


Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR) form the basis of diverse adaptive immune systems directed primarily against invading genetic elements of archaea and bacteria. Cbp1 of the crenarchaeal thermoacidophilic order Sulfolobales, carrying three imperfect repeats, binds specifically to CRISPR DNA repeats and has been implicated in facilitating production of long transcripts from CRISPR loci. Here, a second related class of CRISPR DNA repeat-binding protein, denoted Cbp2, is characterized that contains two imperfect repeats and is found amongst members of the crenarchaeal thermoneutrophilic order Desulfurococcales. DNA repeat-binding properties of the Hyperthermus butylicus protein Cbp2Hb were characterized and its three-dimensional structure was determined by NMR spectroscopy. The two repeats generate helix-turn-helix structures separated by a basic linker that is implicated in facilitating high affinity DNA binding of Cbp2 by tethering the two domains. Structural studies on mutant proteins provide support for Cys7 and Cys28 enhancing high thermal stability of Cbp2Hb through disulphide bridge formation. Consistent with their proposed CRISPR transcriptional regulatory role, Cbp2Hb and, by inference, other Cbp1 and Cbp2 proteins are closely related in structure to homeodomain proteins with linked helix-turn-helix (HTH) domains, in particular the paired domain Pax and Myb family proteins that are involved in eukaryal transcriptional regulation.
INTRODUCTION
Adaptive immune systems based on CRISPR arrays (clustered regularly interspaced short palindromic repeats) provide defence primarily against invading viruses and conjugative plasmids of almost all archaea and many bacteria. They are essentially modular systems involving uptake of foreign DNA as new spacers in CRISPR repeat-spacer arrays—adaptation; processing of CRISPR transcripts into small crRNAs carrying primarily spacer sequences—RNA biogenesis; and the targeting and cleavage of DNA or RNA by protein–crRNA complexes—interference (1–3). In archaea, transcripts from type I and type III CRISPR-based immune systems are produced from strong promoters within the leader adjacent to the first CRISPR repeat (4,5), and they are processed within the repeats at one main site yielding mature crRNAs carrying spacer regions flanked by partial repeat sequences that are used for DNA targeting (6–9). Interference by the diverse type III CRISPR–Cmr/Csm systems requires secondary processing from the 3′-end of crRNAs, removing the repeat region and parts of the spacer sequence, and these smaller crRNAs facilitate RNA or DNA interference (10–13). In a third type II immune system, which is exclusive to bacteria, RNA biogenesis is directed by a trans-acting tracrRNA in combination with the bacteria-specific RNAse III endonuclease (14).
Cbp1Ss is a 17.5 kDa protein that carries three imperfect repeat structures predicted to generate helix-turn-helix (HTH) DNA-binding motifs, and it binds specifically to CRISPR 25 bp repeats of Sulfolobus solfataricus P2 producing a structural distortion at the repeat centre (15). Earlier, Cbp1Ss was proposed to be involved in higher-order structuring of chromosomal CRISPR regions (15), and this hypothesis was reinforced by studies showing retardation of DNA replication through CRISPR-rich chromosomal regions of Sulfolobus acidocaldarius (16). However, the discovery that CRISPR loci were transcribed and processed within the repeats (6,7) raised the possibility of a transcriptional role for Cbp1. More recently, experiments generating knockout mutants of the Cbp1Si protein from Sulfolobus islandicus REY15A, and over-expression of Cbp1 in both S. islandicus and S. solfataricus P2, have provided experimental support for a facilitatory role in the generation of large transcripts (6–7 kb) from CRISPR loci (17). Thus, a Cbp1-minus mutant exhibited a strongly reduced level of intermediate pre-crRNA transcripts, while Cbp1 over-expression produced enhanced yields of longer pre-crRNA transcripts. It was concluded that Cbp1 enhances production of longer CRISPR transcripts by minimizing interference primarily from transcriptional signals accumulated randomly in CRISPR loci from invading genetic elements (9,17,18).
Recognizable homologs of Cbp1 were confined to members of the thermoacidophilic order Sulfolobales (16). These organisms, and their genetic elements, all exhibit A+T-rich genomes (∼64%), which increases the likelihood of the uptake of potential hexameric TATA-like promoter motifs and T-rich terminator motifs in the spacers (9,18–20), and this may explain the widespread presence of this protein in these archaeal thermoacidophiles.
Here, we describe a second type of CRISPR repeat-binding protein, denoted Cbp2, which contains two imperfect internal repeats. Cbp2 occurs exclusively in members of the crenarchaeal thermoneutrophile order Desulfurococcales, which tend to grow optimally at higher temperatures than the Sulfolobales and generally also carry A+T-rich genomes. The Cbp2 protein of the hyperthermophile Hyperthermus butylicus, which can grow at up to 108°C (21,22), was selected for this study. The solution structure of the protein was determined by NMR spectroscopy, and the results presented are consistent with Cbp2 being co-functional with Cbp1 and being involved in transcriptional modulation of CRISPR loci.
MATERIALS AND METHODS
Expression and characterization of Cbp2
DNA was isolated from Hyperthermus butylicus cells (DSMZ, Braunschweig, Germany) using a DNeasy kit (Qiagen, Hilden, Germany). The cbp2Hb gene was amplified by PCR using primers Cbp2Hb-F 5′-TTTGGAATTCCATATGTTGCCCTCCGTTAACG-3′ and Cbp2Hb-R 5′-TTTCCGCTCGAGCTTGAGTCCAAGCTTTTTCAGTGC-3′ and Pfu DNA polymerase. The product was inserted into a pET28a vector (Novagen, Madison, WI, USA) using restriction enzymes XhoI and NdeI (Fermentas, St. Leon-Rot, Germany), and the stop codon was removed and a hexameric C-terminal His-tag was added. Construct integrity was confirmed by sequencing prior to transforming into Escherichia coli BL21 (DE3) cells. Protein expression was induced by growing cells to A600 = 0.6 and adding 0.5 mM isopropylthio-β-d-galactoside (IPTG). Cells were disrupted by sonicating in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole, 0.1 mM phenylmethylsulphonyl fluoride, 0.1 mM EDTA, 1% Triton 100), and the lysate was cleared by ultracentrifugation (Beckman and Coulter, CA, USA) for 15 min at 30 000 rpm. The supernatant was incubated for 15 min at 70°C to precipitate E. coli proteins, centrifuged in an Eppendorf centrifuge for 10 min at 12 000 rpm and DNA was precipitated by adding 0.65% phenylethyleneimine (PEI) and centrifuging for 30 min at 10 000 rpm. The supernatant was incubated with Ni-NTA agarose beads (Qiagen) equilibrated with lysis buffer, and beads were washed with 20 ml 10 mM imidazole and again with 5 ml each of buffer containing 20 mM, and then 50 mM imidazole, before eluting the protein with buffer containing 250 mM imidazole. Co-purified fragments of E. coli DNA were removed by repeated treatment with 0.65% phenylethyleneimine.
