

Abstract
It may be possible to achieve insulin sensitivity through the recently identified mitochondrial target of thiazolidinediones (mTOT), thereby avoiding peroxisome proliferator–activated receptor-γ (PPAR-γ)-dependent side effects. In this phase IIb clinical trial, 258 patients with type 2 diabetes completed a 12-week protocol with 50, 100, or 150 mg of MSDC-0160 (an mTOT modulator), 45 mg pioglitazone HCl (a PPAR-γ agonist), or a placebo. The two active treatments lowered fasting glucose levels to the same extent. The decreases in glycated hemoglobin (HbA1c) observed with the two higher doses of MSDC-0160 were not different from those associated with pioglitazone. By contrast, fluid retention as evidenced by reduction in hematocrit, red blood cells, and total hemoglobin was 50% less in the MSDC-0160–treated groups. There was also a smaller increase in high-molecular-weight (HMW) adiponectin with MSDC-0160 than with pioglitazone (P < 0.0001), suggesting that MSDC-0160 produces less expansion of white adipose tissue. Thus, mTOT modulators may have glucose-lowering effects similar to those of pioglitazone but without the adverse effects associated with PPAR-γ agonists.
Diabetes remains undertreated throughout the developed and developing worlds, and its incidence continues to grow, contributing significantly to rising health-care costs.1,2 It is generally accepted that reducing insulin resistance, and thereby increasing insulin sensitivity, would be an ideal way to treat this disease, given that insulin resistance is a key factor in the early pathogenesis of type 2 diabetes.3,4,5 However, only three insulin sensitizers have ever been approved for clinical use, and two of these have been removed from the market or have had their clinical use severely restricted because of various safety concerns.6 The third one, pioglitazone, although clearly different from the others, has seen its clinical use curtailed because of its side effects, which include weight gain and volume expansion, and concerns about congestive heart failure and bladder cancer.7 We have suggested that this field has been held back because of the singular focus on the hypothesis that insulin sensitizers must activate peroxisome proliferator–activated receptor-γ (PPAR-γ).8 Recently, we showed that insulin sensitivity can be achieved through PPAR-γ-independent mechanisms9 and provided evidence that such PPAR-independent effects may involve control of oxidative metabolism through modulation of a mitochondrial target that regulates metabolism of pyruvate.10 Compounds that work primarily through this mechanism, mitochondrial target of thiazolidinediones (mTOT), should be able to produce insulin-sensitizing pharmacology without the side effects associated with agents that are PPAR-γ agonists.
The prototype mTOT modulator we have examined is MSDC-0160 (Figure 1). This compound and its major hydroxymetabolite have 20-fold lower PPAR-γ activation effects, yet the compound displays insulin-sensitizing pharmacology in animal models (ref. 11; Supplementary Figure S1 online). In the present study, we compared the effects of three dosages of MSDC-0160 with those of placebo, using 45 mg pioglitazone HCl (the highest approved dosage of that compound in type 2 diabetes) as the reference, over a 12-week treatment period. The primary questions to which we sought answers were (i) will treatment with the mTOT modulator MSDC-0160 be able to achieve the same level of glucose control as treatment with the PPAR-γ-agonist pioglitazone? and (ii) will it be able to achieve this action in patients without producing PPAR-γ-like side effects that can be measured in a 12-week trial, namely fluid accumulation and weight gain?
Figure 1.

Structures of medications and establishing of dosages. (a) Structures of pioglitazone and major active metabolites, and of MSDC-0160 and its major metabolite, the R-alcohol. (b) Pharmacokinetics of MSDC-0160 (parent plus hydroxymetabolite) in normal volunteers after the first and seventh doses. The compound was given once daily for 7 consecutive days, and the complete profile was generated on both days 1 (first dose) and 7. Trough levels were measured every day before the next dose. (c) Formulated tablets for use in the phase IIb trial were evaluated in normal volunteers. Data are shown for the combined area under the curve (AUC) of MSDC-0160 and its major metabolite at the three given doses, depicted by closed squares (q.d., mg/day). The dotted line shows the AUC of pioglitazone and its active metabolites (~60,000 ng ∙ hr/ml) after dosing with 45 mg of pioglitazone HCl (Actos). The three doses selected for the phase IIb study are shown by arrows.
