionomycin). There were no significant differences between the Ca2+ addback responses in the thapsigargin-treated and control neurons (Fig 7). The increase in [Ca2+]cyt upon re-introduction of extracellular Ca2+ was uniformly slow and minor in the neurites of vagal neurons, unlike the SOCE responses in somal experiments in Fig 5. Furthermore, Gd3+ (10μM) failed to block the minor Ca2+ addback responses in vagal neurites (data not shown). We conclude that there is no SOCE at the nerve terminal.
Fig 7.

Mean +/− SEM Ca2+ responses of vagal neurites to thapsigargin (10 μM, black, n=7) and vehicle (red, n=7) during changes in [Ca2+]extracellular. Blocked line denotes the 60-s application of agonist. All neurites responded to KCl (75mM) applied immediately before ionomycin.
3. Discussion
The ER is a major store of Ca2+ within the mammalian cell. Free cytosolic Ca2+ ions are pleiotropic biological effectors and, as such, require robust homeostatic control. The ER possesses the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pump that pumps Ca2+ from the cytoplasm into the ER. The ER also expresses ryanodine receptors, which are activated by ryanodine or high [Ca2+]cyt to open Ca2+ permeable channels in the ER. By inhibiting the SERCA pump with thapsigargin or by activating the ryanodine receptor with caffeine, it is possible to induce the release of Ca2+ specifically from the ER. In our studies of vagal dissociated neurons, thapsigargin evoked a Ca2+ transient, consistent with numerous reports of a functional ER within the soma of sensory neurons (Cohen et al., 1997; Cordoba-Rodriguez et al., 1999; Gover et al., 2007; Nicolson et al., 2002; Solovyova et al., 2002; Taylor-Clark et al., 2005).
Cytosolic Ca2+ regulation by the ER is dependent on multiple ER and plasmalemmal proteins, in particular the mechanism underlying SOCE (Parekh and Putney, 2005). Recent studies have supported a role of SOCE in neuronal Ca2+ signaling (Steinbeck et al., 2011), although no studies in vagal neurons have been published. SOCE can be visualized using cytosolic Ca2+ imaging during the removal and re-introduction of extracellular Ca2+. Under these conditions ER depletion of Ca2+ activates the ER Ca2+ sensor STIM1 leading to the gating of plasmalemmal Ca2+-permeable Orai (1, 2 or 3) channels (Varnai et al., 2009). Upon re-introduction of extracellular Ca2+, robust Ca2+ influx occurs through the store-operated Orai channel. Using the same techniques, we investigated SOCE in vagal neurons and neurites in vitro. Removal then re-introduction of extracellular Ca2+ had little effect on cytosolic Ca2+. However, following store depletion with either thapsigargin (1 or 10μM) or caffeine (10mM), re-introduction of Ca2+ evoked robust Ca2+ influx (“Ca2+ addback response” or SOCE) in approximately 25–40% of vagal sensory neurons. This is consistent with previous reports of SOCE in subsets of DRG neurons (Gemes et al., 2011; Shideman et al., 2009; Usachev and Thayer, 1999). Both caffeine and thapsigargin modulate other proteins apart from ER channels (Nehlig et al., 1992; Shmigol et al., 1995), however the similarity in the SOCE responses observed in the caffeine and thapsigargin studies presented here is consistent with a common SOC mechanism. Further characterization of these vagal neurons with AITC or capsaicin (both of which activate nociceptive sensory neurons) showed that SOCE could occur in both nociceptive and non-nociceptive neurons.
Orai channels are inhibited by SKF96365 and by extracellular trivalent lanthanoid ions (Shuttleworth, 2012; Varnai et al., 2009). We tested the hypothesis that SOCE in vagal neurons was mediated by Orai channels. Surprisingly 10μM SKF96365 had no effect on SOCE, although 10μM Gd3+ completely inhibited SOCE, suggesting that SOCE in vagal neurons was not mediated by Orai channels. To support these findings, we performed single neuron RT-PCR and investigated the expression of Orai 1, 2 and 3 mRNA. Consistent with our Ca2+ imaging data, we found virtually no expression of Orai in individual vagal sensory neurons. RT-PCR analysis of the whole vagal ganglia, however, yielded significant expression of all three Orai channels. This is unsurprising given the heterogeneous population of non-neuronal cells within sensory ganglia (mast cells, fibroblasts, glial cells, blood vessels, etc). We conclude that SOCE in vagal neurons is not mediated by Orai channels. At present, the published evidence supporting a role of Orai in sensory neuron SOCE is equivocal (Gemes et al., 2011; Shideman et al., 2009). Shideman et al (2009) demonstrated SKF96365 inhibition of SOCE in DRG lacking γ-secretase activity. In another study by Gemes et al (2011), SOCE in DRG neurons was blocked by La3+ but not by 50μM SKF96365. In addition, these authors presented western blot and quantitative RT-PCR analysis of whole ganglia demonstrating Orai1 (and STIM1) expression. Gemes et al (2011) concluded that SOCE in DRG was mediated by Orai. We believe that the lack of SKF96365-sensitivity of SOCE, and the lack of neuron-specific analysis of Orai expression in the Gemes et al (2011) study reduces confidence in their conclusions regarding the role of Orai in sensory neurons. A cautious interpretation of the data would suggest that SOCE occurs in a population of sensory neurons through an unknown mechanism that does not involve Orai channels.
