

Abstract
Plasmodium falciparum (P. falciparum) is responsible for the majority of life-threatening cases of human malaria, causing 1.5-2.7 million annual deaths. The global emergence of drug-resistant malaria parasites necessitates identification and characterization of novel drug targets and their potential inhibitors. We identified the carbonic anhydrase (CA) genes in P. falciparum. The pfCA gene encodes anα-carbonic anhydrase, a Zn2+-metalloenzme, possessing catalytic properties distinct from that of the human host CA enzyme. The amino acid sequence of the pfCA enzyme is different from the analogous protozoan and human enzymes. A library of aromatic/heterocyclic sulfonamides possessing a large diversity of scaffolds were found to be very good inhibitors for the malarial enzyme at moderate-low micromolar and submicromolar inhibitions. The structure of the groups substituting the aromatic-ureido- or aromatic-azomethine fragment of the molecule and the length of the parent sulfonamide were critical parameters for the inhibitory properties of the sulfonamides. One derivative, that is, 4- (3, 4-dichlorophenylureido)thioureido-benzenesulfonamide (compound 10) was the most effective in vitro Plasmodium falciparum CA inhibitor, and was also the most effective antimalarial compound on the in vitro P. falciparum growth inhibition. The compound 10 was also effective in vivo antimalarial agent in mice infected with Plasmodium berghei, an animal model of drug testing for human malaria infection. It is therefore concluded that the sulphonamide inhibitors targeting the parasite CA may have potential for the development of novel therapies against human malaria.
Keywords: Malaria, Plasmodium falciparum, Carbonic anhydrase, Carbonic anhydrase inhibitor, Aromatic/heterocyclic sulfonamides, Antimalarial agents , Drug target, Parasitic disease
1. Introduction
Malaria, currently one of the most deadly diseases in human, is caused by the genus Plasmodium, classified in phylum Apicomplexa of subkingdom protozoa[1]. With more than 3 billion people are at risk of Plasmodium falciparum (P. falciparum) infection, it is estimated that the disease afflicts 450 million and kills 1.5-2.7 million people each year, most of these are children under 5 years old in sub-Saharan Africa[2]–[4]. P. falciparum is responsible for the majority of deaths[2],[4]. There are now 5 Plasmodium species that infect humans, including P. falciparum, Plasmodium Vivax (P. vivax), Plasmodium falciparum (P. falciparum), Plasmodium ovale(P. ovale), and a new comer Plasmodium knowlesi(P. knowlesi)[3],[5]. Whereas P. vivax is responsible for 25%-40% of the estimated cases of malaria worldwide, with seldom fatal and often relapses after a primary infection has cleared[6]. P. malariae is found worldwide with relatively low frequency, while the least common parasites are P. ovale and P. knowlesi[5]. In addition to the lack of effective vector control and vaccine, the limitation and toxicity of antimalarial drugs in current use, and the spread of drug-resistant malaria parasites accompanied by a worldwide resurgence of malaria highlights the need to develop quickly more effective and less toxic new antimalarial drugs with different mechanism of action[7],[8]. This includes the emergence of resistance to artemisinin, the world's best drug to eradicate malaria[9]. In general, drug screening procedures have rarely been applied to this disease, and there is a paucity of information on a number of metabolic pathways that can be exploited for chemotherapy[10]. A better understanding of biochemical differences between the parasite and human, based on the first draft of P. falciparum and other human malaria parasite genome sequences, may provide new targets for intervention in the disease[5],[6],[11].
In this review, we will highlight the identification and characterization of a Zn2+-metalloenzymeα-carbonic anhydrase of human malaria parasite P. falciparum (pfCA). Aromatic/heterocyclic sulfonamides behave effective inhibitors of the pfCA with the 4-(3, 4-dichlorophenylureido)thioureido-benzenesulfonamide (compound 10) as the most effective inhibitor for in vitro P. falciparum growth. The compound 10 shows in vivo antimalarial activity against Plasmodium berghei (P. berghei) infection in mice. Thus, the sulfonamide CA inhibitors may have the potential for the development of novel antimalarial drugs.