To express individual protein repeat domains, primers Cbp2Hb-F and Cbp2HbN-R-5′-TTTCCGCTCGAGCCTATGTTGCCGGTACCTACCCTTC-3′ were used for the N-domainC7S/C28S and Cbp2HbC-F-5′-TTTGGAATTCCATATGGAAGGGTAGGTA CCGGCAACATAGG-3′ and Cbp2Hb-R were used for the C-domain. Site-directed mutagenesis used the QuikChange protocol (Stratagene, La Jolla, CA) with forward primers 5′-GCCCTCCGTTAACGACAGTCTAGACATAGTCGAGAAGC-3′ for Cbp2HbC7S and 5′-GAGATTGCTAAGCGATCGAACAATAGCATGAGCACTG-3′ for Cbp2HbC28S. Reverse primers carried complementary sequences to these primers. To generate Cbp2HbC7S/C28S, the Cbp2HbC7S construct was used and modified using primers for Cbp2HbC28S.
For NMR studies, cells were grown in M9 minimal medium using 15(NH4)2SO4 and [13C6]-glucose as sole sources of nitrogen and carbon, respectively, to produce 15N, 13C-labelled proteins.
DNA binding
A 134 bp CRISPR DNA region from H. butylicus carrying a repeat-spacer-repeat sequence (CRISPR-2rHb) containing spacer 2 from the leader of CRISPR locus 2 (22) and short flanking regions was amplified by PCR using primers Hb2rptF 5′-CAGCAATTCCAGCAGCAG-3′ and Hb2rptR 5′-AAACTTCGCAAGGCTGTACC-3′. The 25 bp single repeat DNA (CRISPR-1rHb) was generated using primers Hb1rpt-F 5′-CTTGCAATTCTCTTTTGAGTTGTTC-3′ and Hb1rpt-R 5′- GAACAACTCAAAAGAGAATTGCAAG-3′. For competition assays, a 148 bp CRISPRSs-2r DNA was amplified from S. solfataricus P2 DNA using primers 5′-CTCCGCAACTTCATCAATAGTG-3′ and 5′-SsGAGTTGCGGGCACTTTATGACAG-3′ (17). Primers were annealed in 10 mM Tris-HCl, 50 mM NaCl, 1 mM EDTA (pH 7.5), heated to 95°C and cooled slowly to room temperature. DNA fragments were [32P] 5′-end labelled using T4 polynucleotide kinase (Fermentas). Protein–DNA complexes were produced by incubating in 10 mM Tris-Cl, pH 7.6, 150 mM KCl, 10% glycerol for 20 min at 50°C, cooling and adding loading buffer (10 mM Tris-Cl, pH 7.6, 1 mM EDTA, 50% glycerol, 0.5% bromophenol blue) prior to electrophoresing in 8% polyacrylamide gels containing 89 mM Tris-Cl, 25 mM taurine, 0.5 mM EDTA, pH 8.9 and autoradiography.
Isothermal titration calorimetry (ITC) experiments were carried out using a VP-ITC device (MicroCal). Cbp2HbC7S/C28S N- and C-terminal domains with the 12 bp conserved downstream CRISPR repeat fragment of H. butylicus were generated from primers 5′-TTTGAGTTGTTC-3′ and 5′-GAACAACTCAAA 3′ purchased in annealed form (TAGC, Copenhagen, Denmark). All solutions were degassed and dialysed extensively against NMR buffer (20 mM KH2PO4, 10 mM KCl, pH 6.5). Calorimetric data were analysed using MicroCal ORIGIN software (23).
NMR spectroscopy and structure analysis
NMR spectra were recorded on either a Varian Unity Inova 750-MHz or 800-MHz (equipped with a cryoprobe) spectrometer at 25°C. Samples typically contained 1 mM protein in 20 mM KH2PO4, 10 mM KCl, pH 6.5, 10% D2O, 0.02% NaN3. For DNA-binding studies, the Cbp2 protein or separate protein domains were added to dsDNA, heated to 50°C and allowed to equilibrate for 60 min before recording 1H,15N-HSQC spectra. Proton chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) and heteronuclei indirectly using the gyromagnetic ratios. All NMR spectra were processed with NMRPipe (24) and analysed using CcpNMR analysis (25). Backbone resonances were assigned using 2-D 1H,15N-HSQC, 3-D HNCO, HNCA, HN(CA)CO, HN(CO)CA, CBCANH and CBCACONH spectra (26–28). Side chain resonances were assigned using [15N/13C]-resolved HSQC-TOCSY, HCCH-TOCSY and [15N/13C]-edited HSQC-NOESY experiments (29). Nuclear Overhauser effects (NOEs) for inter-proton distance restraints were obtained from [15N] or [13C]-resolved 3-D HSQC-NOESY spectra, both in the aliphatic and aromatic areas, and automatically assigned using CYANA 2.1 with manual inspection (30). Dihedral angle restraints were generated using Cα/Cβ chemical shifts and the program TALOS (31). For each round of calculations, a total of 100 structures were calculated with CYANA 2.1 using a simulated annealing protocol, and after several rounds of iterations, the 20 lowest energy structures were chosen to represent the final ensemble. None of the structures in the final ensemble had NOE violations of >0.5 Å or angle violations of >5°. The structures were checked using the web interface to CING (http://nmr.cmbi.ru.nl/cing/Home.html). Chemical shift differences between DNA-free and DNA-bound Cbp2Hb domains were calculated as: Δδ (ppm) = (Δδ (1H)2 + 0.154 Δδ (15N)2)1/2 (32).
Identification of structural homologs
An initial search was made using the algorithm FATCAT (flexible structure alignment by chaining alignment fragment pairs allowing twists) (33). It yielded >800 similar structures when introducing an upper limit in P < 0.02 (where P < 0.05 indicates significantly similar structures). This set was reduced to 103, mostly globular domains containing an HTH motif, by choosing structures with a root mean square deviation (RMSD) value <3.0 Å from the input structure and where at least 80% of Cbp2 Cα atoms exhibited equivalent positions in the candidate structure. Fourteen of these exhibited bipartite structures. FATCAT detects hinges and performs twists around hinges to improve alignments, and our criterion for structural significance allowed a maximum of one twist in the putatively flexible linker region, which limited the matching set to eight structures.
RESULTS
Cbp2 is a homolog of the CRISPR DNA repeat-binding protein Cbp1
Potential homologs of Cbp1Ss (Sso0454), the CRISPR DNA repeat-binding protein of S. solfataricus P2 (17), were identified by BLAST searches against sequence databases at GenBank (http://blast.ncbi.nlm.nih.gov/Blast) and EBI (http://www.ebi.ac.uk/genomes/archaea.html). Apart from positive matches obtained for several members of the Sulfolobales, including those described earlier (17), five additional matches to smaller proteins were identified for some members of the hyperthermophilic Desulfurococcales. However, whereas the Cbp1 proteins each carried three imperfect repeat sequences, the smaller matching proteins contained two imperfect repeats and were named CRISPR DNA repeat-binding protein 2, Cbp2. As for cbp1 genes, cbp2 genes are present in single genomic copies, irrespective of the number or classes of CRISPR loci present, and they are uncoupled from CRISPR loci and their related gene cassettes encoding Cas, Cmr and Csm proteins.
The protein sequences are aligned for Cbp2 (Figure 1A), and the results indicate two repeats separated by a basic linker region. The structure resembles that of the Cbp1 proteins from selected, phylogenetically diverse, members of the Sulfolobales (Figure 1B). The results show a number of highly conserved amino acids within the repeats some of which are shared between proteins Cbp2 and Cbp1. These conserved residues are shaded in Figure 1A and B, and they demonstrate the relatedness of the two N-terminal repeats and the two C-terminal repeats of Cbp1 and Cbp2 proteins. A phylogenetic tree was also constructed for the Cbp2 repeats in which the N- and C-terminal repeats, respectively, show greater conservation between different organisms than to one another (Figure 1C). This indicates that the two repeats have evolved independently and is consistent with their having different structural roles. Equivalent tree building results, illustrating the uniqueness of the repeats, were obtained for the three repeats of the Cbp1 proteins (Supplementary Figure S1).
