

Abstract
The flavocytochrome cellobiose dehydrogenase (CDH) is a versatile biorecognition element capable of detecting carbohydrates as well as quinones and catecholamines. In addition, it can be used as an anode biocatalyst for enzymatic biofuel cells to power miniaturised sensor–transmitter systems. Various electrode materials and designs have been tested in the past decade to utilize and enhance the direct electron transfer (DET) from the enzyme to the electrode. Additionally, mediated electron transfer (MET) approaches via soluble redox mediators and redox polymers have been pursued. Biosensors for cellobiose, lactose and glucose determination are based on CDH from different fungal producers, which show differences with respect to substrate specificity, pH optima, DET efficiency and surface binding affinity. Biosensors for the detection of quinones and catecholamines can use carbohydrates for analyte regeneration and signal amplification. This review discusses different approaches to enhance the sensitivity and selectivity of CDH-based biosensors, which focus on (1) more efficient DET on chemically modified or nanostructured electrodes, (2) the synthesis of custom-made redox polymers for higher MET currents and (3) the engineering of enzymes and reaction pathways. Combination of these strategies will enable the design of sensitive and selective CDH-based biosensors with reduced electrode size for the detection of analytes in continuous on-site and point-of-care applications.
Keywords: Biosensors, Carbohydrates, Catecholamines, Cellobiose dehydrogenase, Electron transfer, Nanomaterials
Introduction
The last few years have witnessed tremendous development in bioelectrochemistry largely owing to the increased knowledge of making nanostructured electrodes surfaces [1–4] in combination with the understanding of how to bring about controlled architectures/orientation of biomolecules on such surfaces [5–10] and also the closer collaboration between (bio)electrochemists and biochemists/molecular biologists [11–22]. Additionally, research on and the foreseen need for practical applications, e.g. in electrochemical biosensors [23], biofuel cells [24–26] and bioelectrosynthesis [27], have also speeded up the research activities in this area. The drive to make bioelectrochemical devices/systems as simple as possible has put a focus on how to bring about efficient electron transfer reactions between the biologically derived material and electrodes without the need for additional and possibly leaching (and toxic) chemicals [28–32].
Cellobiose dehydrogenase (CDH) is a flavocytochrome [33] belonging to the restricted number of oxidoreductases that in their native wild-type form show efficient direct electron transfer (DET) between the active site and an electrode surface. CDH does this because it consists of two separate domains with different structures and inherent properties joined together by a polypeptide linker region. The larger flavodehydrogenase domain (DHCDH) is catalytically active, whereas the smaller cytochrome domain (CYTCDH) contains haem b as a cofactor and acts as an electron transfer protein between DHCDH and a terminal, macromolecular electron acceptor. By 1991 Hill and co-workers [34] had divided the oxidoreductases, from a bioelectrochemical point of view, into two different groups—intrinsic and extrinsic—and characterised them as follows [34]:
Catalytic reaction between an enzyme and its substrate takes place within a highly localised assembly of redox-active sites. There need be no electron transfer pathways from these sites to the surface of the enzyme, where, it is presumed, it would interact with an electrode. For such intrinsic redox enzymes, electrode reactions may require (1) that the sites of the catalytic reaction be close to the protein surface, (2) that the enzyme can deform without loss of activity, (3) that the electrode surface projects into the enzyme, (4) that electron pathways be introduced by modification of the enzyme. With the extrinsic redox enzymes, there is usually another protein involved in transporting electrons and therefore an electron transfer pathway exists within the enzyme connecting the active sites to an area on the surface where the ancillary protein binds. If this area could be disposed toward an electrode, it would be possible for the enzyme electrochemistry to be obtained.
From a structural point of view CDH [35, 36] is obviously an extrinsic redox enzyme, where the CYTCDH acts as a built-in mediator [37]. What further supports this is that in several recent reports it has been shown that copper-dependent polysaccharide monooxygenase (PMO) is likely to be the physiological redox partner of CDH which can therefore explain the role of CYTCDH (Fig. 1) [38–41].
Fig. 1.
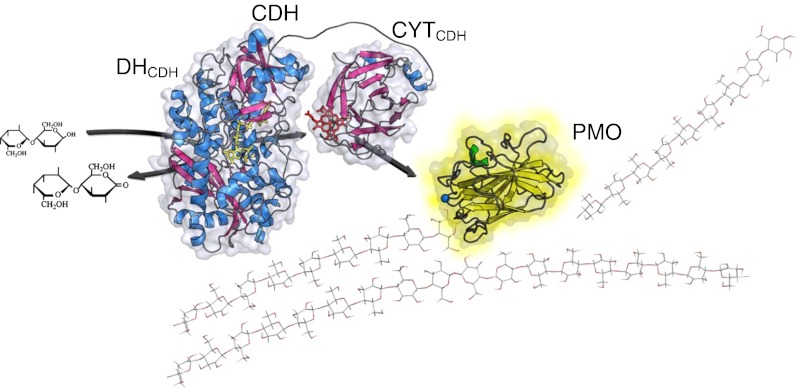
Proposed in vivo function of CDH. Electrons from the oxidation of cellobiose or higher cellodextrins are acquired by CDH, which donates them to the surface-exposed type-2 copper centre of PMO to activate molecular oxygen for the cleavage of cellulose [38–41]. Initial studies show that the electron transfer via the CYTCDH is quite efficient [62]
In 2010 we reviewed the basic electrochemical properties of CDH [19]; however, since then, a series of investigations on the biochemistry and bioelectrochemistry of various CDHs [42–44] have been pursued as well as one on the fundamentals of the intramolecular electron transfer (IET) between the two domains of CDH [45]. Furthermore a series of genetic work has been done to improve the glucose-oxidising properties of CDH (Ortiz et al. submitted) [46], in particular, as well as nanostructuring of both carbon [47] and gold-based electrodes [48, 49] to improve the loading and orientation of CDH on the electrode surface and thus also current densities. Especially in the field of biofuel cells [49–51] great progress has been made on the spatial arrangement of CDH on electrodes (Ortiz et al. submitted) [47–49, 52, 53] and CDH-based biosensors [54–58]. Such progress has prompted this new review on the bioelectrochemistry of CDH. One should also note that CDH has been used to make gold nanoparticles (AuNPs) and with the use of scanning electrochemical microscopy it was possible also to localise such AuNPs on surfaces with the help of CDH [59].
Occurrence and classification of CDH
Cellobiose dehydrogenase is secreted by white- and brown-rot, phytopathogenic as well as composting fungi from the dikaryotic phyla Basidiomycota and Ascomycota under cellulolytic culture conditions [60]. The CDH enzyme family is a heterogenous group of proteins with protein sequences between 749 and 816 amino acids long and sequence identities as low as 35 %. Phylogenetic analyses of these sequences showed several well-supported branches. Basidiomycete CDH sequences (from the Atheliales, Corticiales and Polyporales) form the branch of class I CDHs. Class II consists only of sequences of ascomycete origin (Sordariales, Xylariales and Hypocreales). This class of CDHs partitions into two subclasses: class IIA CDHs and class IIB CDHs. For members of the Eurotiales, Helotiales and Pleosporales, CDH-encoding sequences of a separate phylogenetic branch were found in sequenced genomes. The secretion of these class III CDHs has not yet been confirmed [42, 60, 61]. The molecular architecture of CDHs from class I and II are slightly different. Whereas class I CDHs have supposedly a carbohydrate binding site on the DHCDH, class IIA CDHs carry a small C-terminal family 1 carbohydrate binding module (CBM1), which is missing in class IIB CDHs.
The widespread appearance of CDH in fungi and the fact that it constitutes a considerable fraction of the lignocellulolytic enzymes secreted by these fungi (0.5–12 %) implies that CDH has an important function in wood degradation [62]. Recently the composition of lignocellulolytic enzyme cocktails and the up- and downregulation of single constituents were studied using transcriptomic and proteomic analyses [63–66]. Results from studies on the well-known ascomycete fungus Neurospora crassa and on the white-rot basidiomycete model organism Phanerochaete chrysosporium revealed that CDH genes are up-regulated during growth under cellulolytic conditions [66–68]. Important fungal producers of CDH are summarised in Table 1.
Table 1.
Production of CDH by fungi
| Fungal producer of CDH | Phylum | Volumetric activity (U L−1) | Activity assayc | Reference |
|---|---|---|---|---|
| Phanerochaete chrysosporium | B | 66 (600a) | Cellobiose, cyt c, pH 4.5 | [139, 140] |
| Pycnoporus cinnabarinus | B | 355 | Cellobiose, DCIP, pH 4.5 | [141] |
| Sclerotium rolfsii | B | 7400 (15000b) | Lactose, cyt c, pH 4.5 | [69, 142] |
| Trametes villosa | B | 580 | Lactose, cyt c, pH 3.5 | [142] |
| Trametes versicolor | B | 2030 | Cellobiose, cyt c, pH 3.5 | [143] |
| Ceriporiopsis subvermispora | B | 170 | Lactose, DCIP, pH 6.0 | [70] |
| Thielavia heterothallica | A | 47 | Cellobiose, cyt c, pH 4.5 | [144, 145] |
| Corynascus thermophilus | A | 4000 | Lactose, DCIP, pH 5.0 | [42] |
| Neurospora crassa | A | 100 | Lactose, DCIP, pH 5.5 | [42] |
| Chaetomium sp. INBI 2-26(−) | A | 190 | Cellobiose, DCIP, pH 6.5 | [146] |
A ascomycete, B basidiomycete, DCIP 2,6-dichloroindophenol
aWhen supplemented with bovine calf serum
bWhen using increased concentrations of peptone or certain amino acids
cActivities can vary when using different assays based on other carbohydrate substrates (electron donors) and electron acceptors. The pH used in the assay is identical or close to the pH optimum
Structure and function
CDH is an extracellular fungal flavocytochrome (cellobiose:acceptor 1-oxidoreductase, EC 1.1.99.18) and is a monomeric protein consisting of two domains connected by a flexible linker of about 20 amino acids. The molecular mass ranges from 85 up to 101 kDa depending on the degree of glycosylation, which can account for up to 16 % of the molecular mass [19, 42, 69, 70]. The DHCDH is a member of the glucose-methanol-choline oxidoreductase (GMC) family [61, 71]. The available structure of P. chrysosporium DHCDH (PDB identifiers 1KDG and 1NNA [36]) is peanut shaped with dimensions of 72 × 57 × 45 Å. The average molecular mass of DHCDH is ∼60 kDa without glycosylation, which can contribute up to ∼10 % of the total mass. The isoelectric point of DHCDH is low and varies for different enzymes but is usually around 5 [60, 72, 73]. Oxidation of carbohydrates is catalysed by the non-covalently bound FAD cofactor in DHCDH. The CYTCDH structure available from P. chrysosporium (PDB identifiers 1D7B and 1D7C [35]) is formed by two ellipsoidal antiparallel β-sheets with dimensions of 47 × 36 × 47 Å. The average molecular mass is ∼22 kDa without glycosylation and the isoelectric point of the CYTCDH is very low, around 3. The haem cofactor is coordinated by Met 65 and His 163. This unusual ligation of haem b in CYTCDH causes a relatively low redox potential, which is about 100–160 mV vs. normal hydrogen electrode (NHE) at pH 7.0 [62, 74, 75]. The N-terminal CYTCDH is connected to DHCDH via a flexible linker, which keeps the two domains in close contact and allows IET between them. Hence CDH can transfer reducing equivalents from an electron donor (e.g. cellobiose) via its two redox centres to different types of electron acceptors. Reoxidation of CDH can occur either directly at the (reduced) DHCDH domain by transfer of reduction equivalents to a two-electron acceptor (e.g. quinones) or, alternatively, electrons can be sequentially shuttled from the reduced FAD to the haem b cofactor, followed by consecutive reduction of two one-electron acceptors (ferric iron complexes, cytochrome c) [60, 72, 73].
Recently a novel electron acceptor for CDH was identified. The PMOs are copper-dependent carbohydrate active enzymes (CAZy) of family GH61, which can receive reducing equivalents from CDH and subsequently cleave cellulose by an oxidative mechanism. This interaction could be the key for the elucidation of the biological function of CDH and might end decades of speculation (Fig. 1) [38–41].
Catalytic properties of CDH
Preferred substrates of all CDHs are the β-1,4-linked di- and oligosaccharide breakdown products of cellulose—cellobiose or cellodextrins. Lactose has a very similar structure and is, although certainly not a natural substrate, also readily converted by CDHs [60]. A slight difference is observed between class I CDHs, which strongly discriminate glucose turnover, whereas some class II CDHs can also oxidise other mono- and disaccharides although with lower catalytic efficiencies. This difference might be an adaption to different fungal habitats and substrates [42, 45]. Catalysis takes place in the active site of DHCDH, where two electrons and two protons are subtracted from the anomeric carbon atom of a substrate sugar residue in the reductive cycle, which results in a fully reduced FAD [76]. In the oxidative cycle the FADH2 in the DHCDH reduces electron acceptors such as 2,6-dichloroindophenol (DCIP), o- or p-benzoquinone and their derivatives, the 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) cation radical, triiodide, strongly complexed iron ions, or oxygen. Alternatively, electrons can be donated to the haem b. Some electron acceptors like weakly complexed iron ions, cyt c, PMO and electrode surfaces depend on the action of the CYTCDH. Cyt c is one of only a few one-electron acceptors which solely act with CYTCDH and therefore is a good tool to estimate the IET (Fig. 2). The pH plays an important role in this reaction cascade. In class I CDHs the catalytic reaction at the active site and IET work only under acidic pH conditions. In class II CDHs three different groups of CDH with different IET behaviours were distinguished according to their pH-dependent interaction with cyt c: acidic, intermediate or neutral/alkaline IET optima [42] (Fig. 3). The same IET behaviour was found for these CDHs on polarised graphite electrodes [45].