Results
Summary of preliminary studies
MSDC-0160 has been evaluated through 90 days of toxicology studies in preclinical species, including mice, rats, and cynomolgus monkeys. Single-dose and multiple-dose studies in normal volunteers established the dosage range to be tested in the efficacy trials. The results of these trials have not yet been published, but the phase IIa study results using bulk drug have been presented.12 Figure 1a shows the structures of pioglitazone and its three active metabolites as well as of MSDC-0160 and its major hydroxymetabolite. Figure 1b shows the kinetics of MSDC-0160 and its major metabolite during a 7-day dosing period. The compound and its metabolite achieve steady-state levels within several days, and each has a half-life of ~12 h; by comparison, metabolites III and IV of pioglitazone have half-lives of ~24 h.13 Figure 1c shows the relative areas under the concentration–time curve at various dosages of the formulation of MSDC-0160 to be used in the IIb trial. On the basis of these data, dosages of 50, 100, and 150 mg of MSDC-0160 were selected for comparison with 45 mg of pioglitazone HCl (Actos). These produce areas under the curve that bracket the exposure levels produced by 45 mg Actos.
12-Week phase II trial
The summary of treatment-emergent adverse events in the trial is shown in Supplementary Table S1 online; the disposition of all patients is shown in Supplementary Table S2 online. No safety issues of concern were uncovered in this trial. The primary efficacy evaluation was the change in fasting plasma glucose level from baseline to end point, relative to the placebo group (Supplementary Table S3 online). There was a significant reduction in plasma glucose levels in the MSDC-0160 100-mg group (least-squares mean difference of −18.4 mg/dl; P = 0.0057), the MSDC-0160 150-mg group (least-squares mean difference of −28.9 mg/dl; P < 0.0001), and the pioglitazone group (least-squares mean difference of −31 mg/dl; P < 0.0001). The data from this study were then used to compare the relative effects of the active treatments in the patients who completed the 12-week study per protocol.
The characteristics of patients completing the 12-week trial per protocol are shown in Table 1. There were no differences in the baseline parameters across the five treatment groups. Approximately 80% of the patients were on stable metformin treatment and remained on it throughout the trial. The rest of the patients (~20%) were not receiving antidiabetes medications. The trial population had an average starting glycated hemoglobin (HbA1c) of ~8%, average fasting plasma glucose of ~170 mg/dl, and average time since diagnosis of ~6 years. Both genders were represented, and the average age of the cohort was ~55 years.
Table 1. Demographics.
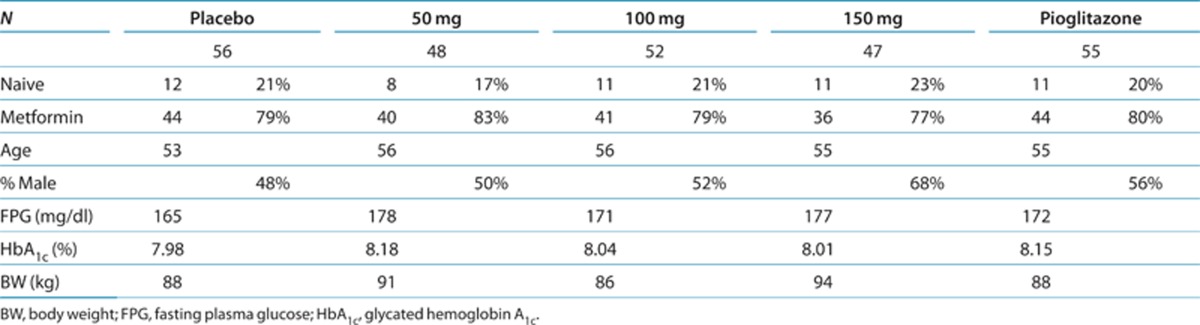
Figure 2 shows the time course of change in HbA1c over the duration of the trial. Figure 2a shows the time course of the average HbA1c levels during the double-blind treatment in the five groups of patients, and Figure 2b shows the placebo-adjusted end point (least-squares mean change from baseline with SE bars) for the active groups at 12 weeks. As shown in Figure 2a, patients in the placebo group demonstrated a progressive worsening of glucose control despite the fact that 79% of them were continuing their current regimens of metformin treatment. There was an improvement in glucose control in all the active treatment groups relative to the placebo group. Treatment with the two highest doses of MSDC-0160 produced reductions in HbA1c that were not different from those obtained with 45 mg pioglitazone HCl (Figure 2b).