Figure 1.

Properties of Cbp2 and Cbp1 protein repeats. (A) Alignment of Cbp2 sequences for members of the Desulfurococcales: H. butylicus (Hbut-0986), Staphylothermus hellenicus (Shel-1510), Staphylothermus marinus (Smar-106), Thermogladius cellulolyticus (Tcel-1281) and Pyrolobus fumarii (Pfum-1392). (B) Multiple alignments of Cbp1 proteins showing three repeats and two linkers. Sequences are aligned for seven representative species of the Sulfolobales: S. solfataricus P2 (Ssol-0454), S. islandicus REY15A (Sisl-1547), S. acidocaldarius DSM639 (Saci-0449), Sulfolobus tokodaii 7 (Stok-0170), Metallosphaera sedula DSM5348 (Msed-2177), Acidianus brierleyi (Abri-2346) and Acidianus hospitalis W1 (Ahos-0975). Amino acid residues: (asterisk) fully conserved; (colon) strongly similar properties and (dot) weakly similar properties. Boxed amino acids are conserved in the N-terminal and C-terminal repeats of Cbp2 and Cbp1. Alignments were made using ClustalW2 (34). Protein repeats were detected using RADAR (35) and repeat limits were inferred from secondary structure predictions. (C) Distance tree showing clustering of the individual Cbp2 repeats where the organisms are labelled by four-letter prefixes and the protein repeats numbered from the N-terminus. Constructed using MEGA4, with neighbour joining and bootstrap values (36).
Cbp2 binds specifically to CRISPR DNA repeat sequences
In preliminary experiments Cbp2 was shown to co-purify with heterogeneous DNA fragments from E. coli. When Cbp2 was complexed with a large CRISPR DNA fragment, the E. coli DNA was displaced and a discrete slower moving band was observed in an agarose gel (Supplementary Figure S2A). In contrast, when Cbp2 was incubated with a similarly sized fragment of pUC18 DNA, E. coli DNA was not displaced from Cbp2, and no discrete slower bands were seen (Supplementary Figure S2B). This suggested that Cbp2–CRISPR DNA binding was specific and, therefore, E. coli DNA was first removed from Cbp2 by repeated treatment with phenylethyleneimine prior to complexing with CRISPR repeat substrates.
Purified Cbp2Hb yielded a single strong band in an SDS-polyacrylamide gel with an estimated size consistent with the predicted molecular weight of the monomeric protein (∼13 kDa), and a weak band corresponding in size to a dimer was also generally observed (Figure 2A). Purified Cbp2Hb was incubated with CRISPR-2rHb, carrying a repeat-spacer-repeat structure from H. butylicus CRISPR locus 2 and short flanking sequences (see Materials and Methods). Band-shift electrophoresis experiments demonstrated that at increasing protein:DNA molar ratios, multiple slower migrating products were produced reaching a maximum of three bands at 5- to 6-fold molar excess of protein (Figure 2B). When the experiment was repeated with a single CRISPR-1rHb repeat, a maximum of two slower migrating products were observed at a 4- to 5-fold molar excess of protein (Figure 2C). The gel patterns resemble those obtained earlier for Cbp1Ss-CRISPR-2rSs complexes of S. solfataricus, where the two faster complex bands were attributed to one and two protein copies bound, respectively, per two DNA repeat substrate, while the upper band was inferred to result from additional protein binding, possibly linking Cbp1 proteins bound on adjacent repeats (14). The presence of a second slower upper band for the Cbp2Hb-CRISPR-1rHb DNA complex (Figure 2C) is also consistent with the occurrence of additional protein–protein interactions. No binding activity of Cbp2Hb was detected with single-stranded DNA and RNA substrates carrying single CRISPR repeat sequences (Supplementary Figure S2C and D).
Figure 2.
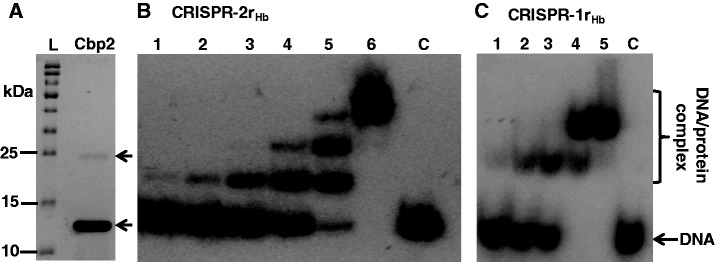
Purification and DNA binding of Cbp2Hb. (A) Electrophoresis of purified Cbp2Hb in an 12.5% polyacrylamide gel containing 0.1% SDS run in 25 mM Tris-Cl, pH 8.6, 192 mM glycine, 0.1% SDS, and staining with Coomassie brilliant blue. L—protein size ladder. The arrow indicates the putative protein dimer. (B) Cbp2Hb was incubated with 8 nM [32P] 5′-end labelled CRISPR-2rHb DNA at a 1, 2, 3, 4, 5 and 6 molar protein excess in lanes 1 to 6, respectively. Lane C—DNA substrate alone. (C) Cbp2Hb was incubated with 8 nM [32P] 5′-end labelled CRISPR-1rHb DNA with a 1, 2, 3, 4 and 5 molar protein excess in lanes 1 to 5, respectively. Lane 6—DNA substrate alone. In (B) and (C) complexes were formed in 10 mM Tris-Cl, pH 7.6, 150 mM KCl, 2 mM DTT, 10% glycerol at 50°C for 20 min and run in 8% polyacrylamide gels.
Among crenarchaea, most CRISPR repeats carry the sequence 5′-GAAAC/G-3′ at the leader distal (downstream) end (37) that was predicted to be a potential binding site for some CRISPR-associated (Cas) proteins (38). Therefore, we compared CRISPR repeats in members of the Desulfurococcales and Sulfolobales encoding Cbp2 and Cbp1 proteins, respectively. The Logo plot results are consistent with the earlier observations, showing that the downstream ends of the repeats are most conserved for organisms of both orders (Figure 3A). Moreover, an alignment of the repeat sequences of H. butylicus and S. solfataricus underlines the higher conservation of the downstream halves of the repeats (Figure 2B). Therefore, we tested for the capacity of Cbp2Hb to bind specifically to CRISPR-2rSs DNA. A band-shift assay showed that single and double protein–DNA complex bands were formed at a 3 and 4 molar protein excess, respectively (Figure 3C). A similar result was observed for the homologous Cbp2Hb–CRISPR-2rHb DNA complex (Figure 2B).
Figure 3.