Fig. 2.

Electron transfer in CDH from the substrate to various terminal electron acceptors. One- and two-electron acceptors (EA) can be reduced directly by the FADH2 in the DHCDH. Alternatively, electrons can be transferred by IET to the haem b in the CYTCDH, which works as a relay for the reduction of macromolocular electron acceptors like PMO, cyt c or an electrode
Fig. 3.

Comparison of pH profiles of basidiomycetous class I and ascomycetous class II CDHs. In the left column acidic class I CDH from Ceriporiopsis subvermispora (CsCDH) is shown above CDH from Trametes villosa (TvCDH) and Phanerochaete chrysosporium (PcCDH). The right column shows class II CDHs from Myriococcum thermophilum (MtCDH), Hypoxylon haematostroma (HhCDH) and Corynascus thermophilus (CtCDH) with intermediate and neutral pH optima. DCIP (grey lines) indicates the activity of DHCDH, whereas cyt c (black lines) is used to determine the pH dependency of the IET. For further information, see [42, 43, 45]
Recombinant production of CDH
Although many lignocellulose-degrading fungi produce CDH in reasonable amounts their cultivation and subsequent protein purification are difficult and time consuming. Therefore, several cdh genes have been cloned and expressed in recombinant expression hosts. Li et al. [77] were the first to report recombinant expression of CDH by homologous overexpression of CDH in P. chrysosporium. The expression level was rather low (600 U L−1), cultivation time was long (9 days) and genetic manipulation was difficult and time consuming. Therefore, Yoshida et al. [78] established the methylotrophic yeast Pichia pastoris as a heterologous expression system for P. chrysosporium CDH. Owing to the high reported expression levels, easy genetic manipulation and the ability of Pichia to perform eukaryotic post-translational modifications, it was used as an expression host for several basidiomyceteous and ascomeceteous CDHs during the following years (Table 2). Additionally, two ascomeceteous CDHs from Thielavia terrestris and Humicola insolens were recombinantly expressed in a fungal system, viz. Aspergillus oryzae [79, 80]. Several attempts to express CDH in the bacterial expression system Escherichia coli failed because of the different post-translational modifications of the two domains. However, the expression of the DHCDH of P. chrysosporium was recently reported [81].
Table 2.
Recombinant production of CDH and DHCDH by recombinant expression hosts
| Fungal producer of CDH | Expression host | Volumetric activity (U L−1) | Activity assay | Reference |
|---|---|---|---|---|
| Phanerochaete chrysosporium | P. chrysospor. a | 600 | Cellobiose, cyt c, pH 4.5 | [77] |
| Phanerochaete chrysosporium | P. pastoris | 1800 | Cellobiose, cyt c, pH 4.5 | [78] |
| P. chrysosporium DHCDH | E. coli | 733b | Cellobiose, DCIP, pH 5.0 | [81] |
| Pycnoporus cinnabarinus | P. pastoris | 7800 | Cellobiose, DCIP, pH 5.0 | [147] |
| Trametes versicolor | P. pastoris | 5218 | Cellobiose, cyt c, pH 4.2 | [148] |
| Myriococcum thermophilum | P. pastoris | 2150 | Lactose, DCIP, pH 5.0 | [82] |
| Neurospora crassa (CDH IIA) | P. pastoris | 1700 | Lactose, DCIP, pH 5.0 | [62] |
| Neurospora crassa (CDH IIB) | P. pastoris | 410 | Lactose, DCIP, pH 5.0 | [62] |
| Corynascus thermophilus | P. pastoris | 376 | Lactose, DCIP, pH 5.5 | [75] |
| Thielavia terrestris | A. oryzae | NG | Cellobiose, DCIP, pH 6.0 | [79] |
| Humicola insolens | A. oryzae | NG | Cellobiose, DCIP, pH 7.0 | [80] |
NG data not given
aHomologous overexpression
bValue refers to cell-free extract after cell disruption and not to cultivation volume
The advantages of recombinant protein expression are a fast, reliable and efficient enzyme production and the possibility to generate genetically engineered enzymes. However, some drawbacks have to be considered. A sub-stoichiometric occupation of the catalytic sites with the cofactor FAD in C. thermophilus CDH results in a generally lower specific activity of CDHs expressed in P. pastoris [75]. The fact that Pichia and Aspergillus produce several glycoforms of the recombinant protein results in an inhomogeneous enzyme preparation in respect to molecular mass and degree of glycosylation. Depending on the cultivation conditions and employed media a varying amount of proteolytically cleaved DHCDH appears during purification [75, 82].
Direct electron transfer
The electron transfer pathways between the DHCDH and an electrode can occur in principle along three different routes as illustrated in Figs. 2 and 4. In the first reaction the sugar substrate, an aldose, is oxidised at the C1 position (only the β-anomer is a substrate for CDH) into its corresponding lactone and concurrently the FAD in the active site of the DHCDH is fully reduced to FADH2-DHCDH, reaction (1):
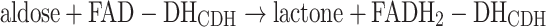 |
1 |
The reoxidation of FADH2-DHCDH can be accomplished by a 2e−, 2H+ acceptor such as quinone (Q) or an equivalent aromatic redox compound according to reaction (2):
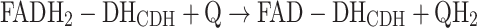 |
2 |
The reduced quinone, QH2, will be reoxidised at the electrode if the applied potential (E
app) is set higher than the formal potential of the Q/QH2 redox couple, 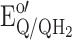 , reaction (3):
, reaction (3):
| 3 |
Alternatively, a 1e−, non-H+ acceptor, e.g. an Os3+ complex (Os3+), accepts the electrons sequentially from the FADH2-DHCDH, whereby Os2+ complex (Os2+) and the enzyme-stabilised semiquinone of the bound FAD, FADH•-DHCDH, are formed in reaction (4):
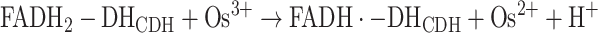 |
4 |
This reaction is then followed by the second electron transfer to a second Os3+, whereby the fully oxidised DHCDH is regained, reaction (5):
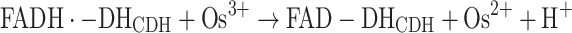 |
5 |
Fig. 4.

Electron transfer between both CDH domains and the terminal electron acceptor. DET depends on CYTCDH as an electron shuttle, whereas MET (blue spheres indicate soluble mediators or polymeric redox centres) transfers electrons directly from DHCDH to the electrode surface
The two Os2+ formed will be reoxidised at the electrode if E
app is set higher than the formal potential of the Os3+/2+ redox couple, 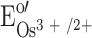 , reaction (6):
, reaction (6):
| 6 |
The electrons can also be transferred from FADH2-DHCDH to the Fe3+−CYTCDH sequentially in an IET process according to reaction (7):
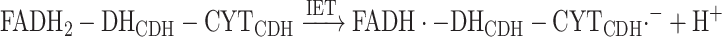 |
7 |
This first electron transfer step is followed by reoxidation of the reduced CYTCDH, Fe2+−CYTCDH, by an e− acceptor such as Os3+ or cytochrome c (or by the electrode, see below); however, the second electron from the DHCDH will then be subsequently transferred to the CYTCDH, reaction (8):
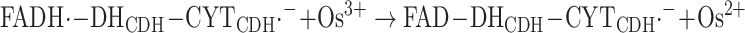 |
8 |
Finally the last electron will be transferred from the CYTCDH to a second 1e− acceptor molecule, reaction (9):
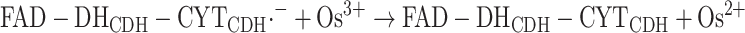 |
9 |
When CDH is immobilised on the electrode surface and in the absence of any competing e− acceptors, the reoxidation of the reduced enzyme can be summarised as follows, reaction (10):
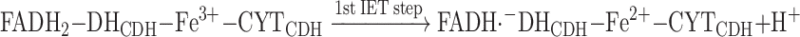 |
10 |
This step is followed by a first electron transfer (ET) step to the electrode, which is immediately followed by a second IET step delivering the second electron from the DHCDH to the CYTCDH, reaction (11):
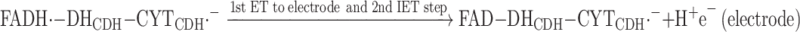 |
11 |
Finally the second electron is then delivered to the electrode, reaction (12):
 |
12 |
It is basically the DET properties of CDH that form the basis for the bioelectrochemical interest in this redox enzyme, ever since it was first documented [83]. The rather strict selectivity of the originally investigated class I basidiomycete CDHs for cellodextrins and lactose [60, 84] mainly found application in lactose biosensors for the dairy industry [55, 58, 85] or cellobiose biosensors that can be used to follow cellulose hydrolysis caused by cellulose-hydrolysing enzymes [54, 86]. However, when it was realised that the recently discovered ascomycete CDHs (class IIA and IIB) could also have high turnover rates for glucose (and a series of other mono- and oligosaccharides) and especially at neutral pH values (in contrast to class I CDHs that work best under slightly acidic conditions) an increased interest in CDH for application in biosensors and biofuel cells appeared [30, 42, 44, 49–51, 56, 57, 87–92].
Optimisation of direct electron transfer
Two approaches are used to optimise DET currents from CDH to electrodes. The first approach is based on new electrode materials, nanostructures and chemically modified electrode surfaces to either enhance the effective surface area available for CDH binding, or to increase the DET rate by suitable orientation of the enzyme on the surface. The second, biochemical approach uses modifications of the enzyme or the reaction cascade to increase the current of CDH-based electrodes. The following sections investigate the published work in the area of direct CDH/electrode interaction.
Novel nanostructures
The desire to increase the current density of bioelectrodes based on DET has led to the construction and use of nanostructured architectures so that a higher loading of the redox enzyme and a higher probability of correct orientation for DET can be obtained. Both the use of nanostructures based on the drop-casting deposition technique of nanomaterials such as single-walled carbon nanotubes (SWCNTs) [47, 89–91] (Ortiz et al. submitted) [93] or gold nanoparticles (AuNPs) [48, 49, 51] onto the electrode surface of Au, glassy carbon or graphite electrodes, as well as prefabricated carbon nanotube (CNT)-modified screen-printed electrodes [55, 57, 58] has been used and in all instances the use of nanomaterials resulted in an improved current density. An overview of the nanostructured materials used is presented in Table 3. For all the nanomaterials and immobilisation techniques tested only bioelectrocatalysis from CYTCDH was observed.
Table 3.