The soma is not the location of signal transduction for sensory nerves in vivo. Vagal sensory fibers innervate the majority of the viscera with free nerve terminals. Electron microscopy of these 1–2 micro-thick fiber terminals are densely packed with mitochondria, but lack overt ER-like structures (Hung et al., 1973; Kikuchi, 1976; Neuhuber, 1987; von During and Andres, 1988). Despite these reports, we found evidence of a functional ER at vagal nerve growth cone/neurites. Using a 3 day culture of dissociated vagal neurons, we were able to investigate ER in the axons and neurites of the newly-grown fibers. ER tracker fluorescence was present throughout the axons and terminal structures. In addition, thapsigargin evoked Ca2+ transients in both axons and terminal structures. These data suggest that a functional ER extends into terminal regions of vagal sensory neurons in vitro.
The presence of ER at the peripheral terminals of vagal nerves would suggest that, as at the soma of neurons, these stores contribute to calcium signaling and homeostasis at the nerve terminal. Nevertheless we were unable to demonstrate SOCE in the nerve terminals of 3-day cultured vagal neurons. Reducing extracellular Ca2+ caused a reduction in neurite cytosolic Ca2+ (similar to soma), but re-introduction of Ca2+ (with and without store depletion through the application of thapsigargin) failed to evoke rapid Ca2+ influx. Neurite cytosolic Ca2+ did slowly increase to (and beyond) baseline levels upon Ca2+ re-introduction, but such responses are inconsistent with the rapid and substantial Ca2+ influx in SOCE (and were not blocked by Gd3+). It is possible that SOCE within the neurites was masked by Ca2+ uptake by local mitochondria (see Figure 5), although the relatively low Ca2+ affinity of mitochondrial uptake mechanisms (Gunter and Sheu, 2009) would suggest that a complete buffering of SOCE would be unlikely. We therefore conclude that SOCE is unlikely to contribute to signal transduction and excitability modulation at the nerve terminal. Instead, SOCE at the neuronal soma may play a modulatory role in Ca2+-regulated gene transcription, apoptosis and ER stress (Chan et al., 2006; Ng et al., 2009; Ong et al., 2007).
In conclusion, we present evidence of ER store-operated Ca2+ entry (SOCE) occurring in a subset of vagal neurons, but surprisingly, this mechanism is neither mediated by Orai channels, nor present at the nerve terminal.
4. Experimental Procedures
All experiments were approved by the University of South Florida Institutional Animal Care and Use Committee.
4.1 Dissociation of mouse vagal ganglia
Male mice were killed by CO2 asphyxiation followed by exsanguination. Vagal ganglia were immediately isolated and enzymatically dissociated from wild-type C57BL/6J and TRPA1−/− mice (Jackson Laboratory, Maine), using previously described methods (Taylor-Clark et al., 2008). Isolated neurons were plated onto poly-D-lysine and laminin-coated coverslips, incubated at 37°C. For acute 24 hour cultures, neurons were incubated in L-15 (supplemented with 10% FBS). For 3 day cultures, neurons were incubated in neurobasal medium (supplemented with B27; 1 mM glutamine; 50 U/mL penicillin/streptomycin; nerve growth factor, brain-derived neurotrophic factor, glial-derived neurotrophic factor, and neurotrophin-3 (all 20 ng/mL); and 20ug Fluoro-deoxy-uridine).