Figure 2.

Effect of 12 weeks of treatment on hemoglobin A1c (HbA1c). Data show the HbA1c changes from baseline (as a percentage) for each of the five treatment groups. (a) The change in HbA1c at each visit during the double-blind treatment period. Variations are not shown on the time curve but are similar to the data shown for the terminal 12-week time point shown in (b). The abscissa marks the time points of the visits (actual time points are baseline and weeks 2, 4, 8, and 12 during the double-blind treatment). (b) The placebo-adjusted least-squares change and SE at the 12-week end point for each of the treatment groups. The baseline HbA1c values and the number per group are shown below the bar chart. P values are given for the least-squares placebo-adjusted change from baseline. LS, least squares.
Although MSDC-0160 produced reductions in the levels of circulating glucose similar to those produced by pioglitazone, no dose level of MSDC-0160 was able to produce a decrease in the concentration of total hemoglobin (an index of volume dilution) similar to those produced by pioglitazone (Figure 3). The time course of the pioglitazone-induced reduction in total circulating hemoglobin is shown in Figure 3a, and the relative effects of the treatments on placebo-adjusted changes at the 12-week end point are shown in Figure 3b. The data show that at no point was MSDC-0160 able to produce the same reduction in total hemoglobin that pioglitazone did. As expected, these data mirror the changes in total red blood cells and hematocrit in the corresponding samples (Supplementary Figure S2 online). The presence of edema was systematically evaluated during the course of the study, using a scale of 0 to 4+. Edema was noted in 11.4% of the patients in the placebo group; in 11.8, 13.0, and 5.7% of the MSDC-0160 50-, 100-, and 150-mg groups; respectively; and in 8.5% of the pioglitazone group. The edema was mostly rated as mild (1 or 2); however, in two patients receiving MSDC-0160 and one receiving pioglitazone the edema was rated as 3, and in another patient receiving MSDC-0160 it was rated as 4 (Supplementary Table S4 online). Peripheral edema was reported as an adverse event in three subjects: one in the MSDC-0160 100-mg group and two in the MSDC-0160 150-mg group. All the cases were mild and resolved while on treatment with no specific action taken. All three subjects completed the 12-week study.
Figure 3.

Effect of 12 weeks of treatment on total hemoglobin as a marker of fluid retention. Data show the change in total hemoglobin (g/dl) from baseline for each of the five treatment groups. (a) The change at each visit during the double-blind treatment period. (b) The placebo-adjusted least-squares change (mean and SE) at the 12-week end point (the last time point in (a)). The starting values of total hemoglobin and the number per group are shown below the bar chart. P values under the bars depict the least-squares change from baseline. P values below the panel show the differences between the MSDC-0160 groups and the pioglitazone group. LS, least squares.
There were also differences between the effects of MSDC-0160 and pioglitazone with respect to changes in high-molecular-weight (HMW) adiponectin (Figure 4). The time courses of the effects of each of the five treatments on the change from baseline in HMW adiponectin at 2 and 12 weeks are shown in Figure 4a. The indexes of volume expansion show clear differences between the increases in this parameter caused by MSDC-0160 and those caused by pioglitazone. The statistical evaluation of these changes at the 12-week end point is shown in Figure 4b. Although MSDC-0160 treatment caused significant increases in HMW adiponectin, no dose of this compound was able to produce the same magnitude of increase as that with pioglitazone treatment. Importantly, these differences between treatment effects occurred under conditions in which glucose was being lowered to the same extent in the two experimental groups.
Figure 4.

Effect of 12 weeks of treatment on high-molecular-weight (HMW) adiponectin. Data show the change in HMW adiponectin from baseline for the five treatment groups. (a) The percentage change after 2 weeks and at the end point (12 weeks) for the five groups. (b) The placebo-adjusted least-squares change (mean and SE) at the 12-week end point. The starting values of HMW adiponectin (ng/ml) and the number per group are shown below the bar chart. P values above the bars depict the change from baseline. P values below the panel show the differences between the MSDC-0160 groups and pioglitazone. HMWA, HMW adiponectin; LS, least squares.