Nano- and surface modifications of CDH-based electrodes
| Electrode nanomodification | Modification method | ET | Surface modification | Surface functionality | Measurement method | pH and substrate concentration | Current density (μA cm−2) | Reference |
|---|---|---|---|---|---|---|---|---|
| GC + SWCNTs; PsCDH | Drop-casting | DET | p-Phenylenediamine | NH2 | CV; 1 mV s−1 | 3.5, 5 mM lactose | 500 | [47] |
| GC + SWCNTs; PsCDH | Drop-casting | DET | p-Aminobenzoic acid | COOH | CV; 1 mV s−1 | 4.5, 5 mM lactose | 150 | [47] |
| Au + AuNP; PcCDH | Drop-casting | DET | ATP/MP + GA | OH | CV; 2 mV s−1 | 4.5, 10 mM lactose | 4.0 | [48] |
| Au + AuNP; PcCDH | Drop-casting | DET | ATP/MBA + GA | COOH | CV; 2 mV s−1 | 4.5, 10 mM lactose | 29.3 | [48] |
| Au + AuNP; PcCDH | Drop-casting | DET | MUNH2/MUOH + GA | OH | CV; 2 mV s−1 | 4.5, 10 mM lactose | 11.6 | [48] |
| Au + AuNP; PcCDH | Drop-casting | DET | MUNH2/MUA + GA | COOH | CV; 2 mV s−1 | 4.5, 10 mM lactose | 15.2 | [48] |
| SPCE + MWCNT; TvCDH | Drop-casting | DET | COOH-functionalised MWCNT | COOH | FIA | 4.5, 0.1 mM lactose | 0.08 | [55] |
| SPCE + MWCNT; PsCDH | Drop-casting | DET | COOH-functionalised MWCNT | COOH | FIA | 4.5, 10 mM lactose | 0.5 | [55] |
| SPCE + MWCNT; PsCDH | Drop-casting | DET | COOH-functionalised MWCNT and GA or PEDGDE | COOH | FIA | 4.5, 10 mM lactose | 5.6 | [55] |
| GE + CNP; CsCDH | Drop-casting | DET | None | Not studied | CV; 2 mV s−1 | 4.5, 50 mM lactose | 297 deglycosylated enzyme ; 240 glycosylated enzyme | [94] |
| Au + SiNP + cyt c; TvCDH | LbL (4 layers) | DET | COOH-functionalised SiNP | 40 nm particles | CV; 5 mV s−1 | 3.5, 5 mM lactose | 4 nAa deglycosylated enzyme | [52] |
| Au + SiNP + cyt c TvCDH | LbL (4 layers) | DET | COOH-functionalised SiNP | 20 nm particles | CV; 5 mV s−1 | 4.5, 5 mM lactose | 28 nAa deglycosylated enzyme; 6 nAa glycosylated enzyme | [52] |
| Au + SiNP + cyt c; TvCDH | LbL (4 layers) | DET | COOH-functionalised SiNP | 15 nm particles | CV; 5 mV s−1 | 5.5, 5 mM lactose | 9 nAa deglycosylated enzyme | [52] |
| Au + SiNP + cyt c; TvCDH | LbL (4 layers) | DET | COOH-functionalised SiNP | 5 nm particles | CV; 5 mV s−1 | 6.5, 5 mM lactose | 8 nAa deglycosylated enzyme | [52] |
| GE + SWCNT PsCDH | Drop-casting | DET | PEGDGE | Not studied | FIA | 5.3, 10 mM lactose | 19.15 | [90] |
| GE + SWCNT; PsCDH | Drop-casting | DET | PEGDGE | Not studied | CV; 1 mV s−1 | 4.5, 5 mM lactose | 90 | [90] |
| GE + SWCNT; PcCDH | Drop-casting | DET | PEGDGE | Not studied | FIA | 5.3, 10 mM lactose | 6.84 | [90] |
| GE + SWCNT; TvCDH | Drop-casting | DET | PEGDGE | Not studied | FIA | 5.3, 10 mM lactose | 4.78 | [90] |
| GE + SWCNT; SrCDH | Drop-casting | DET | PEGDGE | Not studied | FIA | 5.3, 10 mM lactose | 1.36 | [90] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | DET | Co-immobilisation with CNTs | Not studied | CV; 1 mV s−1 | 3.5, 100 mM lactose | 68 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | DET | Co-immobilisation with CNTs | Not studied | LSV; 1 mV s−1 | 4.0, 100 mM lactose | 61.8 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | DET | Co-immobilisation with CNTs | Not studied | LSV; 1 mV s−1 | 4.5, 100 mM lactose | 58 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | DET | Co-immobilisation with CNTs | Not studied | LSV; 1 mV s−1 | 5.0, 100 mM lactose | 45 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | DET | Co-immobilisation with CNTs | Not studied | LSV; 1 mV s−1 | 5.5, 100 mM lactose | 30 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | DET | Co-immobilisation with CNTs | Not studied | LSV; 1 mV s−1 | 6.0, 100 mM lactose | 13 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | MET | Co-immobilisation with CNTs + PEGDGE | Not studied | LSV; 0.2 mV s−1 | 3.5, 100 mM lactose | 300 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | MET | Co-immobilisation with CNTs + PEGDGE | Not studied | LSV; 0.2 mV s−1 | 4.0, 100 mM lactose | 500 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | MET | Co-immobilisation with CNTs + PEGDGE | Not studied | LSV; 0.2 mV s−1 | 4.5, 100 mM lactose | 650 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | MET | Co-immobilisation with CNTs + PEGDGE | Not studied | LSV; 0.2 mV s−1 | 5.0, 100 mM lactose | 700 | [93] |
| GE + SWCNT + CDH; PsCDH | Drop-casting | MET | Co-immobilisation with CNTs + PEGDGE | Not studied | LSV; 0.2 mV s−1 | 6.0, 100 mM lactose | 700 | [93] |
| Au + AuNP; CtCDH | Drop-casting | DET | ATP/MBA + GA | OH | LSV; 2 mV s−1 | 7.4, 5 mM lactose; 100 mM glucose | 40; 26 | [49] |
| Au + AuNP; CtCDH | Drop-casting | DET | ATP/MBA + GA | OH | LSV 2 mV s−1 | 7.4, 5 mM glucose | 7.5 | [51] |
| SPCE + SWCNT; CtCDH | Drop-casting | DET | PEDGDE | Not studied | FIA | 7.4, 300 mM glucose | 18.41 | [57] |
| SPCE + MWCNT; CtCDH | Drop-casting | DET | PEDGDE | Not studied | FIA | 7.4, 300 mM glucose | 15.58 | [57] |
| GE + SWCNT; MtCDH | Drop-casting | DET | PEGDGE | Not studied | FIA | 5.3, 10 mM lactose | 6.16 | [89] |
| GE + SWCNT; MtCDH | Drop-casting | DET | PEGDGE | Not studied | LSV; 1 mV s−1 | 4.5, 100 mM lactose | 5 | [89] |
| GE + SWCNT; MtCDH | Drop-casting | MET | Os-polymer + PEGDGE | Not studied | LSV; 1 mV s−1 | 3.5, 100 mM lactose | 68.4 | [89] |
| GE + SWCNT; MtCDH | Drop-casting | MET | Os-polymer + PEGDGE | Not studied | LSV; 1 mV s−1 | 4.5, 100 mM lactose | 102.6 | [89] |
| GE + SWCNT; MtCDH | Drop-casting | MET | Os-polymer + PEGDGE | Not studied | LSV; 1 mV s−1 | 6.0, 100 mM lactose | 205 | [89] |
| GE + SWCNT | Drop-casting | MET | Os-polymer + PEGDGE | Not studied | LSV; 1 mV s−1 | 7.0, 100 mM lactose | 465 | [89] |
| GE + SWCNT; MtCDH | Drop-casting | MET | Os-polymer + PEGDGE | Not studied | CV; 1 mV s−1 | 8.0, 100 mM lactose | 800 | [89] |
| GE + SWCNT; MtCDH | Drop-casting | MET | Os-polymer + PEGDGE | Not studied | CV; 1 mV s−1 | 7.4, 100 mM lactose; 50 mM glucose | 450; 100 | [89] |
| GC + SWCNTs; CtCDH | Drop-casting | DET | p-Aminobenzoic acid | COOH | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 30; 15 | (Ortiz et al. submitted) |
| GC + SWCNTs; CtCDH | Drop-casting | DET | Aniline | None | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 21; 9 | (Ortiz et al. submitted) |
| GC + SWCNTs; CtCDH | Drop-casting | DET | p-Phenylenediamine | NH2 | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 25; 13 | (Ortiz et al. submitted) |
| GC + SWCNTs; CtCDH | Drop-casting | DET | p-Phenylenediamine + GA | NH2 | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 43; 21 | (Ortiz et al. submitted) |
| GC + SWCNTs; CtCDH | Drop-casting | DET | N,N-Diethyl-p-phenylenediamine | N-(N,N)-Diethyl | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 16; 8 | (Ortiz et al. submitted) |
| GC + SWCNTs; CtCDH | Drop-casting | DET | p-Aminophenol | OH | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 23; 14 | (Ortiz et al. submitted) |
| GC + SWCNTs; CtCDH | Drop-casting | DET | Not used | Not studied | CV; 1 mV s−1 | 7.4, 10 mM lactose; 50 mM glucose | 8; 4 | (Ortiz et al. submitted) |
GC glassy carbon, GE graphite electrode, SPCE screen-printed carbon electrodes, SWCNT single-walled carbon nanotubes, MWCNT multi-walled carbon nanotubes, AuNP gold nanoparticles, SiNP silica nanoparticles, ET electron transfer, NA not applicable, LbL layer by layer, PEGDGE polyethyleneglycol diglycidyl ether, GA glutaraldehyde, CV cyclic voltammetry, MUOH 11-mercapto-1-undecanol, MUA 11-mercapto-1-undecanoic acid, MUNH 2 mercapto-1-undecamine, ATP 4-aminothiophenol, MP 4-mercaptophenol, MBA 4-mercaptobenzoic acid
aArea of the electrode was not given
FIA results obtained through flow injection analysis with the CDH modified electrode mounted in a flow through amperometric cell and the maximum current response was followed after injection of a substrate containing sample volume
LSV results obtained with the CDH modified electrode investigated with linear sweep voltammetry taken the response value as the voltammetric peak current
Carbon-based nanomaterials
Initial investigations with various CDHs on SWCNT- or MWCNT-modified electrodes were based on direct adsorption of CDH on the nanomodified surface [55, 57, 89, 90, 93]. These studies used oxidatively shortened SWCNTs, kept as a suspension in water, without previous modification or commercially available screen-printed electrodes with MWCNT or SWCNT. No previous or further functionalisation of the nanomaterial was done and only in some of the studies was a cross-linker, i.e. polyethyleneglycol diglycidyl ether (PEGDGE) or glutaraldehyde (GA), was used in a following step to stabilise the enzyme–nanoparticle layer on the electrode surface. For screen-printed electrodes the use of a cross-linker increased the electrocatalytical currents in the presence of substrate by one order of magnitude for T. villosa and Phanerochaete sordida CDH-modified MWCNT screen-printed electrodes possibly owing to dissolution of certain components in the paste, thereby exposing a higher surface area for electric communication with the immobilised CDH [55]. In the scope of this type of non-further functionalisation of the carbon nanomaterial, Ortiz et al. [94] increased the specific surface area by drop-casting carbon nanoparticles (CNPs) with a diameter of 27 nm onto graphite electrodes, which increased the electrocatalytical current by 350 times for deglycosylated Ceriporiopsis subvermispora CDH (297 μA cm−2) and more than 500 times for glycosylated C. subvermispora CDH (240 μA cm−2), see Table 3.
Gold nanoparticles and nanomaterials
Drop-casting of citrate-stabilised AuNPs with a diameter of 19 nm onto the surface of Au disk electrodes [95] was used in combination with mixed self-assembled thiol monolayers (SAMs), onto which P. chrysosporium CDH [48] or C. thermophilus CDH [49, 51] was covalently attached. The mixed SAM featured different thiols with amino, carboxy or hydroxyl head groups. The bifunctional cross-linker GA was used to form covalent bonds between the amino-terminated thiols and lysine residues on the enzyme. Cyclic voltammetry (CV) showed clear redox waves in the absence of substrate for all mixed SAMs tested and the heterogeneous electron transfer constant, k s, could be calculated. An enhancement of the electrocatalytical activity of P. chrysosporium CDH of up to 75 times was achieved by this technique [48]. The procedure has been used further to prepare glucose/O2 biofuel cell anodes using C. thermophilus CDH as the bioelement, in combination with cathodes based on adsorbed bilirubin oxidase (BOx) on AuNP-modified Au disks and nanowires [49, 51] (Table 3).
Layer-by-layer nanostructures
A different approach to achieve higher currents was taken by Lisdat et al., who, on the basis of previous work combining cyt c modified electrodes with CDH [96] and other redox enzymes [97–100], constructed a supramolecular structure using a layer-by-layer (LbL) immobilisation technique combining alternate layers of silica nanoparticles (SiNPs) and a mixture of TvCDH and cyt c (Fig. 5) [52]. Various numbers of layers, i.e. 1, 2, 3 and 4, were investigated using both native glycosylated and also deglycosylated TvCDH; the deglycosylated CDH yielded higher currents. The optimal SiNP particle size was 20 nm [52]. An LbL assembly technique was used. First a mixed SAM of MUA/MUOH was used to coat the Au surface and then SiNPs were adsorbed on the partially negatively charged surface, and finally a mixture containing TvCDH and cyt c was added and the nanostructure was built up to four layers [52]. The same type of alkanethiol mixture was used to adsorb cyt c on a SAM of MUOH/MUA on gold wires. The bioelectrochemistry in the presence and the absence of substrate was studied for solutions of TvCDH and CtCDH using this cyt c electrode; significantly, a shift of the optimal pH for electrocatalysis of cellobiose was observed using this approach [53].
Fig. 5.

Schematic representation of a supramolecular [SiNP/CDH · cyt c] architecture prepared on a cyt c monolayer electrode (M). The cyt c monolayer is assembled on a mixed thiol layer (MUOH/MUA). The layer by layer structure is [SiNPs/CDH · cyt c]n (n = 1, 2, 3, 4), where n indicates the number of layers of SiNPs/CDH · cyt c. Reproduced from [52] with permission from The American Chemical Society
Derivatisation of electrode surfaces
In the last few years functionalised SWCNTs have been tested in combination with CDH. It is established that the charge of the surface where CDH is immobilised has a strong effect on the electrochemistry of CDH. Investigations on different SAMs showed a strong effect of the functional group of the used thiol on the efficiency of the electrochemistry of CDH [44, 101–104]. Using the aryl amines p-phenylenediamine (NH2-PD) or p-aminobenzoic acid (COOH-PD) for diazonium activation of the electrode surface, Tasca et al. [47] introduced amino or carboxylic groups, respectively, onto the SWCNTs previously drop-cast on glassy carbon electrodes. In the final step Phanerochaete sordida CDH, belonging to the class I CDHs, was adsorbed onto the modified electrode, where the positively charged –NH2 functionalised surface or the negatively charged –COOH functionalised surface at the actual pH was used during CV measurements. For the –NH2 functionalised surface a current density of 500 μA cm−2 was obtained in the presence of 5 mM lactose, which is the highest current density observed for any CDH-modified electrode based on DET (Fig. 6). For the –COOH functionalised SWCNTs a current density of ∼150 μA cm−2 was obtained. The increased current density of the –NH2 modified surface was attributed to a better orientation of the mainly negatively charged P. sordida CDH (isoelectric point = 5.7) on the electrode. A similar modification procedure using a larger range of diazonium salts on SWCNTs drop-cast on GC was performed by (Ortiz et al. submitted), but in this case using the class II C. thermophilus CDH, which gave current densities of up to 25 μA cm−2 in the presence of 50 mM glucose.