2. Existence of carbonic anhydrase in many protozoan and helminthes parasites
The enzyme carbonic anhydrase has been identified in all organisms so far examined: animals, plants, yeast, archaea and bacteria[12]–[16]. Theα-CA isozymes of mammals, in particular the human and bovine, have been thoroughly investigated[17]–[21]. Using the bioinformatics approach[22],[23], with the amino acid sequence of all available CAs as query, many putative α-CA genes are identified in both protozoan and helminthes parasites surveyed in the public genomic databases. The amino acid sequences of these parasites are not similar to those of the mosquito (Aedes aegypti) and human CA I, II, III and VI isozymes. In silico analyses show the limited information of the putative α-CA genes in all parasite genomes, due in part to the low percentage of amino acid identity among the parasite CAs[24],[25].
In helminthes, the CA genes are identified in a parasitic nematode Ascaris suum (having only one putative gene), a free-living nematode Caenorhabditis elegans (containing six putative genes), and a filarial nematode Brugia malayi (having seven putative genes)[24]–[26]. The deduced amino acid sequences of these parasitic enzymes share very low identity among themselves. The Ascaris CA has the highest identity to the mosquito and human CA III (31%-32%), whereas the Caenorhabditis CA has only 25% identity to the human CA II and III[24],[25].
In protozoan parasites, the CA genes are identified in Plasmodium, Theileria, Trypanosoma, Leishmania and Entamoeba. The Plasmodium CA amino acid sequences have very low identity (<25%) to the mosquito and the human CA I, II, III and VI isozymes. Nevertheless, the malarial CA sequence is closely related to the Theileria enzyme, and is distinct from the other protozoan enzymes. The predicted protein sequences of the malaria parasites, however, exhibit high amino acid identity among themselves, including P. falciparum, Plasmodium chabaudi(P. chabaudi), Plasmodium yoelii(P. yoelii), and Plasmodium berghei(P. berghei)[24],[25].
3. Enzymatic catalysis, functional role and structure of carbonic anhydrase
The CAs, also known as carbonate anhydrases (EC 4.2.1.1) are Zn2+-metalloenzymes, which catalyze the reversible hydration of CO2 in forming the bicarbonate ion (HCO3−) and protons (Eq. 1). This reaction can occur in the absence of a catalyst but it is too slow[17]–[19].
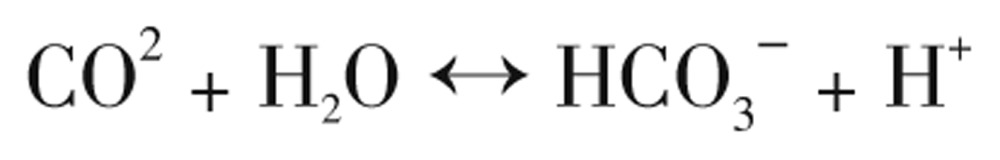 |
(Eq. 1) |
CA was first purified from bovine red cells in 1933[12], followed by the identification of several isozymes ubiquitously distributed in all organisms so far examined[13]–[19]. The CAs in protozoa and helminthes parasites are found sparsely[24]–[28]. Recent studies on the biochemistry and the crystal structure of CAs from various organisms reveal that they evolved independently al least five times, with five genetically distinct enzyme families known to date: the α-, β-, γ-, δ-, and ζ-CAs[29]–[32]. These are the α-CAs (mainly in mammals, vertebrates, some bacteria, algae, protozoa and cytoplasm of plant); the β-CAs (ubiquitously in bacteria, algae and plant chloroplast, many fungi, some archaea); the γ-CAs (predominantly in archaea and some bacteria); whereas the δ- and the ζ-CAs seem to be present only in marine diatoms)[19],[33]. In human tissues, 16 α-CA isozymes and CA-related proteins have been recently identified where they function in diverse essential processes[18], [19], [33]–[37]. In many organisms of vertebrates the CA enzymes so far examined, are involved in essential physiological processes connected with respiration and transport of CO2/HCO3−, pH, CO2 homeostasis, secretion of electrolytes in most tissues/organs, biosynthetic reactions (i.e., fatty acid synthesis, ureagenesis, pyrimidine de novo synthesis, gluconeogenesis, etc.), bone resorption, calcification, tumorigenicity, and many other physiologic or pathologic processes[17]–[19],[25]–[26].Whereas in algae, plants, and some bacteria the CA enzymes play an important role in photosynthesis and other biosynthetic pathways[18]. In diatoms, the δ- and the ζ-CAs play an important role in CO2 fixation [18],[19].