The effects on total body weight are shown in Figure 5a,b. The placebo-treated group tended to lose body weight over the 12-week duration of the study, whereas all the treatment groups tended to show increases in body weight. The statistics for the changes in body weight at the 12-week treatment end point are shown in Figure 5b. Although the increases in body weight in the MSDC-0160 treatment groups tended to be smaller than those in the pioglitazone treatment group, there was no significant difference between the 150-mg MSDC-0160 group and the pioglitazone reference group with respect to this parameter. However, given the differences in the levels of white adipose marker adiponectin (Figure 4), the weight gain associated with the treatments may not be identical in nature. In support of this hypothesis, Figure 5c,d shows that the trend toward increase in waist circumference seen in the pioglitazone comparator group is absent in the MSDC-0160-treated groups.
Figure 5.

Effect of 12 weeks of treatment on body weight and waist circumference. Data show the (a,b) change in body weight and (c,d) waist circumference from baseline for each of the five treatment groups. (a) The change in body weight (kg) at each visit during the double-blind treatment period. (b) The adjusted least-squares change (mean and SE) at the 12-week end point for all five groups. The initial weight (kg) and N are shown below the bars. (c) The change in waist circumference at each visit during the double-blind treatment period. (d) The adjusted least-squares change (mean and SE) at the 12-week end point for all five groups. Data are expressed in cm (mean and SE). In each case, the 12-week time point (b,d) represents the last point on the time curves (a,c). BW, body weight; LS, least squares.
Pioglitazone is known to have positive effects on cholesterol metabolism, which might partly explain its positive impact on cardiovascular outcomes.14,15 Although there are clear differences in this respect between the PPAR-γ agonists approved for clinical use,15,16 it is possible that some aspect of PPAR-γ agonism might contribute to this action, and that compounds designed for less interaction with PPAR-γ might not share this action. Hence, we performed an analysis of the changes in lipoprotein particles during the treatment protocol across the various groups to determine where a compound without significant activation of PPAR-γ might share the positive effects of pioglitazone in this respect. Figure 6a shows the differences in size distribution in the high-density lipoprotein cholesterol subfractions (small, medium, and large) at the 12-week end-point time relative to pretreatment. In comparison to placebo, treatment with either compound tended to reduce the number of the small particles and shift toward a distribution of larger particles. A similar change occurred in the low-density lipoprotein (LDL) subfractions (Figure 6c). This resulted in an increase in the average sizes of both high-density lipoprotein and LDL particles (Figure 6b). In contrast to their effects on high-density lipoprotein, the treatments reduced the sizes of the very-low-density lipoprotein (VLDL) subfractions (Figure 6d). In general, the effects of MSDC-0160 were similar to those of pioglitazone, except that the magnitude of the changes in LDL and VLDL were smaller. This is consistent with the finding that treatment with pioglitazone produces a significant decrease in circulating triglycerides, an effect that did not occur with MSDC-0160 treatment (Supplementary Figure S3 online).
Figure 6.

Effect of 12 weeks of treatment on lipoprotein particle size. Data show the change in the distribution of HDL particles (a: small, medium, large), LDL particles (c: small or large), and VLDL particles (d: small, medium, large). The data (mean and SEM) are presented as change from baseline for each of the five groups. The legend in the center panel presents the number of individuals in each group. (b) The changes in average size (nm) of HDL and LDL particles. Data are expressed as mean and SEM for the numbers shown in parentheses in the central panel. HDL, high-density lipoprotein; VLDL, very low-density lipoprotein.
Discussion
This study demonstrates that MSDC-0160, a prototype mTOT modulator with little ability to bind to or activate PPAR-γ, is able to lower glucose to the same extent as the PPAR-γ agonist pioglitazone over 12 weeks of treatment in patients with type 2 diabetes. Importantly, at the exposure levels required for lowering glucose to this extent, there was a markedly lower effect of volume dilution as detected by a reduction in circulating total hemoglobin and blood cells (Figure 2 as compared with Figure 3). However, little edema was seen in any of the groups, and the potential for differences vs. pioglitazone in this respect requires investigation in larger trials of longer duration.
Also, a comparison of these treatment groups shows that the ability to lower glucose can be decoupled from the extent of elevation of HMW adiponectin (Figure 4 as compared with Figure 2). This is important, given that it is often believed that an increase in white adipose tissue is a necessary sign to show that an insulin sensitizer is exerting appropriate effects by moving lipids out of the peripheral tissues and into white adipose tissue. The hypothesis generally posited for insulin-sensitizing pharmacology predicts that at least some of the PPAR-γ-driven side effects would have to occur, especially the expansion of white adipose tissue, in order to increase insulin sensitivity. The current results, however, argue to the contrary and support the need for continued evaluation of molecules designed in line with the newer hypothesis, i.e., that the mitochondrial metabolic modulation should be maintained at the expense of activation of PPAR-γ.
