

SUMMARY
Multicentric osteolysis with arthropathy (MOA; MIM 605156) is an inherited osteolyses and arthritis syndrome resulting from loss of matrix metalloproteinase 2 (MMP-2). We recently demonstrated that Mmp2–/– mice represent a unique model for the study of the human disease, sharing many features of the human syndrome including skeletal dysplasia and defects in osteoblast behavior. We therefore sought to explore the secondary molecular effects of MMP-2 loss, which coexist with the underlying skeletal and osteoblast phenotypes. We used quantitative real-time RT-PCR (qRT-PCR) to measure osteoblast-related gene expression through ex vivo osteoblast differentiation of bone marrow stromal cells (BMSC) from Mmp2−/− and Mmp2+/+ mice. We used western blot to measure osteopontin (OPN) serum levels and immunohistochemical staining to examine bone expression. MMP-2 expression was inhibited in SaOS2 cells using siRNA, and decreased MMP-2 expression at both RNA and protein levels was confirmed by qRT-PCR and western blot, respectively. Mmp2−/− BMSC induced to differentiate into osteoblasts were shown to significantly upregulate OPN and bone sialoprotein (BSP) expression levels compared with controls. Transcriptional upregulation was maintained in vivo, as demonstrated by increased levels of OPN in serum and bone in Mmp2−/− mice. These effects are generalizable because siRNA-mediated inhibition in cultured cells also upregulated OPN and BSP. OPN and BSP are known to affect MMP-2 expression and activity but have not previously been shown to be regulated by MMP-2. Identification of this newly defined circuitry provides insight into the potential molecular landscape underlying the MOA phenotype and highlights a pathway that might play a role in normal bone homeostasis.
INTRODUCTION
The ‘vanishing bone’ or inherited osteolyses and arthritis syndromes represent a heterogeneous group of skeletal disorders that provide a unique opportunity to define the molecular pathways underlying skeletal growth and development (Mosig et al., 2008). We originally demonstrated that multicentric osteolyses with arthropathy (MOA; MIM 605156) is caused by mutations in the matrix metalloproteinase 2 gene (MMP2) (Al Aqeel et al., 2000; Martignetti et al., 2001). Affected individuals have marked age-related bone density loss, craniofacial defects and articular cartilage destruction (Al Aqeel et al., 2000; Inoue et al., 2006; Martignetti et al., 2001; Mosig et al., 2007). Mmp2−/− mice have recently been shown by others and us to share key phenotypic features with human patients (Inoue et al., 2006; Mosig et al., 2007). Potentially identifying the cellular basis of MMP-2-mediated skeletal defects, we have shown that the murine defects are associated with developmentally restricted changes in osteoblast, osteocyte and osteoclast numbers in vivo and that cultured Mmp2−/− bone marrow cells are deficient in osteoblast and osteoclast differentiation (Inoue et al., 2006; Mosig et al., 2007). In addition, targeted MMP-2 inhibition using siRNA in cultured cell lines suppresses osteoblast proliferation (Mosig et al., 2007). Intriguingly, the MMP-2 deficiency phenotype is counterintuitive because loss of a functional protease would be predicted to lead to a decrease in resorption and, therefore, to bone overgrowth. Thus, the Mmp2−/− mouse provides an excellent experimental model for investigating the molecular changes contemporaneous with the unexpected skeletal phenotype.
Osteopontin (OPN, SPP1) and bone sialoprotein (BSP, IBSP) are structurally related RGD-containing members of the SIBLING (small integrin-binding ligand N-linking glycoprotein) family, which are highly expressed in bone extracellular matrix (Alford and Hankenson, 2006). Their expression can be regulated on multiple levels, including through the activation of the transcription factor Runx2 (also known as Cbfa1), the Notch signaling pathway and the vitamin D receptor (Alford and Hankenson, 2006; Cowles et al., 1998; Merry et al., 1993; Noda et al., 1990; Sato et al., 1998; Shen and Christakos, 2005; Sommer et al., 1996). Although structurally related, temporally distinct expression patterns between OPN and BSP most probably exist due to their opposing functions in mineralization. OPN suppresses crystal growth and is expressed throughout differentiation, then downregulated during mineralization phases. BSP is expressed late in osteoblast differentiation and is necessary for mineralization (Ganss et al., 1999; Giachelli and Steitz., 2000; Gordon et al., 2007). OPN and BSP are also expressed by osteoclasts under the control of nuclear factor κB (NF-κB) (Alford and Hankenson, 2006; Cowles et al., 1998; Merry et al., 1993; Noda et al., 1990; Sato et al., 1998; Shen and Christakos, 2005; Sommer et al., 1996) and facilitate the adhesion, proliferation, migration, differentiation and activity of osteoblasts, osteoclasts and their precursors (Asou et al., 2001; Bellahcène et al., 2000; Liu et al., 2004; Shapses et al., 2003). The cellular effects of OPN and BSP are transduced through their binding to a number of partners in the bone matrix and on cell surfaces, often via their RGD domains (Alford and Hankenson, 2006). For example, OPN binding to integrins can activate intracellular signaling pathways and induce cell response (Liu et al., 1997; Teti et al., 1998). Alternatively, OPN can bind to growth factors or their binding proteins, thus secondarily influencing cell behavior (Nam et al., 2000). Furthermore, simultaneous binding by BSP to integrin and MMP-2 functionally localizes MMP-2 to the cell surface and facilitates migration of bone marrow stromal cells (BMSCs) (Karadag and Fisher, 2006). Indeed, and most relevant to our study, OPN and BSP activities have also been linked to increased expression and activation of MMP-2 (Mi et al., 2006; Philip et al., 2001; Teti et al., 1998; Wai et al., 2005).