All these CA families have been crystallized and characterized in detail, except the δ-CAs. The α-, β-, and γ-CAs families bear no significant amino acid sequence identity and exhibits structural differences, except that their active sites function with a single zinc II ion (Zn2+) essential for catalysis[17],[18]. However, the γ-CAs are probably Fe2+ enzyme (but they are active also with bound Zn2+ or Co2+ ions)[19]. The catalytic and inhibition mechanism ofα-CAs is well understood[17]–[19]. X-ray crystallographic data of the human CA II, a representative of the α-CAs, show that Zn2+ is located at the bottom of a 15 A° deep active site cleft, being coordinated by three histidine residues (His 94, His 96 and His 119) and a water molecule/hydroxide ion (OH−). The histidine cluster (His 64, His 4, His 3, His 17, His 15, and His 10) is critical importance in the catalytic cycle of the enzyme (Figure 1). The overall enzyme-catalyzed reaction of CA is illustrated (Figure 2). The active form of the enzyme is maintained with hydroxide bound to Zn2+ (Figure 2A). This strong nucleophile attacks the CO2 molecule bound in a hydrophobic pocket in its neighborhood (Figure 2B), leading to the formation of HCO3− coordinated to Zn2+ (Figure 2C). The HCO3− is then displaced by a water molecule and liberated into solution, leading to the acid form of the enzyme, which is catalytically inactive with water coordinated to Zn2+ (Figure 2D). The basic form A is then regenerated by a proton transfer reaction from the active site to its environments, e.g, the active site His 64.
Figure 1. Three dimensional structure of active site of human CA II[18].

The Zn2+ ion is situated in the active-site cleft (shown as a pink sphere), and is coordinated by His94, His96 and His119 (shown in green) and a water/hydroxide ion. The histidine cluster involving in the proton-shuttling processes between the active site and the environment are also evidenced. Amino acid residues 92 and 131 involved in the binding of many sulfonamide inhibitors are shown in yellow.
Figure 2. Catalytic mechanism for a carbonic anhydrase-catalyzed reaction of H2O and CO2[25].

Details are described in text for four steps of the overall enzymatic reaction.
The type of α-CAs kinetic mechanism reveals a zinc hydroxide catalysis that also extends to the β- and γ-CA families[31],[38]. This can be summarized as,
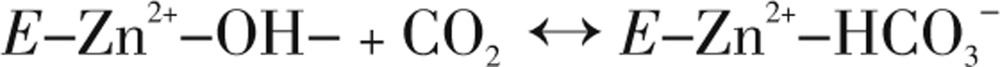 |
(Eq. 2) |
 |
(Eq. 3) |
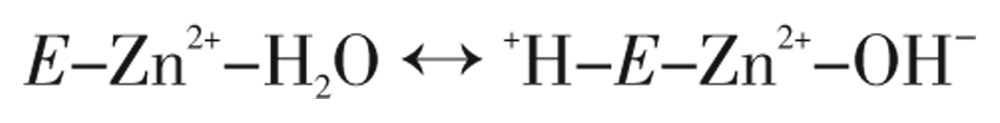 |
(Eq. 4) |
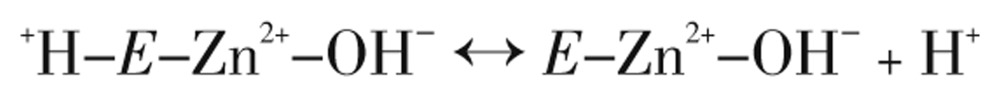 |
(Eq. 5) |
where E is the CA enzyme, and reaction 5 (Eq. 5) is the rate-limiting step in enzymatic catalysis, that is, the proton transfer that regenerates the zinc hydroxide species of the enzyme[18].