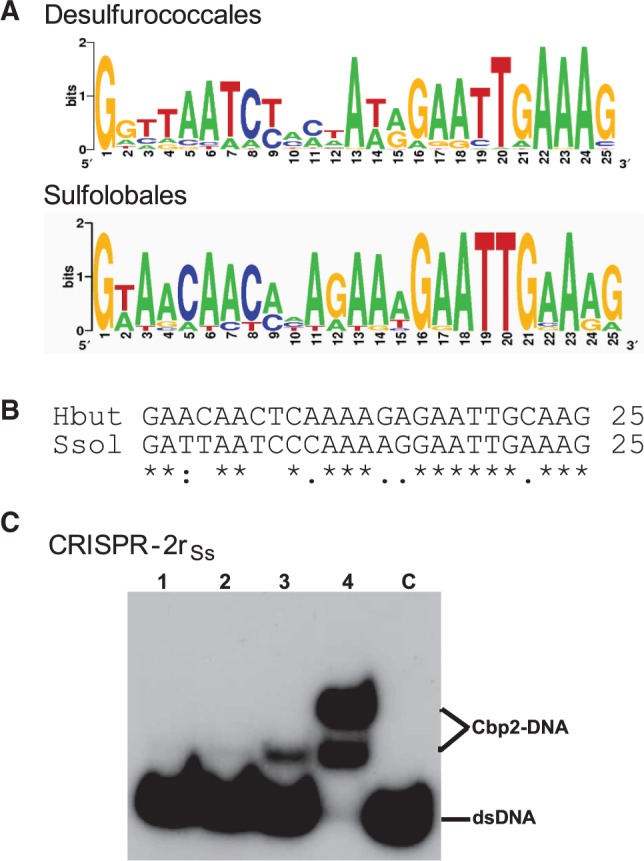
CRISPR repeat sequence conservation and Cbp2 binding dependence. (A) Logo-plot of the CRISPR repeats of Desulfurococcales—4 organisms, 13 repeats: H. butylicus (1), S. hellenicus (4), S. marinus (4) and P. fumarii (4) (CRISPR repeats are not available for T. cellulolyticus), and of Sulfolobales—16 organisms, 36 repeats (number of different repeats for each organism given in brackets): S. solfataricus P2 (3), S. solfataricus 98/2 (3), S. tokodaii (3), S. acidocaldarius (2), A. hospitalis (4), A. brierleyi (4), M. sedula (3) and S. islandicus strains REY15A (1), HVE 10/4 (2), L.S.2.15 (2), Y.N.15.51 (1), M.16.27 (1), Y.G.57.14 (1), M.14.25 (2), M.16.4 (2), L.D.8.5 (2). Logo plots were obtained using available software (http://weblogo.berkeley.edu/). (B) CRISPR repeat alignment for H. butylicus and S. solfataricus P2. (C) Cbp2Hb binding to CRISPR-2rSs DNA. Cbp2Hb was incubated with 8 nM [32P] 5′-end labelled CRISPR-2rSs DNA at a 1, 2, 3 and 4 molar protein excess in lanes 1 to 4, respectively. Lane C—DNA substrate alone.
Cbp2 has a simple bipartite helix-turn-helix structure
Secondary structure predictions for Cbp2Hb (39) suggested that each repeat can generate an HTH structure, separated by a flexible basic linker. Cbp2Hb protein mutants and protein fragments were cloned and expressed in E. coli in order to resolve the Cbp2Hb structure by NMR spectroscopy at atomic detail. The wild-type protein carries two cysteine residues in the N-domain repeat (Cys7 and Cys28). Residues flanking the cysteines apparently undergo chemical exchange broadening because no backbone amide resonance signals were observed for residues 5–9 and 26–33. Chemical exchange broadening also seemed to affect the linker residues 52–60 because no resonances were observed for these residues. To facilitate NMR analysis, mutants were prepared with cysteine to serine replacements at positions 7 and 28 (Cbp2HbC7S/C28S). Moreover, protein fragments constituting the individual repeat domains, Cbp2Hb N-domainC7S/C28S (residues 1–59) and Cbp2Hb C-domain (residues 51–103) that overlap by eight residues, were also expressed.
The 1H,15N-HSQC spectrum of Cbp2HbC7S/C28S exhibited dispersed resonances characteristic of a folded protein. Furthermore, the 1H,15N-HSQC spectra of the individual N- and C-domains are highly similar to the spectrum of the intact protein, indicating that the domains do not interact (Figure 4A and B). Therefore, we attempted to determine the structure of the separate domains. Assignments were achieved for >96% of all backbone resonances (N, C, Cα, Cβ), in both domains, and side chain resonances were also partially or fully assigned. From chemical shift assignments, and using the program TALOS (31), 74 and 83 dihedral angle restraints were generated for the N- and C-domains, respectively. From NOESY spectra, a total of 1392 1H-1H NOEs were obtained that yielded useful distance information amounting to ∼15 restraints per residue. Using CYANA, we selected from a set of 100 structures of the whole protein using distance restraints generated for the N- and C-domains. Then an ensemble of the 20 lowest energy structures was selected to represent the final structures (Figure 4C). The 20 structures aligned with a backbone RMSD of 0.41 (±0.16) and 0.43 (±0.15) for the N- and C-domains, respectively, and heavy atom RMSD of 0.93 (±0.012) and 1.08 (±0.14), respectively. These structural statistics are summarized in Table 1.
Figure 4.

Structural features of Cbp2HbC7S/C28S. 1H,15N-HSQC spectra of Cbp2HbC7S/C28S (black) overlaid with (A) the N-domain construct (blue) and (B) the C-domain construct (red). A signal is observed for all 1H–15N covalent bonds in the protein which are mainly backbone amide groups. The signal has two chemical shifts (one for 15N, y-axis and one for 1H, x-axis) that are highly sensitive to the local chemical environment of each atom, and the spectrum thus provides a fingerprint of the protein fold. The almost complete spectral overlap of the isolated domains and the full-size protein indicates that the domains do not stably interact with each other in the full-size protein since that should result in an observable difference in chemical shifts of the residues that form the interface between the domains. (C) Ensemble of 20 lowest energy structures of N- and C-domains of Cbp2, calculated with CYANA. (D) Lowest energy structure of Cbp2HbC7S/C28S with the N-domain (blue) and C-domain (red) connected by a flexible linker (green). Helices are labelled H1 to H6 and the N- and C-termini are indicated. (E) Conserved amino acids in Cbp2Hb that form hydrophobic cores (see Figure 1A). The conserved ‘phs’ pattern (charged, hydrophobic, small) is formed for the N/C-domains by E23/81, I24/82 and A25/83. Moreover, I24/82 defines the hydrophobic core with V11, V35 and L39 in the N-domains by E23/81, I24/82 and A25/83, respectively. Moreover, I24/82 defines the hydrophobic core with V11, V35 and L39 in the N-domain (blue), and with I69, I93 and L97 in the C-domain (red). Side chains are shown in green.
Table 1.
Structural statistics and restraint information for Cbp2Hb
| Restraints and statistics | N-domain (1–51) | C-domain (61–103) |
|---|---|---|
| Experimental restraints | ||
| Number of structures | 20 | 20 |
| Number of distance restraints | 794 | 598 |
| Sequential | 385 | 319 |
| Medium range | 253 | 186 |
| Long range | 156 | 93 |
| Restraints per residue | 15 | 14 |
| Dihedral angle restraints | 74 | 83 |
| Restraints violations | ||
| NOE violations >0.5 Å | 0 | 0 |
| Dihedral angle violations >5° | 0 | 0 |
| Deviations from ideal geometry (RMSD) | ||
| Impropers (°) | 0.321 ± 0.001 | 0.289 ± 0.001 |
| Bond lengths (Å) | 1.021 ± 0.001 | 1.031 ± 0.001 |
| Bond angles (°) | 0.195 ± 0.001 | 0.216 ± 0.001 |
| RMSD of atomic positions (Å)a | ||
| Backbone atoms | 0.41 ± 0.16 | 0.43 ± 0.15 |
| Heavy atoms | 0.93 ± 0.012 | 1.08 ± 0.14 |
| Ramachandran plot (%)a | ||
| Most favoured regions | 95.7 | 94.7 |
| Additionally allowed regions | 4.3 | 5.3 |
| Generously allowed regions | 0.0 | 0.0 |
| Disallowed regions | 0.0 | 0.0 |
aCalculated using iCING (http://nmr.cmbi.ru.nl/icing/iCing.html#file).