TRANSLATIONAL IMPACT.
Clinical issue
Multicentric osteolysis with arthropathy (MOA; MIM #605156) is an autosomal dominant ‘vanishing bone’ and arthritis disorder caused by loss of matrix metalloproteinase 2 (MMP-2), a protease that degrades several matrix proteins, growth factors, binding proteins and receptors. The finding that complete loss of MMP-2 activity causes this osteolytic disorder remains counterintuitive: why does loss of a protease result in bone destruction rather than bone overgrowth? Recent work by these authors with MMP-2-deficient mice showed that the features of MOA, which are partially recapitulated in the mouse, are due to a loss of osteoblast growth and integrity. However, the precise molecular etiology of MMP-2 loss, and the role of MMP-2 in normal bone regulation and growth, were unclear.
Results
Here, the authors examined the osteoblastic differentiation of MMP-2-deficient cells in vitro to investigate the secondary molecular changes that occur in MMP-2-deficient osteoblasts, with the aim of gaining insight into the overall molecular features of MMP-2 deficiency. They found that loss of MMP-2 results in an osteoblastic gene expression pattern that is notable for upregulation of osteopontin (OPN) and bone sialoprotein (BSP), which are two members of the family of extracellular matrix, non-collagenous proteins known as SIBLING proteins. OPN upregulation was also detected in vivo in mouse serum and on the bone surface. Finally, they showed that OPN and BSP upregulation are specifically and directly caused by loss of MMP-2, as siRNA-mediated inhibition of MMP-2 in SaOS2 cells caused upregulation of OPN and BSP at both the mRNA and protein level.
Implications and future directions
These studies identify previously unidentified details of the circuitry linking MMP-2 with OPN and BSP. Although previous studies have demonstrated that OPN and BSP can upregulate MMP-2, the reverse effect has not been reported. Future work will be required to establish whether increased levels of OPN and BSP themselves contribute to the pathogenesis of MOA, and whether these proteins therefore represent promising therapeutic targets.
Given the shared genetic and phenotypic features present in MOA and the Mmp2−/− mice, we hypothesized that genetic analysis of MMP-2-deficient murine osteoblasts would provide an excellent model system for investigating the molecular pathways that might be altered in both the mouse model and human disease. Therefore, we examined the gene expression profile of Mmp2−/− osteoblasts and, through siRNA-mediated gene silencing, verified these findings on both the RNA and protein levels in MMP-2 wild-type cultured cells. On the basis of these findings, we provide the first evidence that loss of MMP-2 results in the upregulation of OPN and BSP, defining a previously unrecognized candidate circuitry between these matrix-related proteins.
RESULTS
Mmp2−/− BMSC upregulate OPN and BSP expression
To begin exploring the coexistent and downstream effects of Mmp2 deficiency on well-characterized markers of osteoblast differentiation, we directly examined the expression of these genes using parallel cultures of Mmp2−/− and Mmp2+/+ murine BMSCs. mRNA was collected at four time points following induction of osteoblast differentiation, and expression levels were compared using quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR). Candidate markers included Cbfa1, OPN, BSP, collagen type 1α, parathyroid hormone receptor (PTHR), MMP-9, TIMP-2 and LRP5. We have previously demonstrated that Mmp2−/− cells are deficient at producing alkaline phosphatase-positive colonies (Mosig et al., 2007). Using this panel of osteoblast differentiation markers, no differences were noted between Mmp2−/− and control cells for the majority of genes. By marked contrast, at 7 and 14 days and 14 and 21 days after addition of differentiation media, OPN and BSP expression were significantly upregulated, respectively, in Mmp2−/− cells (Fig. 1A,B). For both OPN and BSP, expression levels were increased on average threefold (P<0.05). No other significant changes were observed in our interrogated candidate genes between the different genotypes. This included the major MMP-2 substrate, collagen I, and its endogenous inhibitor, TIMP-2 (Fig. 1C).
Fig. 1.

Upregulation of OPN and BSP mRNA in Mmp2−/−bone marrow stromal cells. (A-C) Mmp2-/- and Mmp2+/+ bone marrow stromal cells were extracted and grown in the presence of ascorbic acid and dexamethasone. RNA was collected at days 0, 4, 7, 14 and 21 during differentiation. No differences between Mmp2-/- and Mmp2+/+ cells were noted early in differentiation. (A) OPN was upregulated about threefold at 7 and 14 days in Mmp2-/- cells compared with Mmp2+/+ cells. (B) BSP was upregulated about threefold at 14 and 21 days in Mmp2−/− cells compared with Mmp2+/+ cells. (C) Expression levels of collagen I (COLA1), CBFA, TIMP-2, LRP5 and PTHR were equivalent in Mmp2−/− and Mmp2+/+ cells throughout (data shown at 0 and 14 days). Values represent the average mRNA fold change over Mmp2+/+ day 0 levels of three separate experiments performed in triplicate; error bars represent standard deviations; *P<0.05.