We recently identified the mitochondrial target of insulin sensitizers as a well-conserved, but previously unrecognized, protein complex (mTOT) in the inner mitochondrial membrane, both in islets and in peripheral tissues.10 Key proteins in this complex form a recognition site for entry of pyruvate into the mitochondria.17,18 This regulatory machinery is therefore at the crossroads of the control of oxidative metabolism.19 Although the need for further elucidation remains, it is not difficult to imagine how metabolic redox signals—regulated by the change in the way pyruvate is handled by the mitochondria—coordinate cellular responses that regulate many cellular functions from cell cycle/differentiation to insulin sensitivity. This study is important because the clinical results show that compounds that focus on this mechanism of action, mTOT modulation, are able to achieve significant lowering of glucose without the liabilities—side effects—associated with PPAR-γ agonists. Consequently, modulation of mTOT might provide a way forward to treat type 2 diabetes.
Henry et al.7 recently reviewed the current understanding of insulin sensitization. There is now consensus that the first three insulin sensitizers that were approved and gained market experience were not identical. Pioglitazone (the only one of the three that remains on the market) clearly proved to have advantages in the long run, yet the side effects and concerns related to PPAR-γ agonism have limited its use. It had previously been suggested that it may be reasonable to explore other ways to increase insulin sensitivity.8 The current observations support the need for further exploration of the potential of mTOT-modulating molecules. This study suggests that mTOT modulators will share the useful pharmacology of pioglitazone, including positive effects on lipoprotein metabolism (Figure 6) that are thought to be important in its protective effects on cardiovascular outcomes.6
Our 12-week study demonstrates that mTOT modulation can deliver comparable efficacy without the liabilities of PPAR-γ agonism that are typically observed in studies of this duration. Longer-term studies are needed to further define the potential of this new approach to treating insulin resistance and to determine whether there will be significant benefits to the use of this compound or other mTOT-modulating molecules in the treatment of type 2 diabetes.
Methods
Preliminary studies. Phase I and bioequivalence trials (MSDC-C001, MSDC-C002, and MSDC-005) to establish dosing were conducted in normal volunteers at the Jasper Clinic (Kalamazoo, MI) after approval by the local institutional review board. MSDC-0160 and formulated tablets of the compound were produced by USV Limited (Daman, India) and were compared with commercial pioglitazone HCl in blinded gelatin capsules (8 × 22 mm). The concentration levels of MSDC-0160 and its major metabolite, and of pioglitazone and its major metabolites, were measured using high-performance liquid chromatography–mass spectrometry at MPI Research (Mattawan, MI) and Medpace (Cincinnati, OH).
Twelve-week clinical study. This trial (NCT01103414) was a multicenter study (26 US sites) coordinated by Medpace. The study consisted of a 2-week, single-blind, placebo lead-in, followed by baseline data collection and then by a 12-week, double-blind, outpatient treatment period. Inclusion criteria for entry into randomization included a fasting plasma glucose ≥126 mg/dl, HbA1c ≥7% and ≤10%, and insulin C-peptide >1 ng/ml. The study population included men and women 18–75 years. During the placebo lead-in, patients self-administered one capsule of single-blind study medication (placebo) once daily. At the end of the placebo lead-in period, eligible patients were randomized to one of five treatment groups based on a stratification criterion of current metformin use (yes/no) to receive any one of the following treatments: MSDC-0160 50 mg, MSDC-0160 100 mg, MSDC-0160 150 mg, pioglitazone 45 mg, or placebo.
During the 12-week study period, patients self-administered one capsule of (double-blind) study medication once daily. All medications were blinded by overencapsulation of the dose, and the patients were instructed that all doses were to be taken in the morning after an overnight fast and at least 30 min before the morning meal. Safety analysis was completed on an intent-to-treat basis, and no safety concerns were uncovered (summary Supplementary Table S1 online).