Fig. 6.

a CV of a P. sordida CDH COOH-PD/SWCNT-GC electrode in the presence of 5 mM lactose (red dashed line) and in the absence of substrate (black line) at pH 4.5. b CV of a P. sordida CDH NH2-PD/SWCNTs-GC electrode in the presence of 5 mM lactose (red dashed line) and in the absence of substrate (black line) at pH 3.5. Scan rate 1 mV s−1. Reproduced from [47] with permission from The American Chemical Society
Thiol-modified gold electrodes
Reports on facile electrochemistry of CDH trapped between a gold electrode modified with a thiol-based SAM and covered with a permselective membrane have appeared since the beginning of the 1990s [87, 101–104]. When such electrodes were investigated by CV they revealed clear non-turnover DET between the haem b cofactor of the CYTCDH in the absence of substrate, whereas in the presence of substrate a clear catalytic current was observed. The interaction between CDH and the thiol-based SAM-modified electrode results in facile electrochemistry which is governed by a combination of different effects: (i) hydrophilic/hydrophobic interactions between the SAM and the enzyme that control the orientation on the electrode surface, e.g. interactions between the CDH cellulose binding domain with –OH terminated alkanethiols, as the cellulose binding domain has a strong affinity for naturally occurring hydroxyl groups present on cellulose [60], but also electrostatic interactions between oppositely charged SAMs and the surface of the enzyme; (ii) a dependency of the rate of ET on the distance between the electrode and the enzyme, described by the Marcus theory [105, 106]; and (iii) the compactness of the SAM, e.g. a densely packed SAM will slow down ET with the electrode.
Recently a series of investigations were performed on the interaction between various CDHs on gold and AuNP-modified solid gold electrodes [44, 48, 49, 51, 94]. In one study the effect of mutations close to the active site of study two different class II CDHs was investigated (Ortiz et al. submitted). In another study two class I CDHs were investigated [94], the focus being the effect of glycosylation/deglycosylation on the efficiency of DET. An exhaustive electrochemical investigation was performed on a number of class II CDHs (Neurospora crassa, C. thermophilus, Humicola insolens) and variants for improved glucose oxidation of C. thermophilus and H. insolens CDH using different aliphatic and aromatic thiols with varying head group functionalities (−NH2, –COOH and –OH) and varying spacer lengths (2, 6 and 11 carbons) or a phenyl group [44] (Ortiz et al. submitted). For all CDHs tested electrodes with SAMs based on –OH functionalised thiols exhibited high electrocatalytical currents. In the case of N. crassa CDH the electrocatalytical currents became higher and the separation between the anodic and cathodic peak potentials (ΔE p) of the CYTCDH decreased when the –OH functionalised alkanethiol increased in length (Fig. 7) [44]. For C. thermophilus CDH the effect was the opposite and even more pronounced (Ortiz et al. submitted). H. insolens CDH showed in general small and poorly defined catalytical currents, but quasi-reversible CVs. The highest electrocatalytical responses for H. insolens, N. crassa and C. thermophilus CDH were found for mercaptohexanol, mercaptobenzoic acid and mercaptoethanol-based SAMs, respectively. N. crassa CDH also showed catalytic currents and a small ΔE p for the thiols containing a phenyl group as spacer. On the other hand C. thermophilus CDH showed poor electrochemistry with such thiols. In another recent paper the electrochemistry of P. chrysosporium CDH was compared using four different thiols with –OH and –COOH head groups and different spacers [48]. In accordance with previous reports, the highest electrocatalytical currents for this class I CDH was found for mercaptoundecanol (MUOH). Interestingly, a lower current density was found for the equivalent thiol having a –COOH functionality (mercaptundecanoic acid, MUA). The reason could be electrostatic repulsion between the negatively charged CDH and the –COO− group. Harreither et al. [45] studied the E°′ and the electrocatalytical activity of C. thermophilus CDH using thioglycerol at different pHs. The formal potential found, 100 mV vs. NHE, is the lowest reported so far for all investigated class II CDHs [45]. It can be concluded that the choice of the thiol is critical. Clear effects of the spacer length and the functional head group are observed.
Fig. 7.

Voltammetric responses of Au-mixed monolayer based SAM-Neurospora crassa CDH modified electrodes in absence (dashed line) and in presence (solid line) of 5 mM lactose. Experimental conditions: scan rate, 10 mV/s; supporting electrolyte, 50 mM citrate buffer pH 5.5. Reproduced from [44] with permission from Revue Roumaine de Chimie (Roumanian Journal of Chemistry)
When native, glycosylated, and deglycosylated CDH from P. chrysosporium and C. subvermispora, denoted PcCDH, dgPcCDH and CsCDH, dgCsCDH, respectively, were tested on an MUOH-based SAM, a higher current density was found for dgCsCDH than for CsCDH, but for dgPcCDH and PcCDH a similar current density was found [94]. The likely reason is that native CsCDH is much more glycosylated than PcCDH so that the effect of deglycosylation is more pronounced for CsCDH.
Mixed thiol SAMs
Since the first bioelectrochemical studies on CDH in the late 1990s most studies have focused on the electrocatalytical activity. CV waves are not really observable on graphite electrodes modified with CDH owing to their high capacitive current. On the other hand, non-covalent interactions between CDH from different sources and gold electrodes or SAM-modified gold electrodes, and where the enzyme was retained behind a permselective membrane, has not shown stable electrochemical CV signals [102–104]. Efforts during recent years have focused on covalently immobilising CDH on alkanethiol-modified Au electrodes to avoid the usage of the permselective membrane [74, 102–104]. Matsumura et al. [48] compared immobilised CDH on four different SAMs based on two mixed thiols, where one of the head groups was –NH2 and the other –COOH or –OH. GA was used to cross-link P. chrysosporium CDH to the amino group of the SAM. Matsumura et al. [48] used this approach to compare 11 different alkyl carbon chain based thiols as well as one incorporating a phenyl group as spacer and the head groups –OH with –COOH [48]. The ratio of the alkanethiol mixture (1:50 v/v) and GA (0.25 %) was optimised. The optimal ratio of the mixture for deglycosylated P. chrysosporium CDH, which has a smaller radius than its glycosylated form, was 1:40. This suggests that the surrounding glycosyl residues and the radius of the CDH have an effect [94]. This immobilisation protocol was also used in a 3D mesoporous structure created by drop-casting AuNPs on the surface of Au disk electrodes based on previous work by Murata et al. [95]. The k s values for AuNP/SAM/GA/PcCDH were lower for the mixture of thiols having a phenyl spacer than those based on the 11-carbon alkane spacer. The rate-limiting step of the overall ET was the DET between the CYTCDH and the electrode instead of the IET from DHCDH to CYTCDH as measured by stoped-flow experiments. An overview of the current densities and k s values is given in Table 4.
Table 4.
Performance of P. chrysosporium CDH on mixed SAMs
| Mixed SAMs | Polycrystalline gold | AuNPs | SPR | |||
|---|---|---|---|---|---|---|
| J (μA cm−2) | J (μA cm−2) | E°′ (mV) | ΔE (mV) | k s (s−1) | Γ (pmol mm−2) | |
| 4-ATP/4-MP | 0.26 | 4.0 | 161.7 | 14.7 | 59.8 | 5.79 |
| 4-ATP/4-MBA | 0.40 | 29.3 | 161.5 | 14.6 | 52.1 | 5.71 |
| MUNH2/MUOH | 0.34 | 11.6 | 161.8 | 14.7 | 154.0 | 5.67 |
| MUNH2/MUA | 0.49 | 15.2 | 161.3 | 14.6 | 112.0 | 5.65 |
Current densities, J, surface coverage, Γ, k s and E°′ for covalently attached CDH onto mixed SAMs. J for polycrystalline gold and AuNPs were obtained at 300 mV vs. NHE. Formal potentials, E°′, and peak separations, ΔE, at 0.2 V s−1
ATP 4-aminothiophenol, MP 4-mercaptophenol, MBA 4-mercaptobenzoic acid, MUNH 2 mercapto-1-undecamine, MUOH 11-mercapto-1-undecanol, MUA 11-mercapto-1-undecanoic acid, SPR surface plasmon resonance
The same immobilisation procedure was applied for immobilising C. thermophilus CDH on AuNP-modified Au disk and Au wire electrodes [49, 51]. The optimum GA amount (1 %) was higher than when using planar disk electrodes (0.25 %) [48], perhaps caused by the nanoparticle curvature but also because of using another CDH. A one-compartment mediator-less glucose/O2 biofuel cell (BFC) working under physiological conditions was established using such a bioanode based on C. thermophilus CDH in combination with a biocathode based on BOx [49]. The BFC had an open-circuit voltage of 0.68 V and a maximum power density of 15 μW cm−2 at a cell voltage of 0.52 V in phosphate buffer and an open-circuit voltage of 0.65 V and a maximum power density of 3 μW cm−2 at a cell voltage of 0.45 V in human blood. The estimated half-lives of the biodevices were greater than 12, less than 8 and less than 2 h in a sugar-containing buffer, human plasma and blood, respectively. The basic characteristics of mediator-less sugar/oxygen BFCs were significantly improved compared with previously designed biodevices [50, 87], because of the usage of 3D AuNP-modified electrodes.
Influence of cations on the activity of CDH
A screening of the influence of various metal cations on the activity of CDH was done in 1999 for the basidiomycete Schizophyllum commune in solution [107]. The highest increase in cyt c activity by 8 % was caused by the presence of 2 mM Cu2+ and the highest decrease by 70 % was caused by the presence of 2 mM Ag+, thereby showing the ability of cations to modulate the activity of CDH. A further study with immobilised P. chrysosporium CDH on a spectrographic graphite electrode showed a 50 % increase in the catalytic current for lactose and cellobiose following the addition of 80 mM NaCl to the buffer [108]. The molecular interactions behind this observation are unknown, but it was excluded that an increased CDH/electrode interaction was the reason.
Recently the influence of cations on the activity of two class II CDHs from Myriococcum thermophilum and Humicola insolens and one class I CDH from P. chrysosporium was investigated in more detail in solution and when immobilised on spectrographic graphite electrodes [109]. When immobilised on the graphite electrode the DET current for M. thermophilum CDH was most tunable by addition of CaCl2 in the millimolar range and exhibited up to a fivefold increase in the catalytic currents (Fig. 8a). The DET current of H. insolens CDH-modified electrodes was enhanced fourfold, but for P. chrysosporium CDH only 2.4-fold. The exchange of CaCl2 by MgCl2 tuned the DET current in a similar manner. However, KCl had a much lower effect and enhanced DET currents by a maximum of twofold for M. thermophilum CDH. Activity assays based on cyt c were used to investigate the modulation of the IET of M. thermophilum and H. insolens CDH in solution. Similar dependencies of the cyt c activities on CaCl2 were observed as for the immobilised CDHs. The separately expressed DHCDH from H. insolens showed no cyt c activity and no dependency on any added cations. Similarly, the DCIP activity of H. insolens DHCDH was completely independent of added CaCl2 or KCl. It can be concluded that divalent cations increase either the interaction between the DHCDH and CYTCDH domains or between the CYTCDH domain and the final electron acceptor, which can be cyt c or an electrode.
Fig. 8.

Variation of response current (I) with the applied potential (E) of spectrographic graphite electrodes modified with Myriococcum thermophilum CDH (a, c) and optionally pre-modified with polyethylenimine (PEI) (b, d) in 50 mM sodium acetate buffer pH 5.5 (a, b) or 50 mM TRIS pH 8.0 (c, d) in the absence (white squares) or in the presence of 50 mM CaCl2 (black squares) with 5 mM lactose as substrate. The inset in c shows a magnification of the response curve
Polyethylenimine as a promoter layer
In analogy to the enhancing effect of CaCl2, a current-increasing effect of the branched polycation polyethylenimine (PEI) as a promoter layer was anticipated for the construction of CDH-modified biosensors. Beneficial effects have been shown in several publications covering, for example, cyt c on carbon electrodes [110], the detection of NADH with phenoxazine derivative modified carbon paste electrodes [111, 112] and some other redox enzyme modified carbon paste electrodes [113–117] or improved electrochemistry of human sulfite oxidase on PEI-modified gold nanoparticles [118]. In recent work the influence of PEI as a promoter layer on spectrographic graphite electrodes modified with M. thermophilum CDH was investigated. The pre-modification of a graphite electrode with PEI increased the maximal catalytic current from the oxidation of 3.75 mM lactose by around 13 times at pH 5.5 (pH optimum) and by around 87 times at pH 8.0 (Fig. 8). The modification with PEI also shifted the pH optimum from 5.5 to 8.0, which is interesting for biosensor applications measuring at human physiological pH (7.4). A further addition of 50 mM CaCl2 still shows enhancing effects, but is less pronounced in the presence of PEI. This indicates that PEI acts similarly to CaCl2. Another explanation would be an increased surface loading of M. thermophilum CDH on the PEI-modified electrode owing to strong electrostatic binding of CDH to PEI [19].