Loss of Mmp2 results in OPN upregulation in vivo
After identifying this ex vivo difference, we next sought to explore whether this transcriptional upregulation was also present in vivo. We measured OPN protein levels in the serum of 6-week-old Mmp2+/+, Mmp2+/- and Mmp2−/− mice by western blot. In accord with our mRNA findings in differentiating osteoblasts, OPN levels were upregulated approximately twofold (P<0.05) in Mmp2−/− serum compared with serum from Mmp2+/- and Mmp2+/+ mice (Fig. 2). Of note, Mmp2+/- mice, which have half normal levels of MMP-2 but do not share the skeletal phenotype present in Mmp2−/− mice (Mosig et al., 2007), displayed no measurable difference in OPN levels (Fig. 2).
Fig. 2.
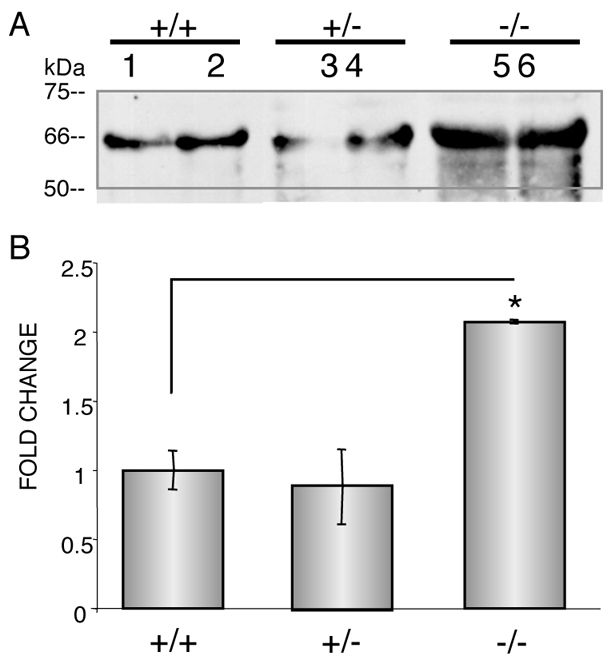
Increased OPN levels in Mmp2−/−serum. (A) Western blot analysis revealed increased levels of OPN in 6-week-old Mmp2−/− serum. Numbers represent animal numbers: 1 and 2 are Mmp2+/+, 3 and 4 are Mmp2+/- and 5 and 6 are Mmp2-/-. (B) Quantification of western blot. Values represent the average level in the two serum samples per genotype; error bars represent standard deviations; *P<0.05.
OPN is known to be expressed in a number of different tissues, and its expression is increased in response to inflammation (Giachelli and Steitz, 2000). We therefore analyzed a panel of serum markers of inflammation in the Mmp2−/− and Mmp2+/+ mice. These markers included CD30L, Fas-L, Fractalkine, GCSF, GM-CSF, IFN-γ, IL-1α, IL1-β, IL-2, IL-3, IL-4, IL-6, IL-9, IL-10, IL-12, IL-13, IL-17, MCP-1, MCSF, MIG, MIP-1α, MIP-1γ, RANTES, SDF-1, TCA-3, TECK, TIMP-1, TIMP-2, TNFα, sTNF-RI and sTNF-RII. No differences in inflammatory responses between the mice was identified (data not shown).
Given that the primary phenotype of the mouse and human affects the skeletal system (Al Aqeel et al., 2000; Inoue et al., 2006; Martignetti et al., 2001; Mosig et al., 2008), we refocused our attention on OPN expression in bone. Immunohistochemical staining of Mmp2−/− femurs, tibias and vertebrae from 4-week-old mice revealed an increase in OPN localization and intensity at the bone surfaces compared with age-matched Mmp2+/+ control samples (Fig. 3). These differences were uniquely developmentally regulated. Interestingly, staining of 4-day-old and 12-week-old bones from Mmp2−/− and Mmp2+/+ mice revealed no difference in the amount of OPN staining (Fig. 3).
Fig. 3.
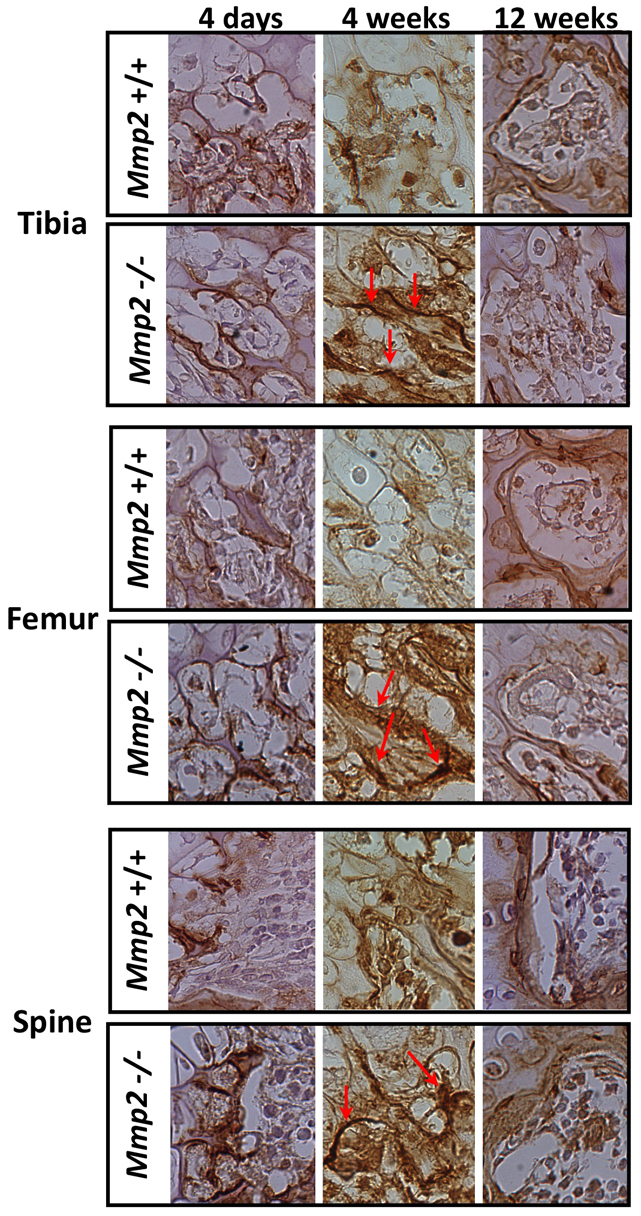
Immunohistology of 4-week-old mice shows increased OPN. OPN concentration and localization to tibia, femur and vertebrae bone surfaces of Mmp2−/− mice. Arrows indicate concentrated regions of OPN staining on bone surface. No differences were seen between genotypes at 4 days or 12 weeks of age. Paraffin-embedded sections were stained with goat anti-murine OPN IgG. Images are representative of staining of two sections per mouse, six mice per genotype.