Both domains yielded well defined secondary structures, each containing three α-helices [N-domain; H1 (V4-D17), H2 (V21-R27), H3 (M32-M45), C-domain; H4 (E63-K75), H5 (V79-L86), H6 (S91-L100)], with a total α-helix content of 68% and a distinct tertiary fold characteristic of HTH motifs. Helices H1 and H2 in the N-domain are antiparallel and almost perpendicular to helix H3, and helices H4, H5 and H6 of the C-domain are packed similarly (Figure 4D). The predicted disordered nature of the basic arginine-rich linker, and the apparently non-interacting domains, suggested that the two domains are arranged like beads on string (Figure 4D).
The Cbp2Hb structure exhibits features of classical HTH motifs where the putative DNA recognition helices constitute helices H3 and H6 (40). The features include the ‘shs’ sequence pattern (‘s’ and ‘h’ denote small and hydrophobic residues, respectively) present in the turns between helices H2 and H3, and helices H5 and H6. The other conserved ‘phs’ pattern (‘p’ a charged residue, ‘h’ is hydrophobic and ‘s’ generally glutamate) was formed by E23/81, I24/82 and A25/83. I24/82 (helix H2). It defines the hydrophobic core together with V11 (helix H1) and V35 + L39 (helix H3) in the N-domain while I69 (helix H4) and I93 + L97 (helix H6) generate this hydrophobic core in the C-domain (Figure 4E). Moreover, the conserved hydrophobic residues of the HTH motifs in both domains, together with at least two other conserved hydrophobic residues in helices H1 and H3, and in helices H4 and H6, face towards the interior producing a typical hydrophobic core that stabilizes the domains (Figure 4E).
Cbp2–DNA interactions
The region linking the two domains of Cbp2Hb is highly basic (Figure 1A). In many bipartite DNA-binding proteins, such linkers are crucial for DNA binding and sequence specificity (41,42). To test for the possible involvement of the Cbp2 linker in DNA binding, we performed trypsin digestion with and without DNA. Cbp2Hb was incubated with trypsin under a variety of conditions and was invariably degraded rapidly (Figure 5A). In contrast, when higher concentrations of trypsin were incubated with Cbp2Hb–CRISPR-2rHb complexes, formed at 4:1 molar ratios, no dissociation was observed (Figure 5B), indicating that the linker region is protected in the protein–DNA complex.
Figure 5.

Trypsin digestion of Cbp2Hb and the Cbp2Hb–CRISPR-2rHb complex. (A) Six microgram Cpb2Hb were incubated with trypsin (specific activity: 806 USP U/mg) in 10 mM phosphate (Na2HPO4/KH2PO4), 137 mM NaCl, 2.7 mM KCl, pH.7.4 for 15 min at 37°C and electrophoresed and stained as in Figure 2A. Lane 1—protein size ladder, lane 2—Cbp2 without trypsin and lanes 3 and 4—treated with 12 and 60 ng of trypsin, respectively. Arrows indicate undegraded and degraded Cbp2Hb. The weak upper bands in lane 2 probably represent Cbp2 oligomers, as in Figure 2A. (B) The Cpb2Hb–CRISPR-2rHb complex (8 nm [32P] 5′-end labelled DNA per sample) was formed at a 4:1 protein:DNA molar ratio and treated with increasing concentrations of trypsin. Lane 1—CRISPR-2rHb, lane 2—Crp2Hb DNA complex, lanes 3 and 4—complex was incubated with 45 and 90 ng trypsin, respectively. Trypsin digestions were performed in 15 mM (NH4)2CO3, 10 mM Mg acetate, pH 7.9 for 15 min at 37°C. Samples were analysed by electrophoresing in 12.5% polyacrylamide gels and autoradiographed (see Materials and Methods).
The thermodynamics of Cbp2Hb binding to repeat DNA was studied using ITC (43). The results for Cbp2Hb and CRISPR-1r were variable possibly owing to complex cooperative intra- and intermolecular protein interactions resulting in a phase transition or simultaneous endo/exo thermic events. Nevertheless, similar experiments were carried out with the N- and C-terminal domains of Cbp2HbC7S/C28S and the conserved 12 bp downstream half of the repeat (Figure 3A and B). The latter was selected as substrate because the heterologous repeat binding result was consistent with this region being important for Cbp2 binding (Figure 3B). Both domains produced evidence of binding, and the estimated binding constants suggest that the N-terminal domain (Kd = 10 nM) has a higher affinity for the DNA substrate than the C-terminal domain (Kd = 120 nM) (Figure 6A). An apparent ∼2 and ∼0.5 binding sites were observed for the N- and C-domain, respectively (see below).
Figure 6.

Cbp2Hb–CRISPR DNA repeat interactions. (A) Isothermal titration calorimetry of the N- and C-terminal domains of Cbp2Hb C7S/C28S with the 12 bp DNA ‘downstream’ conserved CRISPR repeat region of H. butylicus 5′-TTTGAGTTGTTC-3′/5′-GAACAACTCAAA (Figure 3B). A 370 ml syringe stirring at 300 rpm was used to titrate Cbp2 domains into a cell containing 1.4 ml DNA solution at 25°C. Each titration consisted of a preliminary 15 µl injection of the Cbp2 domain into the DNA solution followed by up to 20 subsequent 15 µl injections. Estimated Kd values are given for each domain. (B) 1H,15N-HSQCs of Cbp2Hb and the separate N- and C-domains bound to the 12 bp conserved region of CRISPR-1rHb DNA. 1H,15N-HSQCs of full sized Cbp2Hb (left), the N-domain fragment (middle) and the C-domain fragment (right) overlaid with the 1H,15N-HSQCs of the same constructs bound to the CRISPR-1rHb DNA. Free form spectra are shown in black and the bound form spectra are shown in red.