The X-ray crystallographic structures of many adducts of the CAs with aliphatic, aromatic or heterocyclic sulfonamides have been elucidated, illustrating that the sulfonamide inhibitor is directly bound to Zn2+ of the enzyme through the sulfonamide moiety[18],[19],[30],[39],[40]. The interactions between the bound inhibitor and the enzyme active site are critical for the affinity of this class of inhibitors to the different isozymes and obviously, for the design of novel drugs exploiting the sulfonamide-based structure. At present, at least 25 clinically used drugs have been reported to possess significant CA inhibitory properties [17]–[19]. Thus, CA inhibition and activation has therapeutic applications for treatment of many human diseases, for instance, Alzheimer, glaucoma, edema, obesity, cancer, epilepsy and osteoporosis[18],[19],[33],[34],[38],[40].
4. Basic life cycle, genomics and biochemistry of human malaria parasite
The term ‘malaria’ disease is derived from the Italian word ‘ma'laria’, which means bad air[41]. The disease is transmitted through the bite of an infected female Anopheles mosquito. The life cycle of the parasite is relatively complex, having several stages of its development, in both the mosquito and human hosts. In P. falciparum, the disease symptoms manifested during asexual development, include fever, chills, prostration, severe anemia, delirium, metabolic acidosis, cerebral malaria, multi-organ system failure, coma and death[3],[9].
The genome sequences of human host (size = 2 900 megabase (Mb), having about 31 000 protein-encoding genes) and Anopheles vector for malaria parasites (size = 278 Mb, comprising about 14 000 genes) are well established, whereas the genomics of three human Plasmodium species, including P. falciparum, P. vivax, and P. knowlesi, are now completed and understood in detail[5],[6],[11]. The parasite's genomes are highly polymorphisms, comparing to the human genome. The P. falciparum nuclear genome is composed of 22.8 Mb, distributed into 14 chromosomes ranging in size from 0.64 to 3.29 Mb. The parasite nuclear genome is the most (A+T)-rich sequenced to date, with an average (A+T) composition of 80.6%. The malaria parasite also contains two extra-chromosomal genomes. The mitochondrial genome is a 6 kb linear element with an (A+T) composition of 69%[42]. The circular 35 kb plastid-like genome locating in the apicoplast, a non-photosyntheic organelle, has an (A+T) composition of 86%, and encodes for 30 proteins. Comparative genomics of P. falciparum and a plant Arabidopsis thaliana shows the most similarity[11].
Pre-genomic era of biochemical studies in P. falciparum have been restricted primarily to the intra-erythrocytic stage of the life cycle, because of the difficulty of obtaining suitable quantities of parasite material from the other life-cycle stages. From the genomic analyses, identifying essential metabolic pathways for the parasite survival, 5 268 protein-coding genes are predicted. Of the predicted proteins, 733 (14%) have been identified as enzymes involved in parasite metabolisms. During the intra-erythrocytic stage, the parasite degrades up to 80% of the hemoglobin in the host cell for their amino acid requirement. The degradation occurs in a lysosomal food vacuole[10].The malaria parasite relies on anaerobic glycolysis for ATP production by obtaining glucose from the human host. The essential metabolisms, as well as biochemical pathways that are now elucidated, include the apicoplast fatty acid synthesis, synthesis of heme, isoprenoid and ubiquinone (CoQ), synthesis of amino acids, folate and other vitamin B-coenzymes biosynthesis, glycerolipid metabolism, pentose phosphate pathway, and limited functioning mitochondrial electron transport system[10],[42].
Interestingly, the malaria parasite utilizes purines and pyrimidines as precursors for DNA/RNA synthesis during its multiplication/growth in all stages of the life cycle. The parasite, known as a purine auxotroph, is incapable of de novo purine biosynthesis. It salvages the preformed purine bases/nucleosides from the human host and converts them to their mono-, di- and triphosphates. The parasite can only synthesize pyrimidines de novo from HCO3−, ATP, glutamine, aspartate, and 5-phosphoribosyl-1-pyrophosphate[43]–[53]. These unique properties on both purine and pyrimidine requirement of the parasite are key differences from the human host, in which both functional de novo and salvage pathways of the purine and pyrimidine synthesis exists[46],[48], [54]–[58].