siRNA-mediated knockdown of MMP-2 in osteoblast-like SaOS2 cells upregulates OPN and BSP
Theoretically, unrecognized secondary aspects of murine MMP-2 deficiency (Alford and Hankenson, 2006), could be indirectly responsible for the observed OPN and BSP upregulation. We therefore used the human osteosarcoma cell line SaOS2 to test whether MMP-2 loss caused upregulation of OPN and BSP. We previously generated and tested siRNA molecules (referred to here as siMMP2) that specifically inhibited MMP-2 expression (i.e. did not affect MMP-9 and MMP-14 expression) (Mosig et al., 2007) in both human and mouse cell lines. MMP-2 knockdown resulted in decreased proliferation rates in both cell lines (Mosig et al., 2007). On the basis of these findings, we generated multiple independent SaOS2-siMMP2 stable cell lines using retroviral infection of an siMMP2-containing plasmid and selected two independent clones (clones A and B) for these studies. We achieved ∼70% knockdown of MMP-2 at both the mRNA and protein levels (Fig. 4A). In these genetically engineered cells, OPN and BSP were again upregulated at both the mRNA (>sixfold and >20-fold, respectively; Fig. 4B) and protein (>twofold and 1.7-fold, respectively; Fig. 4C) levels compared with a control cell line transfected with siRNA against luciferase (siLuc).
Fig. 4.

SaOS2 stable siMMP2 clones overexpress OPN and BSP. (A) Approximately 70% knockdown of mRNA encoding MMP-2 (above) and MMP-2 protein activity levels (below) were achieved in isolated clones A and B. E, siLuc-transfected control cell line. (B,C) SiMMP-2 stable clones showed high upregulation of OPN and BSP at mRNA (B) and protein levels (C). Quantification of immunoblots are shown in italics below the blots. Values represent the average of three separate experiments; error bars represent standard deviations; *P<0.05.
DISCUSSION
This report presents the first evidence that loss of MMP-2 expression can directly regulate OPN and BSP expression. The upregulation was present in genetically engineered mice in vivo, in BMSC derived from Mmp2−/− mice and in cultured human SaOS2 cells following siRNA-mediated downregulation of MMP-2. Our combined ex vivo and in vivo studies (Figs 2, 3) demonstrated that MMP-2-deficient bone, and possibly other tissues, overexpress OPN in a temporally specific manner. Taken together, these observations add a new level of recognized crosstalk to the increasingly related biological overlap of MMP-2 with OPN and BSP. It was previously recognized that both OPN and BSP can each increase transcription, activation and functional localization of MMP-2 (Fedarko et al., 2004; Karadag and Fisher, 2006; Liu et al., 2004; Mi et al, 2006; Philip et al, 2001; Teti et al, 1998); however, the converse relationship has not been reported. Another potentially interesting relationship to this newly defined circuitry was recently highlighted using a degradomics approach in which OPN was shown to be a substrate of MMP-2 (Dean and Overall, 2007). Taken together, our results suggest that either increases or decreases in MMP-2 will ultimately affect OPN levels.
The finding that loss of MMP-2 activity results in distinctive skeletal features in humans and mice highlights the importance of the enzyme in skeletal development but leaves the underlying mechanism(s) and etiology of this counterintuitive phenotype unresolved. Our previous work demonstrating that loss of MMP-2 activity leads to defects in osteoblast and osteoclast differentiation ex vivo and that siRNA-mediated reduction of MMP-2 decreases SaOS2 and MC3T3 osteoblastic cell proliferation suggested that the in vivo bone loss is at least partially due to a decrease in osteoblastic abilities (Mosig et al, 2007). In this paper, we have further characterized the osteoblastic deficits caused by MMP-2 loss to include transcriptional dysregulation of OPN and BSP. Interestingly, rather than a broad defect or delay in markers of osteoblast differentiation, this result seems to be specific to OPN and BSP. The expression levels of the other investigated genes (encoding Cbfa1, collagen type1α, PTHR, MMP-9, TIMP-2 and LRP5) were not measurably changed between Mmp2−/− and Mmp2+/+ cells. Furthermore, specific OPN and BSP upregulation was recapitulated in an osteoblast cell line, SaOS2, which was not differentiating. Even in the cell culture environment, MMP-2-deficient cells respond by increasing OPN and BSP expression.
