
Figure 1. Deregulation of the Inflammatory Response Recruited by CXCL10 Following HCV Infection.

Sensing of viral RNA by the innate immune receptors RIG-I and TLR3 following hepatitis C virus (HCV,
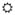 ) infection of the hepatocyte leads to signal transduction through MAVS and TRIF, respectively, activation of transcription factors (NF-κB, IRFs, AP-1, C/EBP-β), and transcription of CXCL10 (Δ) (“CXCL10 Induction”). Secreted CXCL10 forms a chemotactic gradient that recruits immune cells (Natural Killer [NK], CD4+ TH1, and CD8+ Tc cells) and non-parenchymal liver cells (Kupffer cells and Hepatic Stellate Cells [HSCs]) to the site of infection (“Recruitment, Inflammation, and Cell Death”). Upon arriving, these cells produce pro-inflammatory, pro-apoptotic mediators (❖) such as type I interferon (IFN), type III IFN, TNFα, IL-1β, and reactive oxygen species (ROS). This response fails to clear HCV in 80–85% of patients and instead generates persistent inflammation and hepatocyte turnover. It also leads to liver fibrosis through chronic HSC activation, the overproduction of type I collagen, and the inhibition of collagen-degrading matrix metalloproteinases (MMPs) by tissue inhibitor of metalloproteinases (TIMPs) (“Cell Turnover and Fibrosis”). Over several decades, progressive fibrosis can lead to cirrhosis and hepatocellular carcinoma.
) infection of the hepatocyte leads to signal transduction through MAVS and TRIF, respectively, activation of transcription factors (NF-κB, IRFs, AP-1, C/EBP-β), and transcription of CXCL10 (Δ) (“CXCL10 Induction”). Secreted CXCL10 forms a chemotactic gradient that recruits immune cells (Natural Killer [NK], CD4+ TH1, and CD8+ Tc cells) and non-parenchymal liver cells (Kupffer cells and Hepatic Stellate Cells [HSCs]) to the site of infection (“Recruitment, Inflammation, and Cell Death”). Upon arriving, these cells produce pro-inflammatory, pro-apoptotic mediators (❖) such as type I interferon (IFN), type III IFN, TNFα, IL-1β, and reactive oxygen species (ROS). This response fails to clear HCV in 80–85% of patients and instead generates persistent inflammation and hepatocyte turnover. It also leads to liver fibrosis through chronic HSC activation, the overproduction of type I collagen, and the inhibition of collagen-degrading matrix metalloproteinases (MMPs) by tissue inhibitor of metalloproteinases (TIMPs) (“Cell Turnover and Fibrosis”). Over several decades, progressive fibrosis can lead to cirrhosis and hepatocellular carcinoma.