Besides the enhancing effect of PEI on the catalytic currents, the CVs unexpectedly revealed two catalytic redox waves. One starting at −100 mV vs. Ag|AgCl (0.1 M KCl) representing the expected ET from the CYTCDH domain to the electrode [74] and a second catalytic wave, most pronounced at pH 8.0 and starting at around 100 mV, which is visible even in the absence of PEI (Fig. 8c, inset). The origin of the second catalytic wave is unknown, but has been observed before for P. chrysosporium CDH [108] and is currently under investigation. However, the enhancing effect of PEI is also clearly present at potentials below the second catalytic wave. Until now the beneficial effect of PEI was only observed for class II M. thermophilum CDH. The catalytic responses of class II N. crassa CDH and class I P. chrysosporium CDH could not be improved by the pre-modification of graphite electrodes with PEI. This is probably because the latter CDHs already have a more efficient DET and/or IET compared to that of M. thermophilum CDH [43, 88, 119].
Deglycosylation
Deglycosylation of enzymes has been used to facilitate the electron transfer and even to achieve DET from deeply buried prosthetic groups of redox enzymes, even from deeply buried FAD in e.g. Aspergillus niger glucose oxidase (GOx) [120–123]. CDH is a glycoprotein that shows a glycosylation between 8 and 16 % [19] depending on the organism used for expression. The carbohydrate chains are believed to stabilise the tertiary protein structure and increase the solubility of the protein molecule. A voluminous carbohydrate structure on the enzyme surface will act as an insulator for enzymes attached to electrodes [120]. Opposed to what was found for GOx and pyranose dehydrogenase, deglycosylation of CDH did not result in any DET between the FAD in the DHCDH domain and an electrode. However, the catalytic current densities increased two- to threefold in the presence of lactose, for deglycosylated P. chrysosporium (9 % glycosylated) and C. subvermispora CDH (16 % glycosylated) compared with their glycosylated counterparts when adsorbed on spectrographic graphite electrodes (Fig. 9) [94]. The apparent Michaelis–Menten constant 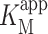 for lactose was also found to decrease by two- to threefold for the deglycosylated CDHs compared to the glycosylated ones. When the enzymes were compared trapped under a permselective membrane and 11-mercaptoundecanol (MUOH) SAM-modified Au disk electrodes, glycosylated and deglycosylated P. chrysosporium CDH showed the same catalytic activity, whereas glycosylated and deglycosylated C. subvermispora CDH showed similar results to that obtained on spectrographic graphite electrodes, i.e. a much higher catalytic current was found when using the deglycosylated form. The E°′ for the deglycosylated CDH was found to shift up by ∼8 mV. In a different study glycosylated and deglycosylated T. villosa CDH were compared when immobilised on an LbL supramolecular architecture stabilised by electrostatic interaction between the layers. Au wire electrodes were first modified with a mixed monolayer of MUOH and MUA. Then successively alternating layers of carboxy-terminated SiNPs and a mixture of cyt c and T. villosa CDH were added and a supramolecular structure was built up to four bilayers. Using this technique, Feifel et al. [52] found a maximum increase of seven times higher faradaic currents in the presence of lactose as substrate for the deglycosylated variant. The results suggest that the increase in current density is attributed to an increase in the amount of molecules packed on the electrodes, i.e. to a more compact packing of CDH on the electrode surface and in the layers.
for lactose was also found to decrease by two- to threefold for the deglycosylated CDHs compared to the glycosylated ones. When the enzymes were compared trapped under a permselective membrane and 11-mercaptoundecanol (MUOH) SAM-modified Au disk electrodes, glycosylated and deglycosylated P. chrysosporium CDH showed the same catalytic activity, whereas glycosylated and deglycosylated C. subvermispora CDH showed similar results to that obtained on spectrographic graphite electrodes, i.e. a much higher catalytic current was found when using the deglycosylated form. The E°′ for the deglycosylated CDH was found to shift up by ∼8 mV. In a different study glycosylated and deglycosylated T. villosa CDH were compared when immobilised on an LbL supramolecular architecture stabilised by electrostatic interaction between the layers. Au wire electrodes were first modified with a mixed monolayer of MUOH and MUA. Then successively alternating layers of carboxy-terminated SiNPs and a mixture of cyt c and T. villosa CDH were added and a supramolecular structure was built up to four bilayers. Using this technique, Feifel et al. [52] found a maximum increase of seven times higher faradaic currents in the presence of lactose as substrate for the deglycosylated variant. The results suggest that the increase in current density is attributed to an increase in the amount of molecules packed on the electrodes, i.e. to a more compact packing of CDH on the electrode surface and in the layers.
Fig. 9.

Dependence of the amperometric response on lactose concentration of a glycosylated Phanerochaete chrysosporium CDH (squares) and deglycosylated P. chrysosporium CDH (circles) and b glycosylated Ceriporiopsis subvermispora CDH (squares) and deglycosylated C. subvermispora CDH (circles) in acetate buffer (pH 4.5). The applied potential was +150 mV vs. Ag|AgCl (0.1 M KCl) and the flow rate of the solution (pH 4.5) was 1 mL min−1. The results were obtained in a flow-injection system. Reproduced from [94] with permission from The American Chemical Society
Mediated electron transfer
Even though a system based on DET is, from a fundamental point of view, more interesting than one based on MET, there are limitations to the current density that can be reached with DET. Even though it should be possible to immobilise multilayers of a redox enzyme with DET properties, it is expected that primarily only the innermost layer of enzyme molecules on the electrode surface will be able to electrochemically communicate with the electrode. However, through the use of nanostructured electrodes and with CDH oriented for improved DET [47–49] the current density can be largely increased compared with that obtained with conventional electrodes. Moreover, as DET between CDH and an electrode is obtained through the CYTCDH, which has an E°′ value much more positive than that of the FAD of the DHCDH [74, 124], in a BFC anode based on MET using a mediator with an E°′ much lower than that of the CYTCDH (and close to that of the DHCDH), both the power output and the current density can be largely improved [89, 91, 93, 125]. An overview of MET approaches with CDH was given in 2010 [19]. Since then mainly two directions have been followed. The first is the combination of CDH with cyt c as a mediator in an LbL approach, as discussed separately above. The second is the further development and application of osmium polymers, which were first applied together with CDH as early as 1992 [126] on the basis of the work by Heller and co-workers [127, 128]. Similar to the use of other sugar-oxidising redox enzymes in biosensors and biofuel cells [129–136], the “wiring” of various CDHs with Os-redox polymers accepting electrons direct from the DHCDH is very promising for obtaining high current densities at low potentials [46, 90, 91, 93, 125]. During the last few years particular focus was given to electrodeposited Os-polymers (Os-EDPs), which contain, in addition to the polymeric backbone and the redox-active Os-complex, either one or more COO− or NH3 + groups. Deposition of those polymers is achieved by local changes of the pH at the electrode surface caused by either electrolysis or change of buffer, resulting in discharged and insoluble Os-polymers that precipitate on the electrode surface. Schuhmann and co-workers [125, 137] synthesised and investigated 50 different Os-EDPs having redox potentials ranging from −430 to +667 mV vs. Ag|AgCl. Os-polymer-mediated electron transfer was shown for CDH, glucose oxidase and PQQ-dependent glucose dehydrogenase suitable for biofuel cell anodes and for laccase and BOx suitable for corresponding cathodes [125, 137]. In a recent paper a bi-enzyme bioanode modified with an Os-DEP, the separate DHCDH of C. thermophilus and pyranose dehydrogenase (PDH) from Agaricus meleagris were investigated for the multiple oxidation of glucose [46]. As CDH oxidises its substrate at the C1 it is only possible to gain two electrons per substrate molecule. However, if CDH is combined with PDH it is possible to obtain up to six electrons per substrate molecule, as PDH oxidises the sugar substrate at either C2 or C3 or both and the product when the substrate is oxidised by CDH is a substrate for PDH and vice versa. Figure 10 outlines how glucose can be oxidised at an anode where C. thermophilus CDH is co-immobilised with Agaricus meleagris PDH yielding six electrons instead of only two.
Fig. 10.

Reaction pathway of glucose oxidation by a bi-enzymatic system consisting of CDH (C1 oxidation, black arrows) and Agaricus meleagris PDH (C2 and C3 oxidation, grey arrows). Reproduced from [46] with permission from Elsevier
However, conventional mediators such as p-benzoquinone (BQ) are still in use as well. When comparing the efficiency of DET for a CDH-modified electrode in relation to MET it is quite convenient to use BQ dissolved in the buffer. This approach has been used previously [88] and was also used for the characterisation of various CDHs [45], especially for N. crassa CDH immobilised on spectrographic graphite electrodes with various substrates at pH 5.2 and 7 [43]. A biosensor for the real-time measurement of cellobiohydrolase activity was established by Cruys-Bagger and co-workers [54] using a BQ-containing carbon paste electrode modified with cross-linked P. chrysosporium CDH. The sensor detected cellobiose with a sensitivity of 87.7 μA mM−1 cm−2, a low detection limit of 25 nM and a response time of ca. 3 s.
Application of CDH in biosensors
CDH was used in two ways in biosensors: either to detect the oxidation of carbohydrates or reduce quinones and catecholamines for signal amplification in an oxidative electrode setup. In a recent review the application of CDH in biosensors until 2010 is described in detail [19]; here we give an overview of developments in the few last years (Table 5). Depending on the source of CDH, specific catalytic properties allow the oxidation of different substrates by different CDHs and therefore different analytes in terms of biosensor applications. Class I CDHs show a very high specificity for β-1,4-linked substrates and are therefore ideal bioelements for the detection of cellobiose and lactose. But it has to be considered that these CDHs work only efficient under acidic pH conditions. Very sensitive lactose sensors based on class I CDH have been developed for use in the dairy industry; these have a detection limit down to 250 nM, which corresponds to 90 μg L−1 [55, 58, 85]. In a different approach a lactose biosensor was developed by the combination of the thermometric signal from lactose oxidation with the amperometric signal of the enzymatic reaction [138]. In contrast to the third-generation biosensor reported by Safina et al. [55], the amperometric signal was based on the reduction of hydroquinone, which is formed during the oxidation of lactose by CDH.
Table 5.
CDH-based biosensors
| Analyte | Detection limit | Sensitivity (μA mM−1 cm−2) | Mediator/enhancera | Electrode modification | Electrode material | CDH | Reference |
|---|---|---|---|---|---|---|---|
| Noradrenaline | 1 nM | 15,800 | Cellobiose | Adsorption | SG | PcCDH | [85] |
| Catechol | 1 nM | 9,500 | Cellobiose | Adsorption | SG | PcCDH | [85] |
| Hydroquinone | 0.75 nM | 11,140 | Cellobiose | Adsorption | SG | PcCDH | [85] |
| l-Adrenaline | 5 nM | 1,140 | Cellobiose | Adsorption | SG | PcCDH | [85] |
| 3-Hydroxylamine hydrochloride | 2.5 nM | 9,160 | Cellobiose | Adsorption | SG | PcCDH | [85] |
| 3,4-Hydroxyphenylacetic acid | 1 nM | 13,440 | Cellobiose | Adsorption | SG | PcCDH | [85] |
| Lactose | 1 μM | 17.8 | No | Adsorption | SG | TvCDH | [85] |
| Lactose | 1 μM | 11.0 | No | Adsorption | SG | PsCDH | [85] |
| Lactose | 1 μM | 1.06 | No | Adsorption | SG | MtCDH | [88] |
| Lactose | 1 μM | 2.8 | No | Adsorption | SCE | MtCDH | [149] |
| Lactose | 1 μM | 0.38 | No | PANI + adsorption | SCE | MtCDH | [149] |
| Lactose | 250 nM | NG | No | Cross-linked | SCE | PsCDH | [55, 58] |
| Lactose | 50 μM | NG | BQ | Covalent binding | CPG | Rec. PcCDH | [138] |
| Cellobiose | 25 μM | 0.8 | No | Adsorption | SG | PcCDH | [150] |
| Cellobiose | 25 μM | 23 | Os-polymer | Entrapment | SG | PcCDH | [150] |
| Cellobiose | 0.5 μM | 1.85 | No | Adsorption | SG | MtCDH | [88] |
| Cellobiose | 25 nM | 87.7 | Hydroquinone | Cross-linked | SG | Rec. PcCDH | [54] |
| Glucose | 1 mM | 0.0068 | No | Adsorption | SG | MtCDH | [88] |
| Glucose | 0.05 mM | 0.22 | No | Cross-linked + SWCNTs | SG | CtCDH | [56] |
| Glucose | 0.01 mM | NG | No | Cross-linked + SWCNTs | SCE | CtCDH | [57] |
SG spectroscopic graphite, SCE screen-printed carbon electrode, PANI polyaniline, CPE carbon paste electrode, Rec. recombinant, NG value not given
aTo increase the signal of catecholamines the enzyme’s carbohydrate substrate was applied for signal amplification by recycling of the analyte
In another recent example, a CDH biosensor was designed to measure the cellobiohydrolase activity on insoluble cellulose [54]. The ability of CDH to oxidise cellobiose—the reaction product of cellobiohydrolase—was used to monitor the transient kinetics of cellobiohydrolase. The approach could be validation by HPLC analysis and represents an interesting real-time method to monitor cellulase activity. The discovery that class II CDHs are able to oxidise the monosaccharide glucose opened the door to designing a third-generation glucose biosensor [88]. Beside their altered substrate specificity, class II CDHs are not restricted to work under acidic pH conditions. Probably the most interesting candidate for a glucose biosensor is the CDH from C. thermophilus because the highest DET rates were found under human physiological pH conditions [45]. The third-generation glucose biosensor based on CDH showed a linear range between 0.1 and 30 mM, which makes it suitable for measuring blood glucose levels [47, 57]. This CDH should allow the construction of a simple glucose biosensor which does not depend on oxygen or any other artificial redox mediators and therefore represents an interesting alternative to established biosensors based on glucose oxidase and glucose dehydrogenase. The main targets for optimising CDH-based biosensors recently focused on the increase in current densities leading to higher sensitivities by designing the interface between the electrode and the enzyme.