Statistical methods. Demographic and baseline characteristics were summarized for all randomized patients and for the patients who had completed the trial per protocol. Age group at screening, gender, ethnicity, race, antidiabetes medication status at screening, statin use at randomization, and body mass index category were summarized as numbers and percentages. Age, duration of type 2 diabetes mellitus, body mass index, and baseline lipid profiles were described using summary statistics (n, mean, SD, median, minimum, and maximum).
All efficacy analyses were performed based on the intent-to-treat population and were repeated on the patients who completed the trial per protocol. The intent-to-treat population consisted of all randomized patients who had taken at least one dose of randomized study medication, and for whom there was a baseline efficacy measurement and at least one postrandomization efficacy measurement. The primary efficacy parameter was the change in fasting plasma glucose from baseline to week 12. Each efficacy parameter was analyzed using an analysis of covariance model with either change or percentage of change as the dependent variable, treatment as a factor, and the baseline value of the parameter. Baseline was defined as the measurement at the week 0 visit. If the week 0 measurement was missing, the last valid measurement before the first dose of randomized double-blind study medication was used. Efficacy parameters were also summarized according to number of patients, mean, median, SD, and minimum and maximum at each visit by treatment group. Given that the purpose of the analysis was to understand the relative effects of the two types of active treatments, only data for patients who completed this trial per protocol were included in the analysis for a comparison among the treatments shown here. Given the exploratory dose-ranging/dose-finding nature of the study, no adjustments for multiple comparisons were made, and a value of P < 0.05 was considered statistically significant.
Plasma concentrations and pharmacokinetic parameters were summarized according to treatment group, using descriptive statistical terms.
All safety analyses were performed on the safety population, which was defined as all randomized patients who had received at least one dose of randomized study medication. Treatment-emergent adverse events were defined as adverse events that occurred for the first time on or after the date of the first dose of double-blind study medication or that had been in existence earlier and worsened during the double-blind treatment period. The numbers and percentages of patients with treatment-emergent adverse events were summarized for each treatment group by system organ class and preferred term (defined by MedDRA version 13.1). Treatment-emergent adverse events associated with double-blind study medications were summarized in the same manner, and also according to system organ class, preferred term, and maximum severity.
Summary statistics were reported for laboratory parameters at baseline, postbaseline visits, and end point by treatment group.
Medpace undertook project management, clinical monitoring, medical monitoring, data management, statistical analysis, and study report preparation. Medpace Reference Laboratories (Cincinnati, OH) performed the clinical laboratory analyses. Medpace Bioanalytical Laboratories (Cincinnati, OH) performed the analyses of pioglitazone and pioglitazone metabolites, and MPI Research performed the analyses of MSDC-0160 and its major hydroxymetabolite; both were by validated liquid chromatography–mass spectrometry methods. Millipore (St Charles, MO) performed the analyses for HMW adiponectin. LipoScience (Raleigh, NC) performed the nuclear magnetic resonance analysis of lipid subfractions.
Author Contributions
J.R.C. wrote the manuscript. J.R.C., J.T.V., W.J.A., W.G.M., J.L., R.Z., and D.G.O. designed the research. A.S. and W.G.M. performed the research. J.R.C., W.J.A., A.S., J.L., R.Z., and D.G.O. analyzed the data.
Study Highlights

Acknowledgments
The authors thank Diane Beuving and Diane Kosis for coordinating the details of this trial, and the following researchers and investigational sites: Michael Azorr, Oregon Clinical Research, Portland, OR; Jolene Berg, Cetero Research, San Antonio, TX; Mark Christiansen, Diablo Clinical Research, Walnut Creek, CA; Steven Chrysant, Oklahoma City, OK; Lisa Cohen, Suncoast Clinical Research, New Port Richey, FL; Martha Hernandez-Illas, Miami, FL; C. Allen Goetsch, Clinical Research Associates, Huntsville, AL; Dean Kereiakes, The Carl and Edyth Lindner Center for Research and Education at Christ Hospital, Cincinnati, OH; Audrey Lacour, Houston, TX; Robert Perry, Pembroke Pines, FL; Sanford Plevin, Suncoast Clinical Research, New Port Richey, FL; Jaime Sandoval, Corpus Christi, TX; Phillip Toth, Indianapolis, IN; Andrew Lewin, National Research Institute, Los Angeles, CA; Terence Hart, Muscle Shoals, AL; Robert Lipetz, Encompass Clinical Research, Spring Valley, CA; William Byars, Mountain View Clinical Research, Greer, SC; Kyle Rasikas, Grand Valley Internal Medicine Specialists, Grand Rapids, MI; Barbara Biggs, Sestron Clinical Research, Marietta, GA; William Randall, Dayton, OH; Azazuddin Ahmed, Apex Medical Research, AMR, Chicago, IL; Cristian Breton, International Research Associates, Miami, FL; Patricia Buchanan, Willamette Valley Clinical Studies, Eugene, OR; James Lassiter, Tiffin, OH; and Gilbert Martinez, Catalina Research Institute, Chino, CA.