Cbp2–DNA binding was investigated at atomic resolution by recording 1H,15N-HSQCs of Cbp2Hb at saturating concentrations of the 25 bp CRISPR-1rHb DNA. The spectra were indicative of specific binding of Cbp2 to DNA with significant chemical shift changes for most of the residues (Figure 6B). Interestingly, the spectra of Cbp2 bound to either the 25 bp repeat or the 12 bp conserved region were highly similar, indicating that the conserved repeat region is the primary binding site (Supplementary Figure S3). However, in line with the ITC results, resonance peaks were broad suggesting that higher order structures were formed and/or that intermediate exchange kinetics occurred on the NMR timescale. 3-D correlation spectroscopy was also attempted but the spectra were of low quality probably due to the slow tumbling time of the protein–DNA complex and/or the exchange process. Moreover, varying the temperature, pH, salt concentration and solvent systems failed to improve spectral quality and therefore, residue assignments for DNA-bound Cbp2 were unsuccessful. However, binding of the separate N- and C-domains to the conserved 12 bp DNA construct could be followed by NMR spectroscopy (Figure 6B). Relative to the intact protein, less pronounced chemical shift perturbations were observed indicating weaker binding for the separate N- and C-domains. Chemical shift differences were plotted as a function of residue for free and DNA-bound N- and C-domains (Supplementary Figure S4). For the N-domain, chemical shift differences were distributed evenly with no discernible pattern while in the C-domain higher than average shift differences were observed mainly for helix 6, the putative DNA binding helix, and for residues E64, V79 and A83. The resonance signal for S91 disappeared, indicating conformational exchange processes. We then analysed the ratio of bound/free protein 1H resonance line-widths. There was a clear global line-width increase upon DNA binding of the N-domain (average 3.4 ± 0.9), whereas the C-domain line-widths were unchanged compared with the free form (average 1.0 ± 0.5) (data not shown). This suggests that the N-domain binds DNA with higher affinity, in agreement with the ITC-derived results. We also observed that the line-widths of the free C-domain were almost twice as large as for the free N-domain (17.9 and 9.2 Hz, respectively), suggesting formation of dimeric structures at the high concentrations used in the NMR experiments, which may affect or even impede DNA binding. This parallels the ITC results because a pre-formed C-domain dimer binding to DNA would give rise to the observed stoichiometry.
Cysteine residues enhance Cbp2 thermostability
Uniquely for Cbp2 and Cbp1 proteins, Cbp2Hb contains two cysteine residues that are juxtapositioned in helices H1 (C7) and H2 (C28) of the N-domain (Figure 7A). Since a disulphide bond could enhance protein thermostability in an organism growing up to 108°C, we compared the binding properties of Cbp2Hb and Cbp2HbC7S/C28S with the CRISPR-2r substrate over the temperature range 50°C–95°C. The results showed that Cbp2Hb bound strongly to the DNA substrate producing two strong complex bands up to 95°C (Figure 7B). In contrast, the Cbp2HbC7S/C28S mutant protein, complexed under similar conditions, showed strongly reduced yields of the upper band above 50°C and decreasing yields of the lower complex band above 75°C. Both of the latter effects are consistent with the putative formation of a disulphide bond enhancing thermal stability (Figure 7C).
Figure 7.
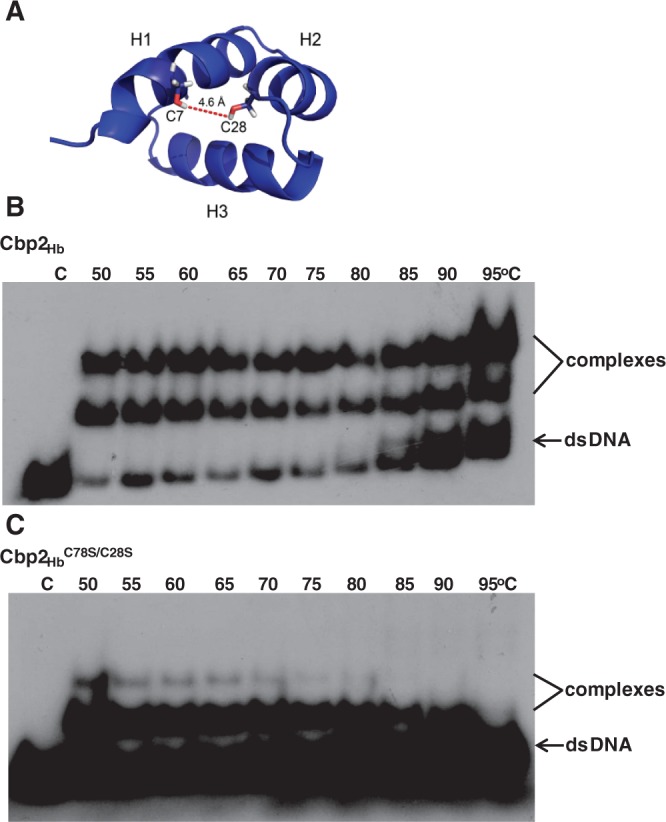
Thermostability and DNA binding dependence on C7 and C28. (A) The N-domain of Cbp2Hb showing positions of C7 and C28 (sticks) and their separation distance. Mobility band shift assays with (B) Cbp2Hb and (C) Cbp2HbC7S/C28S mutant protein incubated with CRISPR-2rHb DNA at increasing temperatures. 40 nM Cbp2Hb or Cbp2HbC7S/C28S were incubated with 10 nM [32P] 5′-end labelled CRISPR-2rHb DNA in 10 mM Tris-Cl, pH 7.6, 150 mM KCl, 10% glycerol for 20 min at 50°C to 95°C and electrophoresed in an 8% polyacrylamide gel. C indicates the CRISPR-2rHb DNA control. Incubation temperatures are indicated for each sample.
Cbp2 shares high structural similarity with homeodomain proteins
Structural similarity searches for Cbp2Hb in public protein databases (see Materials and Methods) demonstrated that despite low sequence conservation, Cbp2 could be classified in the homeodomain superfamily. Proteins containing two HTH domains separated by a flexible linker were compared using the structural alignment algorithm FATCAT (33) (see Materials and Methods). Best matches were obtained with the paired domain (Pax) and Myb (Myeloblastosis) protein structures. Structural superimpositions on representative structures of paired domain (Pax6) and Myb (c-Myb) proteins are illustrated (Figure 8) and the associated statistics are given in Table 2. Since these structures were analysed in protein–DNA complexes (44,45), it is inferred that the Cbp2 domains only undergo minor conformational rearrangements on DNA binding.
Figure 8.

Structural alignment of the N- and C-domains of Cbp2Hb with Pax-6 and c-Myb proteins. Details of the alignment are listed in Table 2. Structures of Pax-6 (PDB 6PAX) and Myb (PDB 1H88) subunits (46,47). The short β-motif at the N-terminus of Pax-6 is omitted and for the tripartite c-Myb protein only the C-terminal R2 and R3 domains are shown.
Table 2.
Structural alignment of Cbp2 with members of paired domain (Pax-6) and Myb (c-Myb) families
| Name | PDB code | Domain | Length (a.a.) | Linker length (a.a.) | Recognition sequence length (bp) | Number of Cαa | r.m.s.db | Seq. id. (%)c | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Cbp2 | 2LVS | – | 103 | 15 | 25 | – | – | – | – |
| Pax-6 | 6PAX | Paired | 133 | 17 | 20 | 92 | 2.73 | 20.4 | 46 |
| c-Myb | 1H88 | Myb | 152 | 10 | 6 | 89 | 2.86 | 15.9 | 47 |
aNumber of equivalent Cα atoms in the alignment.
bRoot mean square deviation of aligned Cα atoms.
cPercentage sequence identity.
Overall, structural alignments were best for the N-domain of Cbp2. The C-terminal helix H6 of Cbp2 aligned well with the corresponding DNA-binding helix of c-Myb but less well with that of Pax-6, which is tilted and longer by one turn, although this tilt could reflect a conformational change of the Pax-6 helix on DNA binding. The flexible linker of Cbp2 and the paired domain proteins are also similar in size and basicity. Moreover, the linker length is approximately proportional to the length of the respective DNA recognition sequence, which for the paired domain proteins is ∼20 bp, similar to ∼25 bp for Cbp2 and Cbp1, while Myb proteins recognize shorter regions (∼8 bp).