Recent advances in CDH-based biofuel cell anode performance
The initial work on BFC applications was based on spectroscopic graphite electrodes modified with adsorbed CDH. The class I CDHs from P. chrysosporium [90, 93], T. villosa [125] and Dichomera saubinetii [87] and the class II CDHs from M. thermophilum [89, 90] and C. thermophilus [50] were used in these initial studies with lactose or glucose as the anodic fuel. In order to increase the current density in some of the studies, SWCNTs were also applied to increase the aspect ratio surface area to achieve higher loadings of adsorbed CDH [89, 90, 93]. Mediating Os-polymers were also applied to achieve a direct contact of the electrode with the DHCDH domain aiming at higher power densities gained at lower potentials [89, 91, 93]. To maximise the open-circuit voltage, it is desirable to have enzyme molecules in DET communication with the electrode, in the absence of redox mediators. Most recently the focus for BFC-based setups using CDH has been on the use of gold nanoparticles (AuNPs). On the basis of the work by Murata et al. [95] on nanomodification of polycrystalline Au electrodes with AuNPs, a covalent immobilisation technique for CDH was developed by Matsumura et al. [48] based on a SAM of mixed thiols, amino and hydroxyl or carboxylic acid terminated, and GA as a cross-linker. The technique was optimised and further used with a C. thermophilus CDH-based anode in combination with a BOx-based cathode to construct a one-compartment, mediator-less BFC working under human physiological conditions with glucose/O2 as the fuel [49]. The performance of the biodevice was evaluated in blood and plasma. In buffer containing glucose, the open-circuit potential was 0.66 V with a maximum power density of 3.3 μW cm−2 at a cell voltage of 0.52 V. The performance was only slightly reduced in the physiological fluids. The half-life was 30 h in buffer, 8 h in blood and 2 h in plasma. The same modification was used to fabricate a device tested in lachrymal fluid (tears) for possible future applications on non-invasive medical devices ex vivo [51]. The CDH/BOx/AuNP-based nanodevice had an open-circuit potential of 0.57 V with a maximum power density of 3.5 μW cm−2 at a comparably low operational voltage of 0.2 V, probably owing to interferences with ascorbic acid and dopamine. At 0.5 V a maximum power density of only 0.8 μW cm−2 was observed because of the low concentration of glucose in the lachrymal fluid. Figure 11 shows the performance of the BFC in the described buffers and physiological fluids.
Fig. 11.

Dependency of the power density on the operating voltage of a glucose/O2 biofuel cell based on Au|AuNP|CtCDH||bilirubin oxidase|AuNP|Au under air-saturated quiescent conditions in a PBS pH 7.4, 5 mM glucose; b human blood; c human plasma; d unstimulated human saliva; e human basal tears. Reproduced from [51] with permission from Elsevier
Conclusions and outlook
CDH is an emerging biocatalyst for biosensors and biofuel cells. Its versatility originates from its unique molecular properties, but also from the catalytic heterogeneity of CDHs from different sources. Differences in the pH optima, the pH optima for IET, the substrate specificity, etc., span a wide range and provide a toolbox of CDHs for different tasks and applications. Over 20 CDHs from different fungi have been characterised so far and recombinant expression techniques guarantee access to these enzymes. An issue of importance with CDH is, however, the microheterogeneity of enzyme preparations, especially glycoforms. Glycosyl residues alter the IET, but also the binding to electrode surfaces and the orientation on the electrode and are therefore an important factor to watch in future experiments. Another critical factor is the loading of FAD in recombinantly produced ascomycete CDHs. The substochioimetric presence of FAD in the DHCDH results in apparently lower turnover numbers and reduces the catalytic current. Alternative expression strategies or expression hosts should be tested for better results. The development of specifically modified electrode surfaces, either by chemical modification or by nanostructures, has tremendously increased the currents achievable with CDH-modified electrodes. A combination of the approaches from materials science and enzyme engineering will have great impact on the development of CDH-modified electrodes with high current densities in the milliampere per square centimetre range. Such high current densities will ultimately allow miniaturisation of CDH electrodes down to single carbon fibre electrodes, which can be used in miniaturised biosensors for continuous on-site or point-of-care measurements. Finally, one should mention the exciting use of ionic liquids in combination with CDH. Ionic liquids are potential candidates as non-aqueous solvents in this context and were shown to improve the shelf-life of CDH bound to AuNPs and carbon nanoparticles [151, 152].
Acknowledgments
The authors thank the following agencies for financial support: the European Commission (project 3D-Nanobiodevice NMP4-SL-2009-229255, project Chebana FP7-PEOPLE-2010-ITN-264772), the nmC@LU, the Swedish Research Council (project 621-2010-5031), the Austrian Science Fund (translational project FWF L395-B11) and the Austrian Academy of Science (APART project 11322) and the New Zealand Ministry of Innovation, Business and Enterprise (formerly the Ministry of Science and Innovation).
Abbreviations
- AuNP
gold nanoparticles
- BQ
p-benzoquinone
- BOx
bilirubin oxidase
- CBM1
carbohydrate binding module (family 1)
- CDH
cellobiose dehydrogenase
- CNP
carbon nanoparticles
- CNT
carbon nanotube
- CV
cyclic voltammetry, cyclic voltammogram
- cyt c
cytochrome c (from horse heart)
- CYTCDH
cytochrome domain of CDH
- DCIP
2,6-dichloroindophenol
- DET
direct electron transfer
- DHCDH
flavodehydrogenase domain of CDH
- E°′
formal potential
- ET
electron transfer
- GA
glutaraldehyde
- IET
intramolecular electron transfer
- LbL
layer by layer
- MET
mediated electron transfer
- MUA
mercapto-1-undecanoic acid
- MUOH
mercapto-1-undecanol
- MWCNT
multi-walled carbon nanotubes
- NHE
normal hydrogen electrode
- Os-EDP
osmium electrodeposition paint
- PBS
phosphate buffered saline
- PEGDGE
polyethyleneglycol diglycidyl ether
- PMO
polysaccharide monooxygenase
- SAM
self-assembled monolayer
- SiNP
silica nanoparticles
- SPR
surface plasmon resonance
- SWCNT
single-walled carbon nanotube
Footnotes
Published in the topical collection Bioelectroanalysis with guest editors Nicolas Plumeré, Magdalena Gebala and Wolfgang Schuhmann.
References
- 1.Deepshikha, Basu T. Anal Lett. 2011;44(6):1126–1171. doi: 10.1080/00032719.2010.511734. [DOI] [Google Scholar]
- 2.Iost RM, Madurro JM, Brito-Madurro AG, Nantes IL, Caseli L, Crespilho FN. Int J Electrochem Sci. 2011;6(7):2965–2997. [Google Scholar]
- 3.Plowman BJ, Bhargava SK, O’Mullane AP. Analyst. 2011;136(24):5107–5119. doi: 10.1039/c1an15657h. [DOI] [PubMed] [Google Scholar]
- 4.Rahman MM, Ahammad AJS, Jin J-H, Ahn SJ, Lee J-J. Sensors. 2010;10(5):4855–4886. doi: 10.3390/s100504855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asefa T, Duncan CT, Sharma KK. Analyst. 2009;134(10):1980–1990. doi: 10.1039/b911965p. [DOI] [PubMed] [Google Scholar]
- 6.Chen D, Wang G, Li JH. J Phys Chem. 2007;111(6):2351–2367. doi: 10.1021/jp065099w. [DOI] [Google Scholar]
- 7.Gooding JJ, Darwish N. Chem Rec. 2012;12(1):92–105. doi: 10.1002/tcr.201100013. [DOI] [PubMed] [Google Scholar]
- 8.Pingarron JM, Yanez-Sedeno P, Gonzalez-Cortes A. Electrochim Acta. 2008;53(19):5848–5866. doi: 10.1016/j.electacta.2008.03.005. [DOI] [Google Scholar]
- 9.Shipway AN, Katz E, Willner I. Chem Phys Chem. 2000;1(1):18–52. doi: 10.1002/1439-7641(20000804)1:1<18::AID-CPHC18>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 10.Xiao YH, Li CM. Electroanalysis. 2008;20(6):648–662. doi: 10.1002/elan.200704125. [DOI] [Google Scholar]
- 11.Alferov S, Coman V, Gustavsson T, Reshetilov A, von Wachenfeldt C, Hägerhäll C, Gorton L. Electrochim Acta. 2009;54(22):4979–4984. doi: 10.1016/j.electacta.2009.03.090. [DOI] [Google Scholar]
- 12.Baronian K, Downard A, Lowen R, Pasco N. Appl Microbiol Biotechnol. 2003;60(1–2):108–113. doi: 10.1007/s00253-002-1108-3. [DOI] [PubMed] [Google Scholar]
- 13.Chelikani V, Downard AJ, Kunze G, Gooneratne R, Pasco N, Baronian KHR. Electrochim Acta. 2012;73:136–140. doi: 10.1016/j.electacta.2011.11.078. [DOI] [Google Scholar]
- 14.Chelikani V, Rawson FJ, Downard AJ, Gooneratne R, Kunze G, Pasco N, Baronian KHR. Biosens Bioelectron. 2011;26(9):3737–3741. doi: 10.1016/j.bios.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Coman V, Gustavsson T, Finkelsteinas A, Von Wachenfeldt C, Hägerhäll C, Gorton L. J Am Chem Soc. 2009;131(44):16171–16176. doi: 10.1021/ja905442a. [DOI] [PubMed] [Google Scholar]
- 16.Durand F, Kjaergaard CH, Suraniti E, Gounel S, Hadt RG, Solomon EI, Mano N. Biosens Bioelectron. 2012;35(1):140–146. doi: 10.1016/j.bios.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasan K, Patil SA, Górecki K, Leech D, Hägerhäll C, Gorton L (2012) Bioelectrochemistry. doi:10.1016/j.bioelechem.2012.05.004 [DOI] [PubMed]
- 18.Kostesha NV, Almeida JRM, Heiskanen AR, Gorwa-Grauslund MF, Hahn-Hägerdahl B, Emnéus J. Anal Chem. 2009;81(24):9896–9901. doi: 10.1021/ac901402m. [DOI] [PubMed] [Google Scholar]
- 19.Ludwig R, Harreither W, Tasca F, Gorton L. Chem Phys Chem. 2010;11(13):2674–2697. doi: 10.1002/cphc.201000216. [DOI] [PubMed] [Google Scholar]
- 20.Patil SA, Hasan K, Leech D, Hägerhäll C, Gorton L. Chem Commun. 2012;48(82):10183–10185. doi: 10.1039/c2cc34903e. [DOI] [PubMed] [Google Scholar]
- 21.Rawson FJ, Garrett DJ, Leech D, Downard AJ, Baronian KHR. Biosens Bioelectron. 2011;26(5):2383–2389. doi: 10.1016/j.bios.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Rawson FJ, Gross AJ, Garrett DJ, Downard AJ, Baronian KHR. Electrochem Commun. 2012;15(1):85–87. doi: 10.1016/j.elecom.2011.11.030. [DOI] [Google Scholar]
- 23.Katz E, Willner I. Chem Phys Chem. 2004;5(8):1085–1104. [Google Scholar]
