
Figure 9.
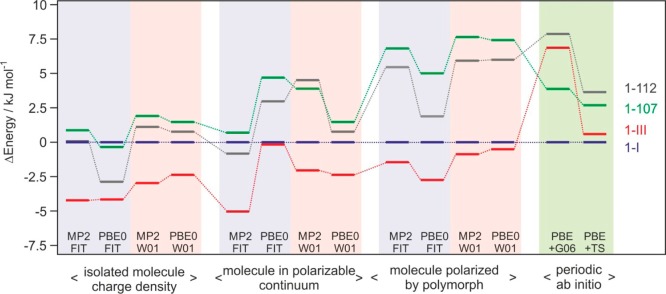
Lattice energy differences of the four methyl paraben polymorphs. Isolated molecule charge density = relaxed structures obtained using the CrystalOptimizer methodology; molecule in polarizable continuum = relaxed structures with average polarization from the PCM model; molecule polarized by polymorph = relaxed structures with the addition of the induction energy; periodic ab initio = density functional theory relaxations with dispersion correction. PBE0 or MP2: PBE0/6-31G(d,p) or MP2/6-31 (d,p) level of theory used for calculating molecular geometry and charge density; FIT or W01: FIT or Williams01 repulsion-dispersion potential parameters; G06 or TS: Grimme or Tkatchenko and Scheffler dispersion corrections. Tie lines have been added to show the changes in relative ordering.