4.2 Fluorescent Ca2+ imaging
Cells were studied for changes in [Ca2+]i with Fura-2AM (4 μM, for 30 min at 37°C). For imaging, the coverslip was placed in a custom-built heated chamber (bath volume of 300 μL) and superfused by gravity at 8 ml/min with Locke solution (35°C; composition (mM): 136 NaCl, 5.6 KCl, 1.2 MgCl2, 2.2 CaCl2, 1.2 NaH2PO4, 14.3 NaHCO3, 10 dextrose (gassed with 95% O2–5% CO2, pH 7.3–7.4)). Changes in [Ca2+]cyt (Fura-2AM) were monitored by sequential dual excitation, 340 and 380 nm (emission 510 nm), measured by digital microscopy (CoolSnap HQ2; Photometrics, Surrey, BC, Canada) and analyzed by specialized software (Nikon Elements; Nikon, Melville, NY, USA). Neurons were visualized using 10X magnification and axons/terminals with 60X. The ratio images were acquired every 6 seconds. KCl (75 mM, 60 sec) was used to confirm voltage sensitivity. Ionomycin (5 μM, 60 sec) was used to obtain a maximal response.
For the analysis of [Ca2+]cyt, we used the excitation ratio 340/380 (to avoid calibrating the ratiometric responses to [Ca2+]cyt for each cell) and related all measurements to the peak positive response in each cell. Cells that had a robust response to the positive control were included in analyses. At each time point for each cell, data was presented as the percentage change in 340/380 ratio (R), and normalized to ionomycin maximum response: responsex = 100 × (Rx−Rbl)/(Rmax−Rbl), where Rx was the 340/380 ratio of the cell at a given time point, Rbl was the cell’s mean baseline 340/380 ratio measured over 60 s, and Rmax was the cell’s peak 340/380 ratio. Data are presented as mean peak response as a % of ionomycin peak.
4.3 Confocal microscopy
3 day cultured neuron-covered glass bottom plates were incubated (37°C) with ER tracker Blue-White (200 nM, for 30 min) and Mitotracker Orange (100 nM, for 30 min) in Neurobasal medium. The plates were then imaged using a Leica TCS SP2 laser scanning confocal microscope: ER tracker (excitation 374 nm, emission 430–640 nm) and MitoTracker (excitation 578 nm, emission 599 nm). To aid visualization, ER tracker Blue-White signals were converted to green using Nikon Elements software.
4.4 Single cell RT-PCR
First strand cDNA was synthesized from single vagal neurons using the SuperScript™ III CellsDirect cDNA Synthesis System (Invitrogen, Carlsbad, California) according to the manufacturer’s recommendations.
Cell Picking
Coverslips of dissociated neurons were constantly perfused by Locke’s solution and identified by using microscopy. Single cells were harvested into a glass-pipette (tip diameter 50–150 μm) pulled with a micropipette puller (Model P-97, Sutter Instrument Co.) by applying negative pressure. The pipette tip was then broken in a PCR tube containing RNAse Inhibitor (RNAseOUT, 2 UμL−1), immediately snap frozen and stored on dry ice. A sample of the bath solution from the vicinity of a neuron was collected from each coverslip for no-template experiments (bath control).
Reverse transcription
Samples were defrosted and lysed (10 min at 75°C). Then, poly(dT), random hexamer primers (Roche Applied Bioscience, Indianapolis, IN, USA), and dNTP’s were added. Samples were put into a BioRad C1000 Thermal Cycler (5 min at 65°C) and cooled on ice (1 min) before adding 5 × Buffer, dithiothrietol, 25mM MgCl2 and RNAseOUT. Half of the volume was reverse transcribed by adding SuperscriptIII RT for cDNA synthesis, whereas water was added to the remaining sample, which was used in the following as a negative (RNA) control. cDNA synthesis was performed in thermal cycler (75 min at 55°C, 5 min at 85°C, and 4°C forever).
Polymerase chain reaction
The PCR reaction mix (final volume of 20 μl) contained 0.5 U HotStar Taq Polymerase (Qiagen, Valencia, CA) supplemented with 2.5 mM MgCl2, PCR buffer, dNTPs, custom synthesized primers for β-Actin (sense: 5′-GTGGGAATGGGTCAGAAGG-3′, anti-sense: 5′-GAGGCATACAGGGACAGCA-3′; calculated product length 302 bp), ORAI1 channel (sense: 5′-GCCAGAGTTACTCCGAGGTG-3′, anti-sense: 5′-TGCAGGCACTAAAGACGATG-3′; calculated product length 210 bp), ORAI2 channel (sense 5′-ACACAGACGCTAGCCACGAG-3′, anti-sense 5′-GAGCTTCCTCCAGGACAGTG-3′; calculated product length 240 bp), or ORAI3 channel (sense 5′-GCTAAGCTCAAAGCCTCCAG-3′, anti-sense 5′-GTGTGGTGACTGGTGGACAG-3′; calculated product length 246 bp) (all intron-spanning and purchased from BioSynthesis), and 1.2 μl of each sample (cDNA, RNA control or bath control, respectively). After an initial activation step at 95°C for 15 min, samples were amplified by 50 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s and extension at 72°C for 1 min followed by a final extension at 72°C for 10 min. Products were then visualized in ethidium bromide-stained 1.2 % agarose gels.