DISCUSSION
Protein Cbp2Hb of H. butylicus was selected for NMR structural studies because of its small size, high thermostability and its simpler bipartite structure than the tripartite Cbp1 proteins. Cbp2 repeats show significant sequence similarity to the N- and C-terminal repeats of Cbp1 and, moreover, Cbp2Hb bound specifically to CRISPR DNA repeats of H. butylicus and of S. solfataricus P2, the substrate for Cbp1Ss (15,17). The similarities of the protein structures and of the CRISPR DNA repeat-binding properties suggest that Cbp2 proteins, like the Cbp1 proteins, facilitate production of longer transcripts from CRISPR loci, by overcoming adverse effects of transcriptional signals taken up randomly in spacers (17,18). Proteins Cbp2 and Cbp1 occur in organisms carrying A+T-rich genomes (63–65%) for which many CRISPR spacers, and occasionally repeats, contain archaeal-type hexameric TATA-like promoter motifs, or T-rich terminator motifs. These motifs can potentially lead to interference of the continuous transcription of CRISPR loci by generating intermitent reverse transcripts (9,19,20), a hypothesis that was reinforced for Cbp1 by experimental studies on Sulfolobus species (17). Exceptionally, H. butylicus has a lower A+T content of 46%, but this may reflect its adaptation to growth at extremely high temperatures of up to 108°C (21).
Cbp2 consists of two well-defined and non-interacting HTH domains separated by a flexible linker. The N-domains, and C-domains, of Cbp2 and Cbp1 are related structurally, each exhibiting four residues conserved in both proteins, while the central domain of Cbp1 is unique (Figures 1A and B and Supplementary Figure S1). For Cbp2, the N-domain residues Y15, G18, K/R22, I24, Y36 and L39, and the C-domain residues I69, Y80, I82, G87, S91, Y94, L97, K98 and K/R99 are conserved (Figure 1A and B). Three of the N-domain residues, Y15, I24 and L39, are important for maintaining the hydrophobic core of Cbp2, while residues I69 and I82 of the C-domain are typical of HTH motifs (40) and are buried in the C-domain core with L97. The resonance signal of conserved residue S91 disappeared on CRISPR-1rHb binding, suggesting that chemical exchange had occurred and, moreover, the conserved residues, Y95, K98 and K99, located in the putative DNA recognition helix 6 of the C-domain, experienced large chemical shift changes consistent with a direct contact with DNA (Supplementary Figure S3). The linkers of the Cbp2 proteins are rich in basic residues but variable in length. Their sequences align best with the linker between repeats 2 and 3 of Cbp1. The linker connecting subunits 1 and 2 of Cbp1 is also basic, but it carries proportionally more hydrophobic residues (Figure 1B).
DNA binding of Cbp2 revealed a complex pattern of interactions where the primary binding site is apparently the conserved 12 bp downstream region of the 25 bp repeat. The combination of ITC and NMR results, and our demonstration that exchanging the N-domain cysteines of Cbp2 weakens DNA binding at higher temperatures, collectively showed that the N-domain has a higher affinity for the DNA. On the other hand, the C-domain may provide specificity through its dimerization properties that could facilitate cooperative binding of two Cbp2 molecules to a repeat DNA sequence.
Cbp2 proteins occur in organisms with higher optimal growth temperatures than those containing Cbp1 proteins, and this may reflect that smaller proteins tend to exhibit higher thermal stability and/or a tendency of hyperthermophiles to evolve minimally sized genomes (22,46). Cbp2Hb of H. butylicus is also the only CRISPR DNA repeat-binding protein carrying two cysteines, and the DNA-binding experiments indicate that their presence strongly enhances DNA binding at high temperatures. This is consistent with the finding that intramolecular disulphide bonds occur more frequently in hyperthermophilic organisms where they can enhance protein thermostability (47,48). The two cysteine residues were estimated, on the basis of the corresponding serine positions, to be separated by 4.6 Å in the Cbp2 structure and to face one another. Thus formation of a disulphide bond of about 2.05 Å (49) would only require a minor backbone movement induced by thermal motion at the high growth temperatures. The structures of Cbp2, with and without a disulphide bond, are therefore likely to be similar, requiring only a small relative movement of helices H1 and H2.
Structural alignment analyses revealed that the bipartite structure of Cbp2Hb corresponds most closely to structures of homeodomain superfamily proteins that carry linker regions. Most significant similarity, based on the structural alignments, domain architecture, linker length and sequence identity, was to the paired domain Pax and Myb proteins. Consistent with our results, the N-terminal domain of the Pax protein produces stronger DNA binding (44). Moreover, the interaction of the similar basic linkers of the Pax protein with the minor groove of the DNA (44) correlates with the inaccessibility of the Cbp2 linker in the DNA complex. Myb family proteins frequently carry three repeats, similar to Cbp1 proteins, which are also each more conserved intermolecularly. In conclusion, the involvement of the two eukaryal protein families in transcriptional regulation (50,51) provides additional support for the putative role of Cbp2, and of Cbp1, in regulating transcription from CRISPR loci in hyperthermophiles of the Desulfurococcales.
ACCESSION NUMBERS
Chemical shifts have been deposited in the biological magnetic resonance data bank under the accession code 18589, and the structural coordinates have been submitted to the protein data bank with the accession code 2LVS.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–4 and Supplementary Reference [36].
FUNDING
The Carlsberg Foundation (to P.O.H., B.B.K. and F.M.P.); Danish Natural Science Research Council (to R.A.G.); State Government of Karnataka, India (to C.S.K.). Funding for open access charge: Danish Natural Science Research Council.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Dr Shiraz A. Shah’s help with bioinformatical analyses of repeat sequences was appreciated, and we thank Dr Kaare Teilum and Susanne Erdmann for insightful discussions. Dr Lars Olsen advised on the ITC experiments and Dr Magnus Kjaergaard is thanked for technical assistance with NMR.