- 24.Meredith MT, Minteer SD. Annu Rev Anal Chem. 2012;5(1):157–179. doi: 10.1146/annurev-anchem-062011-143049. [DOI] [PubMed] [Google Scholar]
- 25.Osman MH, Shah AA, Walsh FC. Biosens Bioelectron. 2011;26(7):3087–3102. doi: 10.1016/j.bios.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Willner I, Yan YM, Willner B, Tel-Vered R. Fuel Cells. 2009;9(1):7–24. doi: 10.1002/fuce.200800115. [DOI] [Google Scholar]
- 27.Rabaey K, Rozendal RA. Nat Rev Microbiol. 2010;8(10):706–716. doi: 10.1038/nrmicro2422. [DOI] [PubMed] [Google Scholar]
- 28.Barton SC, Gallaway J, Atanassov P. Chem Rev. 2004;104(10):4867–4886. doi: 10.1021/cr020719k. [DOI] [PubMed] [Google Scholar]
- 29.Cracknell JA, Vincent KA, Armstrong FA. Chem Rev. 2008;108(7):2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- 30.Falk M, Blum Z, Shleev S. Electrochim Acta. 2012;82:191–202. doi: 10.1016/j.electacta.2011.12.133. [DOI] [Google Scholar]
- 31.Leger C, Bertrand P. Chem Rev. 2008;108(7):2379–2438. doi: 10.1021/cr0680742. [DOI] [PubMed] [Google Scholar]
- 32.Woolerton TW, Sheard S, Chaudhary YS, Armstrong FA. Energ Environ Sci. 2012;5(6):7470–7490. doi: 10.1039/c2ee21471g. [DOI] [Google Scholar]
- 33.Mowat CG, Gazur B, Campbell LP, Chapman SK. Arch Biochem Biophys. 2010;493(1):37–52. doi: 10.1016/j.abb.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Guo LH, Hill HAO (1991) Adv Inorg Chem 36:341–376
- 35.Hallberg BM, Bergfors T, Baeckbro K, Pettersson G, Henriksson G, Divne C. Structure. 2000;8(1):79–88. doi: 10.1016/S0969-2126(00)00082-4. [DOI] [PubMed] [Google Scholar]
- 36.Hallberg MB, Henriksson G, Pettersson G, Divne C. J Mol Biol. 2002;315(3):421–434. doi: 10.1006/jmbi.2001.5246. [DOI] [PubMed] [Google Scholar]
- 37.Ikeda T, Kobayashi D, Matsushita F, Sagara T, Niki K. J Electroanal Chem. 1993;361(1–2):221–228. [Google Scholar]
- 38.Beeson WT, Phillips CM, Cate JHD, Marletta MA. J Am Chem Soc. 2012;134(2):890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]
- 39.Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD. Appl Environ Microbiol. 2011;77(19):7007–7015. doi: 10.1128/AEM.05815-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Beeson WT, Phillips CM, Marletta MA, Cate JHD. Structure. 2012;20(6):1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips CM, Beeson WT, Cate JH, Marletta MA. ACS Chem Biol. 2011;6(12):1399–1406. doi: 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
- 42.Harreither W, Sygmund C, Augustin M, Narciso M, Rabinovich ML, Gorton L, Haltrich D, Ludwig R. Appl Environ Microbiol. 2011;77(5):1804–1815. doi: 10.1128/AEM.02052-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kovacs G, Ortiz R, Coman V, Harreither W, Popescu IC, Ludwig R, Gorton L. Bioelectrochemistry. 2012;88:84–91. doi: 10.1016/j.bioelechem.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Kovacs G, Ortiz R, Coman V, Harreither W, Popescu IO, Ludwig R, Gorton L (2012) Rev Roum Chim 57:361–368
- 45.Harreither W, Nicholls P, Sygmund C, Gorton L, Ludwig R. Langmuir. 2012;28(16):6714–6723. doi: 10.1021/la3005486. [DOI] [PubMed] [Google Scholar]
- 46.Shao M, Nadeem Zafar M, Sygmund C, Guschin DA, Ludwig R, Peterbauer CK, Schuhmann W, Gorton L. Biosens Bioelectron. 2013;40:308–314. doi: 10.1016/j.bios.2012.07.069. [DOI] [PubMed] [Google Scholar]
- 47.Tasca F, Harreither W, Ludwig R, Gooding JJ, Gorton L. Anal Chem. 2011;83(8):3042–3049. doi: 10.1021/ac103250b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsumura H, Ortiz R, Ludwig R, Igarashi K, Samejima M, Gorton L. Langmuir. 2012;28(29):10925–10933. doi: 10.1021/la3018858. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Falk M, Ortiz R, Matsumura H, Bobacka J, Ludwig R, Bergelin M, Gorton L, Shleev S. Biosens Bioelectron. 2012;31(1):219–225. doi: 10.1016/j.bios.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 50.Coman V, Ludwig R, Harreither W, Haltrich D, Gorton L, Ruzgas T, Shleev S. Fuel Cells. 2010;10(1):9–16. doi: 10.1039/b808859d. [DOI] [PubMed] [Google Scholar]
- 51.Falk M, Andoralov V, Blum Z, Sotres J, Suyatin DB, Ruzgas T, Arnebrant T, Shleev S. Biosens Bioelectron. 2012;37(1):38–45. doi: 10.1016/j.bios.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 52.Feifel SC, Ludwig R, Gorton L, Lisdat F. Langmuir. 2012;28:9189–9194. doi: 10.1021/la301290z. [DOI] [PubMed] [Google Scholar]
- 53.Sarauli D, Ludwig R, Haltrich D, Gorton L, Lisdat F. Bioelectrochemistry. 2012;87:9–14. doi: 10.1016/j.bioelechem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Cruys-Bagger N, Ren G, Tatsumi H, Baumann MJ, Spodsberg N, Andersen HD, Gorton L, Borch K, Westh P. Biotechnol Bioeng. 2012;109:3199–3204. doi: 10.1002/bit.24593. [DOI] [PubMed] [Google Scholar]
- 55.Safina G, Ludwig R, Gorton L. Electrochim Acta. 2010;55:7690–7695. doi: 10.1016/j.electacta.2009.10.052. [DOI] [Google Scholar]
- 56.Tasca F, Zafar MN, Harreither W, Nöll G, Ludwig R, Gorton L. Analyst. 2011;136(10):2033–2036. doi: 10.1039/c0an00311e. [DOI] [PubMed] [Google Scholar]
- 57.Zafar MN, Safina G, Ludwig R, Gorton L. Anal Biochem. 2012;425(1):36–42. doi: 10.1016/j.ab.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 58.Glithero N, Clark C, Gorton L, Schuhmann W, Pasco N (2012) Anal Bioanal Chem (in press) [DOI] [PubMed]
- 59.Malel E, Ludwig R, Gorton L, Mandler D. Chem Eur J. 2010;16(38):11697–11706. doi: 10.1002/chem.201000453. [DOI] [PubMed] [Google Scholar]
- 60.Zamocky M, Ludwig R, Peterbauer C, Hallberg BM, Divne C, Nicholls P, Haltrich D. Curr Prot Pept Sci. 2006;7(3):255–280. doi: 10.2174/138920306777452367. [DOI] [PubMed] [Google Scholar]
- 61.Zámocký M, Hallberg M, Ludwig R, Divne C, Haltrich D. Gene. 2004;338(1):1–14. doi: 10.1016/j.gene.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 62.Sygmund C, Kracher D, Scheibelbrandner S, Zahma K, Felice AK, Kittl R, Harreither W, Ludwig R. Appl Environ Microbiol. 2012;78(17):6161–6171. doi: 10.1128/AEM.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phillips CM, Iavarone AT, Marletta MA. J Proteome Res. 2011;10(9):4177–4185. doi: 10.1021/pr200329b. [DOI] [PubMed] [Google Scholar]
- 64.Sun J, Glass NL. PLoS One. 2011;6(9):1–14. doi: 10.1145/2039239.2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun J, Tian C, Diamond S, Louise Glassa N. Eukaryot Cell. 2012;11(4):482–493. doi: 10.1128/EC.05327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tian C, Beeson WT, Iavarone AT, Sun J, Marletta MA, Cate JHD, Glass NL. Proc Natl Acad Sci U S A. 2009;106(52):22157–22162. doi: 10.1073/pnas.0906810106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.MacDonald J, Doering M, Canam T, Gong Y, Guttman DS, Campbell MM, Master ER. Appl Environ Microbiol. 2011;77(10):3211–3218. doi: 10.1128/AEM.02490-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wymelenberg AV, Sabat G, Martinez D, Rajangam AS, Teeri TT, Gaskell J, Kersten PJ, Cullen D. J Biotechnol. 2005;118(1):17–34. doi: 10.1016/j.jbiotec.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 69.Baminger U, Subramaniam SS, Renganathan V, Haltrich D. Appl Environ Microbiol. 2001;67(4):1766–1774. doi: 10.1128/AEM.67.4.1766-1774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harreither W, Sygmund C, Dünhofen E, Vicuña R, Haltrich D, Ludwig R. Appl Environ Microbiol. 2009;75(9):2750–2757. doi: 10.1128/AEM.02320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cavener DR. J Mol Biol. 1992;223(3):811–814. doi: 10.1016/0022-2836(92)90992-S. [DOI] [PubMed] [Google Scholar]
- 72.Cameron MD, Aust SD. Enzyme Microb Technol. 2001;28(2–3):129–138. doi: 10.1016/S0141-0229(00)00307-0. [DOI] [PubMed] [Google Scholar]
- 73.Henriksson G, Johansson G, Pettersson G. J Biotechnol. 2000;78(2):93–113. doi: 10.1016/S0168-1656(00)00206-6. [DOI] [PubMed] [Google Scholar]
- 74.Coman V, Harreither W, Ludwig R, Haltrich D, Gorton L. Chem Anal (Warsaw) 2007;52(6):945–960. [Google Scholar]
- 75.Harreither W, Felice AKG, Paukner R, Gorton L, Ludwig R, Sygmund C. Biotechnol J. 2012;7:1359–1366. doi: 10.1002/biot.201200049. [DOI] [PubMed] [Google Scholar]
- 76.Hallberg BM, Henriksson G, Pettersson G, Vasella A, Divne C. J Biol Chem. 2003;278(9):7160–7166. doi: 10.1074/jbc.M210961200. [DOI] [PubMed] [Google Scholar]
- 77.Li B, Rotsaert FAJ, Gold MH, Renganathan V. Biochem Biophys Res Commun. 2000;270(1):141–146. doi: 10.1006/bbrc.2000.2381. [DOI] [PubMed] [Google Scholar]
- 78.Yoshida M, Ohira T, Igarashi K, Nagasawa H, Aida K, Hallberg BM, Divne C, Nishino T, Samejima M. Biosci Biotechnol Biochem. 2001;65(9):2050–2057. doi: 10.1271/bbb.65.2050. [DOI] [PubMed] [Google Scholar]
- 79.Langston JA, Brown K, Xu F, Borch K, Garner A, Sweeney MD. Biochim Biophys Acta, Protein Proteom. 2012;1824(6):802–812. doi: 10.1016/j.bbapap.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 80.Xu F, Golightly EJ, Duke KR, Lassen SF, Knusen B, Christensen S, Brown KM, Brown SH, Schülein M. Enzyme Microb Technol. 2001;28(9–10):744–753. doi: 10.1016/S0141-0229(01)00319-2. [DOI] [PubMed] [Google Scholar]
- 81.Desriani, Ferri S, Sode K. Biotechnol Lett. 2010;32(6):855–859. doi: 10.1007/s10529-010-0215-y. [DOI] [PubMed] [Google Scholar]
- 82.Zámocký M, Schümann C, Sygmund C, O’Callaghan J, Dobson ADW, Ludwig R, Haltrich D, Peterbauer CK. Protein Expr Purif. 2008;59(2):258–265. doi: 10.1016/j.pep.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 83.Larsson T, Elmgren M, Lindquist SE, Tessema M, Gorton L, Henriksson G. Anal Chim Acta. 1996;331(3):207–215. doi: 10.1016/0003-2670(96)00136-5. [DOI] [Google Scholar]
- 84.Maischberger T, Nguyen TH, Sukyai P, Kittl R, Riva S, Ludwig R, Haltrich D. Carbohydr Res. 2008;343(12):2140–2147. doi: 10.1016/j.carres.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 85.Stoica L, Ludwig R, Haltrich D, Gorton L. Anal Chem. 2006;78(2):393–398. doi: 10.1021/ac050327o. [DOI] [PubMed] [Google Scholar]
- 86.Hildén L, Eng L, Johansson G, Lindqvist S-E, Pettersson G. Anal Biochem. 2001;290(2):245–250. doi: 10.1006/abio.2000.4959. [DOI] [PubMed] [Google Scholar]
- 87.Coman V, Vaz-Dominguez C, Ludwig R, Herreither W, Haltrich D, De Lacey AL, Ruzgas T, Gorton L, Shleev S. Phys Chem Chem Phys. 2008;10(40):6093–6096. doi: 10.1039/b808859d. [DOI] [PubMed] [Google Scholar]