4.5 Chemicals
α,β methylene ATP and SKF96365 were purchased from Tocris Bioscience (Minneapolis, MN). Ionomycin was purchase from LKT laboratories (St. Paul, MN). Thapsigargin was purchased from Alomone Labs (Jerusalem, Israel). Nerve growth factor was purchased from BD biosciences (San Jose, CA). Brain-derived neurotrophic factor, glial-derived neurotrophic factor and neurotrophin-3 were purchased from Prospec (East Brunswick, NJ). Mitotracker and ER tracker were purchased from Invitrogen (Carlsbad, CA). All other chemicals including allyl isothiocyanate, capsaicin, caffeine, dithiotrietol and GdCl3 were purchased from Sigma (St. Louis, MO).
Highlights.
Current evidence of store-operated Ca2+ entry (SOCE) in vagal sensory neurons is lacking.
-
ER store depletion evoked store-operated Ca2+ entry (SOCE) in vagal sensory neurons.
SOCE is inhibited by extracellular Gd3+ ions.
SOCE is not inhibited by Orai inhibitor SKF96365 and Orai mRNA is not present in vagal neurons.
SOCE is not observed in vagal neurites that contain ER.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Justin Shane Hooper, Email: jhooper@health.usf.edu.
Stephen H Hadley, Email: shadley@health.usf.edu.
Adithya Mathews, Email: amathew1@health.usf.edu.
Thomas E Taylor-Clark, Email: ttaylorc@health.usf.edu.
References
- Bahia PK, Suzuki R, Benton DC, Jowett AJ, Chen MX, Trezise DJ, Dickenson AH, Moss GW. A functional role for small-conductance calcium-activated potassium channels in sensory pathways including nociceptive processes. J Neurosci. 2005;25:3489–98. doi: 10.1523/JNEUROSCI.0597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MJ, Undem BJ. Bronchopulmonary afferent nerves. Respirology. 2003;8:291–301. doi: 10.1046/j.1440-1843.2003.00473.x. [DOI] [PubMed] [Google Scholar]
- Chan SL, Liu D, Kyriazis GA, Bagsiyao P, Ouyang X, Mattson MP. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J Biol Chem. 2006;281:37391–403. doi: 10.1074/jbc.M605552200. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Moore KA, Bangalore R, Jafri MS, Weinreich D, Kao JP. Ca(2+)-induced Ca2+ release mediates Ca2+ transients evoked by single action potentials in rabbit vagal afferent neurones. J Physiol. 1997;499(Pt 2):315–28. doi: 10.1113/jphysiol.1997.sp021929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Moore KA, Kao JP, Weinreich D. Calcium regulation of a slow post-spike hyperpolarization in vagal afferent neurons. Proc Natl Acad Sci U S A. 1999;96:7650–7. doi: 10.1073/pnas.96.14.7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–49. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Kao JP, Weinreich D. Calcium homeostasis in trigeminal ganglion cell bodies. Cell Calcium. 2007;41:389–96. doi: 10.1016/j.ceca.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Weinreich D. Role of calcium in regulating primary sensory neuronal excitability. Handb Exp Pharmacol. 2009:563–87. doi: 10.1007/978-3-540-79090-7_16. [DOI] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J, Pomorski P, Wisniewska MB, Kuznicki J. Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS One. 2011;6:e19285. doi: 10.1371/journal.pone.0019285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung KS, Hertweck MS, Hardy JD, Loosli CG. Ultrastructure of nerves and associated cells in bronchiolar epithelium of the mouse lung. J Ultrastruct Res. 1973;43:426–37. doi: 10.1016/s0022-5320(73)90019-1. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–5. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Kikuchi S. The structure and innervation of the sinu-atrial node of the mole heart. Cell Tissue Res. 1976;172:345–56. doi: 10.1007/BF00399517. [DOI] [PubMed] [Google Scholar]
- Lee MG, Macglashan DW, Jr, Undem BJ. Role of chloride channels in bradykinin-induced guinea pig airway vagal C-fibre activation. J Physiol. 2005;566:205–12. doi: 10.1113/jphysiol.2005.087577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Gao SB, Lv CX, Wu Y, Guo ZH, Ding JP, Xu T. Characterization of voltage-and Ca2+-activated K+ channels in rat dorsal root ganglion neurons. J Cell Physiol. 2007;212:348–57. doi: 10.1002/jcp.21007. [DOI] [PubMed] [Google Scholar]
- Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, Gamper N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl-channels. J Clin Invest. 2010;120:1240–52. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Brain Res Rev. 1992;17:139–70. doi: 10.1016/0165-0173(92)90012-b. [DOI] [PubMed] [Google Scholar]
- Neuhuber WL. Sensory vagal innervation of the rat esophagus and cardia: a light and electron microscopic anterograde tracing study. J Auton Nerv Syst. 1987;20:243–55. doi: 10.1016/0165-1838(87)90153-6. [DOI] [PubMed] [Google Scholar]
- Ng SW, Nelson C, Parekh AB. Coupling of Ca(2+) microdomains to spatially and temporally distinct cellular responses by the tyrosine kinase Syk. J Biol Chem. 2009;284:24767–72. doi: 10.1074/jbc.M109.011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson TA, Bevan S, Richards CD. Characterisation of the calcium responses to histamine in capsaicin-sensitive and capsaicin-insensitive sensory neurones. Neuroscience. 2002;110:329–38. doi: 10.1016/s0306-4522(01)00561-9. [DOI] [PubMed] [Google Scholar]
- Ong HL, Liu X, Sharma A, Hegde RS, Ambudkar IS. Intracellular Ca(2+) release via the ER translocon activates store-operated calcium entry. Pflugers Arch. 2007;453:797–808. doi: 10.1007/s00424-006-0163-5. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Tate S, Woolf CJ. Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov. 2009;8:55–68. doi: 10.1038/nrd2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shideman CR, Reinardy JL, Thayer SA. gamma-Secretase activity modulates store-operated Ca2+ entry into rat sensory neurons. Neurosci Lett. 2009;451:124–8. doi: 10.1016/j.neulet.2008.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Kostyuk P, Verkhratsky A. Dual action of thapsigargin on calcium mobilization in sensory neurons: inhibition of Ca2+ uptake by caffeine-sensitive pools and blockade of plasmalemmal Ca2+ channels. Neuroscience. 1995;65:1109–18. doi: 10.1016/0306-4522(94)00553-h. [DOI] [PubMed] [Google Scholar]
- Shuttleworth TJ. Orai3--the ‘exceptional’ Orai? J Physiol. 2012;590:241–57. doi: 10.1113/jphysiol.2011.220574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyova N, Fernyhough P, Glazner G, Verkhratsky A. Xestospongin C empties the ER calcium store but does not inhibit InsP3-induced Ca2+ release in cultured dorsal root ganglia neurones. Cell Calcium. 2002;32:49–52. doi: 10.1016/s0143-4160(02)00094-5. [DOI] [PubMed] [Google Scholar]
- Steinbeck JA, Henke N, Opatz J, Gruszczynska-Biegala J, Schneider L, Theiss S, Hamacher N, Steinfarz B, Golz S, Brustle O, Kuznicki J, Methner A. Store-operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp Neurol. 2011;232:185–94. doi: 10.1016/j.expneurol.2011.08.022. [DOI] [PubMed] [Google Scholar]
- Taylor-Clark T, Undem BJ. Transduction mechanisms in airway sensory nerves. J Appl Physiol. 2006;101:950–9. doi: 10.1152/japplphysiol.00222.2006. [DOI] [PubMed] [Google Scholar]
- Taylor-Clark TE, Kollarik M, MacGlashan DW, Jr, Undem BJ. Nasal sensory nerve populations responding to histamine and capsaicin. J Allergy Clin Immunol. 2005;116:1282–8. doi: 10.1016/j.jaci.2005.08.043. [DOI] [PubMed] [Google Scholar]
- Taylor-Clark TE, McAlexander MA, Nassenstein C, Sheardown SA, Wilson S, Thornton J, Carr MJ, Undem BJ. Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J Physiol. 2008;586:3447–59. doi: 10.1113/jphysiol.2008.153585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol. 1999;519(Pt 1):115–30. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Hunyady L, Balla T. STIM and Orai: the long-awaited constituents of store-operated calcium entry. Trends Pharmacol Sci. 2009;30:118–28. doi: 10.1016/j.tips.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von During M, Andres KH. Structure and functional anatomy of visceroreceptors in the mammalian respiratory system. Prog Brain Res. 1988;74:139–54. doi: 10.1016/s0079-6123(08)63008-3. [DOI] [PubMed] [Google Scholar]