The finding that decreased MMP-2 in bone cells results in the transcriptional upregulation of OPN and BSP, which have in turn been shown to upregulate MMP-2 (Asou et al, 2001; Bellahcène et al, 2000; Liu et al, 2004; Shapses et al, 2003), suggests the existence of a previously unrecognized feedback or compensatory regulatory mechanism. As OPN and BSP have been shown by others to increase cell adhesion, migration and differentiation (Bellahcène et al., 2000; Karadag and Fisher, 2006; Kojima et al., 2004; Liu et al., 2004; Suzuki et al., 2002), MMP-2-deficient cells might upregulate the expression of these molecules in an attempt to recover multiple defects in vivo. For example, overexpression of OPN and BSP facilitates attachment and migration of a number of normal and cancer cell lines (Mi et al., 2006; Philip et al., 2001; Teti et al., 1998), often through an interaction with MMP-2 (Karadag and Fisher, 2006). In the MMP-2-deficient mouse we see temporal upregulation of OPN specifically in the bones of 4-week-old mice, but not in 4-day-old or 12-week-old mice. We have previously demonstrated that much of the murine cortical bone abnormalities seen immediately after birth partially resolve by 4 weeks of age, and more so by 12 weeks (Mosig et al., 2007). It is possible that the upregulation of OPN contributes to this recovery, although further research remains to prove this functional relationship. Taken together with our studies, the coordinated expression and interaction of MMP-2 with BSP and OPN suggests that their self-regulation in bone cells is important. The mechanism(s) through which loss of MMP-2 results in this increase is currently unknown but could represent an important control point in bone cell homeostasis.
The developmental expression pattern of OPN has not been previously investigated and, therefore, our result demonstrating temporally limited overexpression in mouse bone is interesting (Fig. 3). We did not identify differences in the level of expression in Mmp2+/+ mice across the three time-points tested, although there was an increase in OPN expression in Mmp2−/− mice at 4 weeks of age (Fig. 3). Previous data has indicated that Mmp2−/− bones are recovering from initial cortical bone defects at this age (Mosig et al., 2007) and it is therefore possible that OPN and BSP represent crucial modulators of this process. Interestingly, expression of another SIBLING protein, DMP-1, was shown to be decreased at this time-point, probably due to a decreased number of osteocytic processes and canaliculi (Inoue et al., 2006). Alternatively, one could speculate that overexpression of these proteins, although integral for the rescue of some deficits, could accelerate or contribute to the development of other aspects, such as reduced bone mineral density or arthritis.
The increased OPN and BSP levels in MMP-2-deficient cells could begin to provide some mechanistic basis towards understanding the skeletal defects associated with the MMP-2 deficiency syndrome, although the precise mechanism remains to be fully elucidated. Our findings parallel those seen in other experimental systems. For example, similar to our findings of progressive bone loss (Mosig et al., 2007), OPN levels are increased in weight-unloading bone loss experiments, whereas MMP-2 is downregulated (Zhong et al., 2005). The increased levels of OPN might also contribute to development of the arthritic phenotype in MMP-2-deficient humans and mice. The Mmp2−/− mouse has histologic evidence of arthritis as early as 12 weeks of age (Mosig et al., 2007). OPN is recognized as acting as an inflammatory cytokine and increased OPN levels have been observed in osteoarthritis as well as rheumatoid arthritis, where it has been shown to stimulate inflammatory cells (Ohshima et al., 2002; Xu et al., 2005; Yagi et al., 2005). Ultimately, the MMP-2 mouse model could provide a unique model system for exploring the expanded functional and coordinated relationship between MMP-2 and OPN and BSP.
MATERIALS AND METHODS
Mmp2−/− mice
The Mmp2−/− mice used in these studies were originally generated by Itoh and co-workers by targeted deletion of the promoter, 5′-UTR, and exon 1 of the Mmp2 gene (Itoh et al., 1997). Genotyping was done by PCR of genomic DNA extracted from tail snips. Primers amplifying exon 1 were used to confirm the presence of the wild-type allele, and neo cassette primers targeted the null allele: exon 1 forward, 5′-CAGACTTCCCTGGTGGCT-3′ exon 1 reverse, 5′-CACCATCGCCCATCATCAAG-3′ neomycin forward, 5′-AGGATCTCCTGTCATCTCACCTTGCTCC-3′ neomycin reverse, 5′-AAGAACTCGTCACGAAGGCGATAGAAGG-3′. The reaction conditions for exon 1 primers were: 95°C for 10 minutes, followed by 35 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 45 seconds and a final extension at 72°C. Reaction conditions for neomycin primers were identical except for an annealing temperature of 62°C.
All procedures were performed in accordance with an institutionally approved protocol for the use of animals in research.
Cell culture and siRNA silencing
Osteoblasts
Marrow from femurs and tibias of 8-week-old mice was extracted in αMEM containing FBS and penicillin and streptomycin antibiotics. Cells were allowed to adhere and the non-adherent population removed. After 2 days in culture, non-adherent cells were removed and cells re-plated. After 4 more days in culture, adherent cells were isolated and re-plated in osteoblast differentiation medium containing 100 μM ascorbic acid and 5 nM dexamethasone at 30,000 cells/ml, and subsequently collected for mRNA expression analysis as described below. Differentiation assays were repeated three times in triplicate. Although results were replicated in each experiment, the degree of upregulation varied between trials and resulted in large standard deviations (Fig. 1).