- 88.Harreither W, Coman V, Ludwig R, Haltrich D, Gorton L. Electroanalysis. 2007;19(2–3):172–180. doi: 10.1002/elan.200603688. [DOI] [Google Scholar]
- 89.Tasca F, Gorton L, Harreither W, Haltrich D, Ludwig R, Nöll G. J Phys Chem. 2008;112(35):13668–13673. doi: 10.1021/jp805092m. [DOI] [Google Scholar]
- 90.Tasca F, Gorton L, Harreither W, Haltrich D, Ludwig R, Nöll G. J Phys Chem. 2008;112(26):9956–9961. doi: 10.1021/jp802099p. [DOI] [Google Scholar]
- 91.Tasca F, Gorton L, Kujawa M, Patel I, Harreither W, Peterbauer CK, Ludwig R, Nöll G. Biosens Bioelectron. 2010;25(7):1710–1716. doi: 10.1016/j.bios.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 92.Vasilchenko LG, Karapetyan KN, Yershevich OP, Ludwig R, Zamocky M, Peterbauer CK, Haltrich D, Rabinovich ML. Biotechnol J. 2011;6(5):538–553. doi: 10.1002/biot.201000373. [DOI] [PubMed] [Google Scholar]
- 93.Tasca F, Gorton L, Harreither W, Haltrich D, Ludwig R, Nöll G. Anal Chem. 2009;81(7):2791–2798. doi: 10.1021/ac900225z. [DOI] [PubMed] [Google Scholar]
- 94.Ortiz R, Matsumura H, Tasca F, Zahma K, Samejima M, Igarashi K, Ludwig R, Gorton L (2012) Anal Chem 84:10315–10323 [DOI] [PubMed]
- 95.Murata K, Kajiya K, Nukaga M, Suga Y, Watanabe T, Nakamura N, Ohno H. Electroanalysis. 2010;22:185–190. doi: 10.1002/elan.200900323. [DOI] [Google Scholar]
- 96.Fridman V, Wollenberger U, Bogdanovskaya V, Lisdat F, Ruzgas T, Lindgren A, Gorton L, Scheller FW. Biochem Soc Trans. 2000;28(2):63–70. doi: 10.1042/bst0280063. [DOI] [PubMed] [Google Scholar]
- 97.Dronov R, Kurth DG, Möhwald H, Spricigo R, Leimkühler S, Wollenberger U, Rajagopalan KV, Scheller FW, Lisdat F. J Am Chem Soc. 2008;130(4):1122–1123. doi: 10.1021/ja0768690. [DOI] [PubMed] [Google Scholar]
- 98.Lisdat F, Dronov R, Möhwald H, Scheller FW, Kurth DG (2009) Chem Commun (3):274–283 [DOI] [PubMed]
- 99.Spricigo R, Dronov R, Lisdat F, Leimkühler S, Scheller FW, Wollenberger U. Anal Bioanal Chem. 2009;393(1):225–233. doi: 10.1007/s00216-008-2432-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Spricigo R, Dronov R, Rajagopalan KV, Lisdat F, Leimkühler S, Scheller FW, Wollenberger U. Soft Matter. 2008;4(5):972–978. doi: 10.1039/b717694e. [DOI] [PubMed] [Google Scholar]
- 101.Lindgren A. Electrochemistry of heme containing enzymes - fundamentals and applications. Lund: Lund University; 2000. [Google Scholar]
- 102.Lindgren A, Gorton L, Ruzgas T, Baminger U, Haltrich D, Schülein M. J Electroanal Chem. 2001;496(1–2):76–81. [Google Scholar]
- 103.Lindgren A, Larsson T, Ruzgas T, Gorton L. J Electroanal Chem. 2000;494(2):105–113. doi: 10.1016/S0022-0728(00)00326-0. [DOI] [Google Scholar]
- 104.Stoica L, Dimcheva N, Haltrich D, Ruzgas T, Gorton L. Biosens Bioelectron. 2005;20(10):2010–2018. doi: 10.1016/j.bios.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 105.Marcus RA. J Chem Phys. 1965;43(2):679–701. doi: 10.1063/1.1696792. [DOI] [Google Scholar]
- 106.Marcus RA, Sutin N. Biochim Biophys Acta Rev Bioenerg. 1985;811(3):265–322. doi: 10.1016/0304-4173(85)90014-X. [DOI] [Google Scholar]
- 107.Fang J, Huang F, Gao P. Process Biochem. 1999;34(9):957–961. doi: 10.1016/S0032-9592(99)00016-3. [DOI] [Google Scholar]
- 108.Larsson T, Lindgren A, Ruzgas T, Lindquist SE, Gorton L. J Electroanal Chem. 2000;482(1):1–10. doi: 10.1016/S0022-0728(99)00503-3. [DOI] [Google Scholar]
- 109.Schulz C, Ludwig R, Micheelsen PO, Silow M, Toscano MD, Gorton L. Electrochem Commun. 2012;17:71–74. doi: 10.1016/j.elecom.2012.01.031. [DOI] [Google Scholar]
- 110.Lojou E, Bianco P. Electrochim Acta. 2007;52(25):7307–7314. doi: 10.1016/j.electacta.2007.06.003. [DOI] [Google Scholar]
- 111.Domínguez E, Lan HL, Okamoto Y, Hale PD, Skotheim TA, Gorton L, Hahn-Hägerdal B. Biosens Bioelectron. 1993;8(3–4):229–237. doi: 10.1016/0956-5663(93)85038-P. [DOI] [Google Scholar]
- 112.Ladiu CI, Popescu IC, Gorton L. J Solid State Electrochem. 2005;9(5):296–303. doi: 10.1007/s10008-004-0618-6. [DOI] [Google Scholar]
- 113.Gorton L, Jönsson-Pettersson G, Csöregi E, Johansson K, Domínguez E, Marko-Varga G. Analyst. 1992;117(8):1235–1241. doi: 10.1039/an9921701235. [DOI] [Google Scholar]
- 114.Kacaniklic V, Johansson K, Marko-Varga G, Gorton L, Jönsson-Pettersson G, Csöregi E. Electroanalysis. 1994;6(5–6):381–390. doi: 10.1002/elan.1140060505. [DOI] [Google Scholar]
- 115.Lutz M, Burestedt E, Emnéus J, Lidén H, Gobhadi S, Gorton L, Marko-Varga G. Anal Chim Acta. 1995;305(1–3):8–17. doi: 10.1016/0003-2670(94)00573-5. [DOI] [Google Scholar]
- 116.Spohn U, Narasaiah D, Gorton L, Pfeiffer D. Anal Chim Acta. 1996;319(1–2):79–90. doi: 10.1016/0003-2670(95)00485-8. [DOI] [Google Scholar]
- 117.Vijayakumar AR, Csöregi E, Heller A, Gorton L. Anal Chim Acta. 1996;327(3):223–234. doi: 10.1016/0003-2670(96)00093-1. [DOI] [Google Scholar]
- 118.Frasca S, Rojas O, Salewski J, Neumann B, Stiba K, Weidinger IM, Tiersch B, Leimkühler S, Koetz J, Wollenberger U. Bioelectrochemistry. 2012;87:33–41. doi: 10.1016/j.bioelechem.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 119.Stoica L, Ruzgas T, Ludwig R, Haltrich D, Gorton L. Langmuir. 2006;22(25):10801–10806. doi: 10.1021/la061190f. [DOI] [PubMed] [Google Scholar]
- 120.Courjean O, Gao F, Mano N. Angew Chem Int Ed Engl. 2009;48:5897–5899. doi: 10.1002/anie.200902191. [DOI] [PubMed] [Google Scholar]
- 121.Demin S, Hall EAH. Bioelectrochemistry. 2009;76(1–2):19–27. doi: 10.1016/j.bioelechem.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 122.Lindgren A, Tanaka M, Ruzgas T, Gorton L, Gazaryan I, Ishimori K, Morishima I. Electrochem Commun. 1999;1(5):171–175. doi: 10.1016/S1388-2481(99)00033-8. [DOI] [Google Scholar]
- 123.Yakovleva M, Killyeni A, Popescu IO, Ortiz R, Schulz C, MacAodha D, Ó Conghaile P, Leech D, Peterbauer CK, Gorton L. Electrochem Commun. 2012;24:120–122. doi: 10.1016/j.elecom.2012.08.029. [DOI] [Google Scholar]
- 124.Igarashi K, Verhagen M, Samejima M, Schülein M, Eriksson KEL, Nishino T. J Biol Chem. 1999;274(6):3338–3344. doi: 10.1074/jbc.274.6.3338. [DOI] [PubMed] [Google Scholar]
- 125.Stoica L, Dimcheva N, Ackermann Y, Karnicka K, Guschin DA, Kulesza PJ, Rogalski J, Haltrich D, Ludwig R, Gorton L, Schuhmann W. Fuel Cells. 2009;9(1):53–62. doi: 10.1002/fuce.200800033. [DOI] [Google Scholar]
- 126.Elmgren M, Lindquist SE, Henriksson G. J Electroanal Chem. 1992;341(1–2):257–273. [Google Scholar]
- 127.Gregg BA, Heller A. J Phys Chem. 1991;95(15):5970–5975. doi: 10.1021/j100168a046. [DOI] [Google Scholar]
- 128.Heller A. J Phys Chem. 1992;96(9):3579–3587. doi: 10.1021/j100188a007. [DOI] [Google Scholar]
- 129.Heller A. Phys Chem Chem Phys. 2004;6(2):209–216. doi: 10.1039/b313149a. [DOI] [Google Scholar]
- 130.Heller A. Curr Opin Chem Biol. 2006;10(6):664–672. doi: 10.1016/j.cbpa.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 131.Heller A. Anal Bioanal Chem. 2006;385(3):469–473. doi: 10.1007/s00216-006-0326-4. [DOI] [PubMed] [Google Scholar]
- 132.Heller A, Feldman B. Chem Rev. 2008;108(7):2482–2505. doi: 10.1021/cr068069y. [DOI] [PubMed] [Google Scholar]
- 133.Mano N, Mao F, Heller A. ChemBioChem. 2004;5(12):1703–1705. doi: 10.1002/cbic.200400275. [DOI] [PubMed] [Google Scholar]
- 134.Mano N, Mao F, Heller A (2004) Chem Commun (18):2116–2117 [DOI] [PubMed]
- 135.Mao F, Mano N, Heller A. J Am Chem Soc. 2003;125(16):4951–4957. doi: 10.1021/ja029510e. [DOI] [PubMed] [Google Scholar]
- 136.Pothukuchy A, Mano N, Georgiou G, Heller A. Biosens Bioelectron. 2006;22(5):678–684. doi: 10.1016/j.bios.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 137.Guschin DA, Castillo J, Dimcheva N, Schuhmann W. Anal Bioanal Chem. 2010;398(4):1661–1673. doi: 10.1007/s00216-010-3982-3. [DOI] [PubMed] [Google Scholar]
- 138.Yakovleva M, Buzas O, Matsumura H, Samejima M, Igarashi K, Larsson P-O, Gorton L, Danielsson B. Biosens Bioelectron. 2012;31(1):251–256. doi: 10.1016/j.bios.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 139.Bao W, Usha SN, Renganathan V. Arch Biochem Biophys. 1993;300(2):705–713. doi: 10.1006/abbi.1993.1098. [DOI] [PubMed] [Google Scholar]
- 140.Habu N, Igarashi K, Samejima M, Pettersson B, Eriksson K-EL. Biotechnol Appl Biochem. 1997;26(2):97–102. [PubMed] [Google Scholar]
- 141.Temp U, Eggert C. Appl Environ Microbiol. 1999;65(2):389–395. doi: 10.1128/aem.65.2.389-395.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ludwig R, Haltrich D. Appl Microbiol Biotechnol. 2003;61(1):32–39. doi: 10.1007/s00253-002-1209-z. [DOI] [PubMed] [Google Scholar]
- 143.Roy BP, Dumonceaux T, Koukoulas AA, Archibald FS. Appl Environ Microbiol. 1996;62(12):4417–4427. doi: 10.1128/aem.62.12.4417-4427.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Canevascini G, Borer P, Dreyer J-L. Eur J Biochem. 1991;198(1):43–52. doi: 10.1111/j.1432-1033.1991.tb15984.x. [DOI] [PubMed] [Google Scholar]
- 145.Subramaniam SS, Nagalla SR, Renganathan V. Arch Biochem Biophys. 1999;365(2):223–230. doi: 10.1006/abbi.1999.1152. [DOI] [PubMed] [Google Scholar]
- 146.Vasil’chenko LG, Khromonygina VV, Karapetyan KN, Vasilenko OV, Rabinovich ML. J Biotechnol. 2005;119(1):44–59. doi: 10.1016/j.jbiotec.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 147.Bey M, Berrin JG, Poidevin L, Sigoillot JC (2011) Microb Cell Fact 10:113 [DOI] [PMC free article] [PubMed]
- 148.Stapleton PC, O’Brien MM, O’Callaghan J, Dobson ADW. Enzyme Microb Technol. 2004;34(1):55–63. doi: 10.1016/j.enzmictec.2003.08.006. [DOI] [Google Scholar]
- 149.Trashin SA, Haltrich D, Ludwig R, Gorton L, Karyakin AA. Bioelectrochemistry. 2009;76(1–2):87–92. doi: 10.1016/j.bioelechem.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 150.Tessema M, Larsson T, Buttler T, Csöregi E, Ruzgas T, Nordling M, Lindquist SE, Pettersson G, Gorton L. Anal Chim Acta. 1997;349(1–3):179–188. doi: 10.1016/S0003-2670(97)00197-9. [DOI] [Google Scholar]
- 151.Fujita K, Nakamura N, Igarashi K, Samejima M, Ohno H. Green Chem. 2009;11:351–354. doi: 10.1039/b813529k. [DOI] [Google Scholar]
- 152.Fujita K, Nakamura N, Murata K, Igarashi K, Samejima M, Ohno H. Electrochim Acta. 2011;56:7224–7227. doi: 10.1016/j.electacta.2011.03.107. [DOI] [Google Scholar]