REFERENCES
- 1.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011;14:1–7. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrett RA, Vestergaard G, Shah SA. Archaeal CRISPR-based immune systems: exchangeable functional modules. Trends Microbiol. 2011;19:549–556. doi: 10.1016/j.tim.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lillestøl RK, Shah SA, Brügger K, Redder P, Phan H, Christiansen J, Garrett RA. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol. Microbiol. 2009;72:259–272. doi: 10.1111/j.1365-2958.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- 5.Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R. Identification and characterisation of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 2010;75:1495–1512. doi: 10.1111/j.1365-2958.2010.07073.x. [DOI] [PubMed] [Google Scholar]
- 6.Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drungowski M, Elge T, Brosius J, Hüttenhofer A. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc. Natl Acad. Sci. USA. 2002;99:7536–7541. doi: 10.1073/pnas.112047299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang TH, Polacek N, Zywicki M, Huber H, Brügger K, Garrett R, Bachellerie JP, Hüttenhofer A. Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol. Microbiol. 2005;55:469–481. doi: 10.1111/j.1365-2958.2004.04428.x. [DOI] [PubMed] [Google Scholar]
- 8.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wurtzel O, Sapra R, Chen F, Zhu YW, Simmons BA, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang R, Preamplume G, Terns MP, Terns RM, Li H. Interaction of Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011;19:257–264. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Rouillon C, Kerou M, Reeks J, Brügger K, Graham S, Reimann J, Cannone G, Liu H, Albers SV, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol. Cell. 2012;45:303–313. doi: 10.1016/j.molcel.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatoum-Aslan A, Maniv I, Marraffini LA. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl Acad. Sci. USA. 2011;108:21218–21222. doi: 10.1073/pnas.1112832108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng X, Brügger K, Shen B, Chen L, She Q, Garrett RA. Genus-specific protein binding to the large clusters of DNA repeats (short regularly spaced repeats) present in Sulfolobus genomes. J. Bacteriol. 2003;185:2410–2417. doi: 10.1128/JB.185.8.2410-2417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lundgren M, Andersson A, Chen L, Nilsson P, Bernander R. Three replication origins in Sulfolobus species: synchronous initiation of chromosome replication and asynchronous termination. Proc. Natl Acad. Sci. USA. 2004;101:7046–7051. doi: 10.1073/pnas.0400656101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng L, Kenchappa CS, Peng X, She Q, Garrett RA. Transcription of CRISPR loci in Sulfolobus is modulated by the repeat-binding protein Cbp1. Nucleic Acids Res. 2011;40:2470–2480. doi: 10.1093/nar/gkr1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah SA, Hansen NR, Garrett RA. Distributions of CRISPR spacer matches in viruses and plasmids of crenarchaeal acidothermophiles and implications for their inhibitory mechanism. Biochem. Soc. Trans. 2009;37:23–28. doi: 10.1042/BST0370023. [DOI] [PubMed] [Google Scholar]
- 19.Torarinsson E, Klenk HP, Garrett RA. Divergent transcriptional and translational signals in Archaea. Environ. Microbiol. 2005;7:47–54. doi: 10.1111/j.1462-2920.2004.00674.x. [DOI] [PubMed] [Google Scholar]
- 20.Santangelo TJ, Cubonova L, Skinner KM, Reeve JN. Archaeal intrinsic transcription termination in vivo. J. Bacteriol. 2009;191:7102–7108. doi: 10.1128/JB.00982-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zillig W, Holz I, Janekovic D, Klenk HP, Imsel E, Trent J, Wunderl S, Forjaz VH, Coutinho R, Ferreira T. Hyperthermus butylicus, a hyperthermophilic sulfur-reducing archaebacterium that ferments peptides. J. Bacteriol. 1990;172:3959–3965. doi: 10.1128/jb.172.7.3959-3965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brügger K, Chen L, Stark M, Zibat A, Redder P, Ruepp A, Awayez M, She Q, Garrett RA, Klenk HP. The genome of Hyperthermus butylicus: a sulphur-reducing, peptide fermenting, neutrophilic crenarchaeote growing up to 108°C. Archaea. 2007;2:127–135. doi: 10.1155/2007/745987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wass JA. Biotech Software and Internet Report. 2002;3:130–133. [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich ER, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;1:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 26.Feher VA, Zapf JW, Hoch JA, Dahlquist FW, Whiteley JM, Cavanagh J. 1H, 15N, and 13C backbone chemical shift assignments, secondary structure, and magnesium-binding characteristics of the Bacillus subtilis response regulator, Spo0F, determined by heteronuclear high-resolution NMR. Protein Sci. 1995;4:1801–1814. doi: 10.1002/pro.5560040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grzesiek S, Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 28.Yamazaki T, Nicholson LK, Torchia DA, Stahl SJ, Kaufman JD, Wingfield PT, Domaille PJ, Campbell-Burk S. Secondary structure and signal assignments of human-immunodeficiency-virus-1 protease complexed to a novel, structure-based inhibitor. Eur. J. Biochem. 1994;15:707–712. doi: 10.1111/j.1432-1033.1994.tb19987.x. [DOI] [PubMed] [Google Scholar]
- 29.Lee W, Revington MJ, Arrowsmith C, Kay LE. A pulsed field gradient isotope-filtered 3D 13C HMQC-NOESY experiment for extracting intermolecular NOE contacts in molecular complexes. FEBS Lett. 1994;15:87–90. doi: 10.1016/0014-5793(94)00740-3. [DOI] [PubMed] [Google Scholar]
- 30.Güntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 31.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 32.Heidarsson PO, Bjerrum-Bohr IJ, Jensen GA, Pongs O, Finn BE, Poulsen FM, Kragelund BB. The C-terminal tail of human neuronal calcium sensor 1 regulates the conformational stability of the Ca2+-activated state. J. Mol. Biol. 2012;417:51–64. doi: 10.1016/j.jmb.2011.12.049. [DOI] [PubMed] [Google Scholar]
- 33.Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19:246–255. doi: 10.1093/bioinformatics/btg1086. [DOI] [PubMed] [Google Scholar]
- 34.Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through the sequence weighing position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackey AJ, Haystead TA, Pearson WR. Getting more from less: algorithms for rapid protein identification with multiple short peptide sequences. Mol. Cell. Proteomics. 2002;1:139–147. doi: 10.1074/mcp.m100004-mcp200. [DOI] [PubMed] [Google Scholar]
- 36.Kumar S, Dudley J, Nei M, Tamura K. MEGA4: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008;9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lillestøl RK, Redder P, Garrett RA, Brügger K. A putative viral defence mechanism in archaeal cells. Archaea. 2006;2:59–72. doi: 10.1155/2006/542818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007;8:611–617. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;35:197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 41.van Leeuwen HC, Strating MJ, Rensen M, de Laat W, van der Vliet PC. Linker length and composition influence the flexibility of Oct-1 DNA binding. EMBO J. 1997;16:2043–2053. doi: 10.1093/emboj/16.8.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vuzman D, Polonsky M, Levy Y. Facilitated DNA search by multidomain transcription factors: cross-talk via a flexible linker. Biophys. J. 2010;99:1202–1211. doi: 10.1016/j.bpj.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wardleworth BN, Russell RJ, Bell SD, Taylor GL, White MF. Structure of Alba: an archaeal chromatin protein modulated by acetylation. EMBO J. 2002;21:4654–4662. doi: 10.1093/emboj/cdf465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999;13:1263–1275. doi: 10.1101/gad.13.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tahirov TH, Sato K, Itchikawa-Iwata E, Sasaki M, Inoue-Bungo T, Shiina M, Kimura K, Takata S, Fujikawa A, Morii H, et al. Mechanism of c-Myb-C/EBP beta cooperation from separated sites on a promoter. Cell. 2002;108:57–70. doi: 10.1016/s0092-8674(01)00636-5. [DOI] [PubMed] [Google Scholar]
- 46.Kumar S, Nussinov R. How do thermophilic proteins deal with heat? Cell. Mol. Life Sci. 2001;58:1216–1233. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mallick P, Boutz DR, Eisenberg D, Yeates TO. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc. Natl Acad. Sci. USA. 2002;99:9679–9684. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beeby M, O’Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The genomics of disulfide bonding and protein stabilization in thermophiles. PloS Biol. 2005;3:1549–1558. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hazes B, Dijkstra BW. Model building of disulfide bonds in proteins with known three-dimensional structure. Protein Eng. 1988;2:119–125. doi: 10.1093/protein/2.2.119. [DOI] [PubMed] [Google Scholar]
- 50.Ness SA. Myb binding proteins:regulators and cohorts in transformation. Oncogene. 1999;18:3039–3046. doi: 10.1038/sj.onc.1202726. [DOI] [PubMed] [Google Scholar]
- 51.Lang D, Powell SK, Plummer RS, Young KP, Ruggeri BA. Pax genes: roles in development, pathophysiology, and cancer. Biochem. Pharmacol. 2007;73:1–14. doi: 10.1016/j.bcp.2006.06.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.