SaOS2 siMMP2 stable cell lines
Four siMMP2 oligonucleotides (Dharmacon) were screened as previously described (Mosig et al., 2007) and the most specific and efficient were chosen for the development of stable cell lines. Forward and reverse sequences for the MMP-2 shRNA construct were 5-GATCCCCGACAGTGGATGATGCCTTTTTCAAGA-GAAAAGGCATCATCCACTGTCTTTTTGGAAA-3 and 5-AGCTTTTCCAAAAAGACAGTGGATGATGCCTTTTCTCTT -GAAAAAGGCATCATCCACTGTCGGG-3, respectively. Forward and reverse sequences for the Luc shRNA construct (targeting the luciferase gene not present in our parental cell line) were 5-GATCCCCTACTTCGAAATGTCCGTTCTTCAAG-AGAGAACGGACATTTCGAAGTATTTTTGGAAA-3 and 5-AGCTTTTCCAAAAATACTTCGAAATGTCCGTTCTCTCTT-GAAGAACGGACATTTCGAAGTAGGG-3, respectively. Oligonucleotides were ligated into pSUPER.retro vector (Oligoengine, Seattle, WA) as per the manufacturer's protocol. Briefly, forward and reverse oligonucleotides were annealed to form a duplex. The annealed oligos were then ligated into the BglII-HindIII cleavage site within the pSUPER.retro vector prelinearized with the same restriction enzymes. pSUPER.retro-siMMP2 vector or pSUPER.retro-siLuc vector was transfected into Phi-NX (‘Phoenix’) packaging cell line kindly provided by a generous gift from Gary Nolan (Stanford, CA) to produce ecotropic retroviral supernatants. Phoenix cells were cultured in DMEM supplemented with 10% FBS. The day before transfection, Phoenix cells were seeded in 10-cm dishes (3×106 cells per dish) in order to reach 60% confluence at the time of transfection. Cells were transfected with 10 μg of viral vector DNA using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen, San Diego, CA). At 48 hours after transfection, culture medium was filtered through a 0.45-μm filter and the viral supernatant was used for SaOS2 cell infection after addition of 4 μg/ml of polybrene (Sigma). After infection, SaOS2 cells were incubated at 37°C in 5% CO2 for 5 hours, followed by a second round of infection. After a second 5-hour incubation, medium was changed with fresh medium and SaOS2 cells were allowed to recover for 48 hours at 37°C in 5% CO2. Infected cells were selected by adding puromycin (1 μg/ml) and clones were picked and grown under further selection. MMP-2 expression in siLuc- and siMMP2-infected SaOS2 cells was analyzed by qRT-PCR and zymography as described below.
RNA isolation and qRT-PCR
RNA was collected (Qiagen RNeasy Mini Kit) and treated with DNase (Qiagen). A total of 0.5 μg of RNA was reverse-transcribed per reaction using first-strand complementary DNA synthesis with random primers (Iscript, Bio-Rad Laboratories). qRT-PCR was performed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems). Cycle number values were normalized separately against both GAPDH and β-actin and the values confirmed to be equivalent. Values were analyzed as fold change compared with siLuc vector or Mmp2+/+ control. Data shown is the average of three separate experiments performed in triplicate. Statistical significance was determined by comparing fold change using the unpaired, two-tailed, Student's t-test assuming equal variances. Primers used for real-time RT-PCR are given in supplementary material Table S1.
Immunoblot analysis and zymogram
Serum samples were collected from age- and sex-matched mice and SaOS2 cell extracts were harvested in radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology) using a standard protocol. Serum-free conditioned medium was collected and concentrated. Equal amounts of protein (50 μg; determined by the Bio-Rad DC protein quantification assay) were loaded into a pre-cast NuPage 12% Bis-Tris gel (Invitrogen) and separated at 120 V for 90 minutes according to the manufacturer's protocol. Proteins were then transferred using the NuPage semi-dry transfer apparatus (Invitrogen) to nitrocellulose membranes (Bio-Rad) at 40 V for 1 hour using the manufacturer's instructions. Membranes were probed with anti-OPN antibody (#AF808-mouse and #AF1433-human, R&D Systems) or anti-BSP antibody (#AB1854, Chemicon) followed by secondary antibodies to goat (#305-035-003, Jackson ImmunoResearch Laboratories) or rabbit (#NA934V, GE Healthcare UK), respectively. Lumi-light western blotting substrate (Roche) was used for detection. Enhanced chemiluminescent immunoblot images were analyzed by scanning densitometry and quantified with a BIOQUANT NOVA imaging system. Values were expressed as fold change relative to control. For gelatin zymography, 5 μg of conditioned media were run on 10% gelatin zymogram gels (Novex), which were developed and stained according to the manufacturer's instructions, as previously described (Mosig et al., 2007).
Histology and immunohistochemistry
Tibias, femurs and vertebrae were isolated from age-matched Mmp2−/− and Mmp2+/+ mice and fixed in 10% phosphate buffered formalin overnight and decalcified in cold EDTA, as previously described (Mosig et al., 2007). Sagittal sections of 4 μm thickness were stained with antibody to OPN (#AF808-mouse, R&D Systems). The signal was visualized using the anti-goat HRP-DAB Cell and Tissue Staining Kit (CTS008, R&D Systems) and counterstained with hematoxylin. Timing of DAB reactions was optimized to visualize osteoblast and cortical bone staining in Mmp2−/− mice.