J.R.C., J.T.V., W.J.A., A.S., and W.G.M. are all employees and/or part owners of Metabolic Solutions Development Company. The other authors declared no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/cpt
Supplementary Material
References
- Huang E.S., Basu A., O'Grady M., Capretta J.C. Projecting the future diabetes population size and related costs for the U.S. Diabetes Care. 2009;32:2225–2229. doi: 10.2337/dc09-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild S., Roglic G., Green A., Sicree R., King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Abdul-Ghani M.A., Tripathy D., DeFronzo R.A. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–1139. doi: 10.2337/diacare.2951130. [DOI] [PubMed] [Google Scholar]
- Wajchenberg B.L. beta-cell failure in diabetes and preservation by clinical treatment. Endocr. Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- Kim-Muller J.Y., Accili D. Cell biology. Selective insulin sensitizers. Science. 2011;331:1529–1531. doi: 10.1126/science.1204504. [DOI] [PubMed] [Google Scholar]
- Ryder R.E.J. Pioglitazone: an agent which reduces stroke, myocardial infarction and death and is also a key component of the modern paradigm for the optimum management of type 2 diabetes. Br. J. Diabetes Vasc. Dis. 2011;11:113–120. [Google Scholar]
- Henry R.R., Erikson D., Ciraldi T.A. PPARγ agonists and the future for insulin sensitizers. Br. J. Diabetes Vasc. Dis. 2012;12:206–210. [Google Scholar]
- Colca J.R., Kletzien R.F. What has prevented the expansion of insulin sensitisers. Expert Opin. Investig. Drugs. 2006;15:205–210. doi: 10.1517/13543784.15.3.205. [DOI] [PubMed] [Google Scholar]
- Chen Z., et al. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor γ-sparing thiazolidinedione. J. Biol. Chem. 2012;287:23537–23548. doi: 10.1074/jbc.M112.363960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colca J.R., et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)-relationship to newly identified mitochondrial pyruvate carrier proteins PLoS ONE (in press). [DOI] [PMC free article] [PubMed]
- Bolten C.W., et al. Insulin sensitizing pharmacology of thiazolidinediones correlates with mitochondrial gene expression rather than activation of PPAR gamma. Gene Regul. Syst. Bio. 2007;1:73–82. [PMC free article] [PubMed] [Google Scholar]
- Colca J.R., Kletzien R.F., VanderLugt J.T.A PPAR-sparing insulin sensitizer is effective in type 2 diabetic patients without causing weight gain20th World Diabetes Congress D-773. 18–22 October 2009. (Montreal, Québec, 2009
- Eckland D.A., Danhof M.Clinical pharmacokinetics of pioglitazone Exp. Clin. Endocrinol. Diabetes 108suppl. 2), S234–S242.2000 [Google Scholar]
- Winkelmayer W.C., Setoguchi S., Levin R., Solomon D.H. Comparison of cardiovascular outcomes in elderly patients with diabetes who initiated rosiglitazone vs pioglitazone therapy. Arch. Intern. Med. 2008;168:2368–2375. doi: 10.1001/archinte.168.21.2368. [DOI] [PubMed] [Google Scholar]
- Deeg M.A., et al. GLAI Study Investigators Pioglitazone and rosiglitazone have different effects on serum lipoprotein particle concentrations and sizes in patients with type 2 diabetes and dyslipidemia. Diabetes Care. 2007;30:2458–2464. doi: 10.2337/dc06-1903. [DOI] [PubMed] [Google Scholar]
- Rosenson R.S., et al. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011;57:392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- Bricker D.K., et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- Halestrap A.P. The mitochondrial pyruvate carrier: has it been unearthed at last. Cell Metab. 2012;16:141–143. doi: 10.1016/j.cmet.2012.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.