Supplementary Material
Acknowledgments
FUNDING: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Footnotes
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS: R.A.M. designed and performed experiments and wrote the manuscript; J.A.M. designed the research and wrote the manuscript.
SUPPLEMENTARY MATERIAL: Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.007914/-/DC1
References
- Al Aqeel A., Al Sewairi W., Edress B., Gorlin R. J., Desnick R. J., Martignetti J. A. (2000). Inherited multicentric osteolysis with arthritis: a variant resembling Torg syndrome in a Saudi family. Am. J. Med. Genet. 93, 11-18 [DOI] [PubMed] [Google Scholar]
- Alford A. I., Hankenson K. D. (2006). Matricellular proteins: extracellular modulators of bone development, remodeling, and regeneration. Bone 38, 749-757 [DOI] [PubMed] [Google Scholar]
- Asou Y., Rittling S. R., Yoshitake H., Tsuji K., Shinomiya K., Nifuji A., Denhardt D. T., Noda M. (2001). Osteopontin facilitates angiogenesis, accumulation of osteoclasts, and resorption in ectopic bone. Endocrinology 142, 1325-1332 [DOI] [PubMed] [Google Scholar]
- Bellahcène A., Bonjean K., Fohr B., Fedarko N. S., Robey F. A., Young M. F., Fisher L. W., Castronovo V. (2000). Bone sialoprotein mediates human endothelial cell attachment and migration and promotes angiogenesis. Circ. Res. 86, 885-891 [DOI] [PubMed] [Google Scholar]
- Cowles E. A., DeRome M.E., Pastizzo G., Brailey L.L., Gronowicz G.A. (1998). Mineralization and the expression of matrix proteins during in vivo bone development. Calcif. Tissue Int. 62, 74-82 [DOI] [PubMed] [Google Scholar]
- Dean R. A., Overall C. M. (2007). Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics 6, 611-623 [DOI] [PubMed] [Google Scholar]
- Fedarko N. S., Jain A., Karadag A, Fisher L. W. (2004). Three small integrin binding ligand N-linked glycoproteins (SIBLINGs) bind and activate specific matrix metalloproteinases. FASEB J. 18, 734-736 [DOI] [PubMed] [Google Scholar]
- Ganss B., Kim R. H., Sodek J. (1999). Bone sialoprotein. Crit. Rev. Oral Biol. Med. 10, 79-98 [DOI] [PubMed] [Google Scholar]
- Giachelli C. M., Steitz S. (2000). Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol. 19, 615-622 [DOI] [PubMed] [Google Scholar]
- Gordon J. A., Tye C. E., Sampaio A. V., Underhill T. M., Hunter G. K., Goldberg H. A. (2007). Bone sialoprotein expression enhances osteoblast differentiation and matrix mineralization in vitro. Bone 41, 462-473 [DOI] [PubMed] [Google Scholar]
- Inoue K., Mikuni-Takagaki Y., Oikawa K., Itoh T., Inada M., Noguchi T., Park J. S., Onodera T., Krane S. M., Noda M., et al. (2006). A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J. Biol.Chem. 281, 33814-33824 [DOI] [PubMed] [Google Scholar]
- Itoh T., Ikeda T., Gomi H., Nakao S., Suzuki T, Itohara S. (1997). Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J. Biol. Chem. 272, 22389-22392 [DOI] [PubMed] [Google Scholar]
- Karadag A, Fisher L. W. (2006). Bone sialoprotein enhances migration of bone marrow stromal cells through matrices by bridging MMP-2 to alpha(v)beta3-integrin. J. Bone Miner. Res. 21, 1627-1636 [DOI] [PubMed] [Google Scholar]
- Kojima H., Uede T, Uemura T. (2004). In vitro and in vivo effects of the overexpression of osteopontin on osteoblast differentiation using a recombinant adenoviral vector. J. Biochem. 136, 377-386 [DOI] [PubMed] [Google Scholar]
- Liu Y. K., Uemura T., Nemoto A., Yabe T., Fujii N., Ushida T., Tateishi T. (1997). Osteopontin involvement in integrin-mediated cell signaling and regulation of expression of alkaline phosphatase during early differentiation of UMR cells. FEBS Lett. 420, 112-116 [DOI] [PubMed] [Google Scholar]
- Liu S. J., Hu G. F., Liu Y. J., Liu S. G., Gao H., Zhang C. S., Wei Y. Y., Xue Y., Lao W. D. (2004). Effect of human osteopontin on proliferation, transmigration and expression of MMP-2 and MMP-9 in osteosarcoma cells. Chin. Med. J. (Engl.) 117, 235-240 [PubMed] [Google Scholar]
- Martignetti J. A., Aqeel A. A., Sewairi W. A., Boumah C. E., Kambouris M., Mayouf S. A., Sheth K. V., Eid W. A., Dowling O., Harris J., et al. (2001). Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 28, 261-265 [DOI] [PubMed] [Google Scholar]
- Merry K., Dodds R., Littlewood A., Gowen M. (1993). Expression of osteopontin mRNA by osteoclasts and osteoblasts in modelling adult human bone. J. Cell Sci. 104, 1013-1020 [DOI] [PubMed] [Google Scholar]
- Mi Z., Guo H., Wai P. Y., Gao C, Kuo P. C. (2006). Integrin-linked kinase regulates osteopontin-dependent MMP-2 and uPA expression to convey metastatic function in murine mammary epithelial cancer cells. Carcinogenesis 27, 1134-1145 [DOI] [PubMed] [Google Scholar]
- Mosig R. A., Dowling O., DiFeo A., Ramirez M. C., Parker I. C., Abe E., Diouri J., Aqeel A. A., Wylie J. D., Oblander S. A., et al. (2007). Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum. Mol. Genet. 16, 1113-1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig R. A., Dowling O., Martignetti J. A. (2008). MMP2 and the multicentric osteolysis, nodulosis and arthropathy (MONA) and Winchester syndromes. In Inborn Errors of Development: The Molecular Basis of Clinical Disorders of Morphogenesis, 2nd edn (ed. Epstein C. J., Erickson R. P., Wynshaw-Boris A.), pp. 1453-1462 New York: Oxford University Press [Google Scholar]
- Nam T. J., Busby W. H., Jr, Rees C., Clemmons D. R. (2000). Thrombospondin and osteopontin bind to insulin-like growth factor (IGF)-binding protein-5 leading to an alteration in IGF-I-stimulated cell growth. Endocrinology 141, 1100-1106 [DOI] [PubMed] [Google Scholar]
- Noda M., Vogel R. L., Craig A. M., Prahl J., DeLuca H. F., Denhardt D. T. (1990). Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc. Natl. Acad. Sci. USA 87, 9995-9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima S., Yamaguchi N., Nishioka K., Mima T., Ishii T., Umeshita-Sasai M., Kobayashi H., Shimizu M., Katada Y., Wakitani S., et al. (2002). Enhanced local production of osteopontin in rheumatoid joints. J. Rheumatol. 29, 2061-2067 [PubMed] [Google Scholar]
- Philip S., Bulbule A, Kundu G. C. (2001). Osteopontin stimulates tumor growth and activation of promatrix metalloproteinase-2 through nuclear factor-kappa B-mediated induction of membrane type 1 matrix metalloproteinase in murine melanoma cells. J. Biol. Chem. 276, 44926-44935 [DOI] [PubMed] [Google Scholar]
- Sato M., Morii E., Komori T., Kawahata H., Sugimoto M., Terai K., Shimizu H., Yasui T., Ogihara H., Yasui N., et al. (1998). Transcriptional regulation of osteopontin gene in vivo by PEBP2alphaA/CBFA1 and ETS1 in the skeletal tissues. Oncogene 17, 1517-1525 [DOI] [PubMed] [Google Scholar]
- Shapses S. A., Cifuentes M., Spevak L., Chowdhury H., Brittingham J., Boskey A. L., Denhardt D. T. (2003). Osteopontin facilitates bone resorption, decreasing bone mineral crystallinity and content during calcium deficiency. Calcif. Tissue Int. 73, 86-92 [DOI] [PubMed] [Google Scholar]
- Shen Q, Christakos S. (2005). The vitamin D receptor, Runx2, and the Notch signaling pathway cooperate in the transcriptional regulation of osteopontin. J. Biol. Chem. 280, 40589-40598 [DOI] [PubMed] [Google Scholar]
- Sommer B., Bickel M., Hofstetter W, Wetterwald A. (1996). Expression of matrix proteins during the development of mineralized tissues. Bone 19, 371-380 [DOI] [PubMed] [Google Scholar]
- Suzuki K., Zhu B., Rittling S. R., Denhardt D. T., Goldberg H. A., McCulloch C. A., Sodek J. (2002). Colocalization of intracellular osteopontin with CD44 is associated with migration, cell fusion, and resorption in osteoclasts. J. Bone Miner. Res. 17, 1486-1497 [DOI] [PubMed] [Google Scholar]
- Teti A., Farina A. R., Villanova I., Tiberio A., Tacconelli A., Sciortino G., Chambers A. F., Gulino A, Mackay A. R. (1998). Activation of MMP-2 by human GCT23 giant cell tumour cells induced by osteopontin, bone sialoprotein and GRGDSP peptides is RGD and cell shape change dependent. Int. J. Cancer 77, 82-93 [DOI] [PubMed] [Google Scholar]
- Wai P. Y., Mi Z., Guo H., Sarraf-Yazdi S., Gao C., Wei J., Marroquin C. E., Clary B., Kuo P. C. (2005). Osteopontin silencing by small interfering RNA suppresses in vitro and in vivo CT26 murine colon adenocarcinoma metastasis. Carcinogenesis 26, 741-751 [DOI] [PubMed] [Google Scholar]
- Xu G., Nie H., Li N., Zheng W., Zhang D., Feng G., Ni L., Xu R., Hong J., Zhang J. Z. (2005). Role of osteopontin in amplification and perpetuation of rheumatoid synovitis. J. Clin. Invest. 115, 1060-1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi R., McBurney D., Laverty D., Weiner S, Horton W. E., Jr (2005). Intrajoint comparisons of gene expression patterns in human osteoarthritis suggest a change in chondrocyte phenotype. J. Orthop. Res. 23, 1128-1138 [DOI] [PubMed] [Google Scholar]
- Zhong N., Garman R. A., Squire M. E., Donahue L. R., Rubin C. T., Hadjiargyrou M, Judex S. (2005). Gene expression patterns in bone after 4 days of hind-limb unloading in two inbred strains of mice. Aviat. Space Environ. Med. 76, 530-535 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.