5. Gene and protein of malaria parasite α-carbonic anhydrase
In 1998, Sein and Aikawa demonstrated the in situ CA activity for the first time in the P. falciparum-infected red cells by using electron microscopy and CA-specific Hanssen's stain[27]. During 2001-2004, we demonstrated the existence of the CA enzymatic activities in P. falciparum from in vitro culture and in the mouse parasite P. berghei[28],[59].
The P. falciparum-infected red cells contain CA activity about 2 times higher than those of normal and uninfected red cells. The three asexual developmental stages: ring (young), trophozoite (growing) and schizont (mature) show stage-dependent activity, with the specific activity of the CA enzyme increasing as the parasite matures (Table 1). The malarial enzyme belongs to the α-CAs family. The P. falciparum α-CA (pfCA), which is completely inhibited by acetazolamide (AZA), has been purified and extensively characterized. In P. berghei, a 5-fold increase in total activity of the CA enzyme in the infected, compared to the uninfected and normal mouse red cells, has been observed. At least four isozymes (namely, pbCA1, pbCA2, pbCA3, and pbCA4) were demonstrated in P. berghei[59]. The pbCA2 and pbCA3 isozymes are major forms. All four P. berghei CA activities are completely inhibited by AZA. The purified and native pfCA enzyme has a KI value of AZA higher than those of the human CA II and many bacterial CAs. Notably, the yeast CA, the plant CA and the mammalian CA III are peculiarly insensitive to AZA inhibition [60]–[67].
Table 1. Stage-dependent α-carbonic anhydrase activity in three developmental forms of P. falciparum, comparing to normal and infected red cells[28].
| Cell | Activity (unit, nmol/min) |
|
| Unit/109 cells | Unit/mg protein | |
| Normal red cell | 420 ± 35 | 0.07 ± 0.01 |
| Infected red cell (mixed stages) | 750 ± 60 | 0.20 ± 0.03 |
| Parasites | ||
| Ring | 85 ± 6 | 0.13 ± 0.01 |
| Trophozoite | 640 ± 80 | 0.28 ± 0.03 |
| Schizont | 1 540 ± 110 | 0.42 ± 0.04 |
With the available genome sequence for P. falciparum, a search for nucleotide sequences that encode CA enzymes in the malaria parasites was performed. By using the bioinformatics approach, TBLASTN searching of the genome database using protein CA sequences obtained from other organisms yields an open reading frame similar to the -CAs from various organisms, including human. The single copy CA gene was found in P. falciparum, and in three species of mouse malaria parasites; P. chabaudi, P. yoelii and P. berghei. The primary amino acid sequence of the P. falciparum gene has 47%, 42% and 40% identity with P. berghei, P. chabaudi and P. yoelii, respectively. High identity (>80%) was observed among the three rodent malarial parasites, P. chabaudi, P. yoelii and P. berghei. Low identity (<25%) of the malarial sequences are found when compared to the mosquito Aedes and human CA I, II, III and VI sequences. The active site residues responsible for binding of substrate and catalysis are, nevertheless, highly conserved among the four Plasmodium species[24],[25].
At present, only the full-length pfCA gene encoding the major form of P. falciparum CA, has been cloned and functionally expressed in Escherichia coli[59]. The recombinant pfCA protein shows authenticity to the native enzyme purified from in vitro P. falciparum culture[28]. The kinetic parameters including KM, KCAT, KI of the inhibitor AZA are also found to be similar between the native and recombinant enzymes (Table 2). The recombinant protein obtained is used for an in vitro drug-screening test for a mechanism-based drug design, especially for aromatic/heterocyclic sulfonamide CA inhibitors.
Table 2. Comparison of kinetic and inhibitory constants of human red cell CA II, native and recombinant Plasmodium falciparum α-carbonic anhydrase (pfCA).
| Enzyme | KM (mM) | KCAT (min−1) | KI AZA (nM) | KI SFA(mM) |
| Human CA II | 10.1 ± 0.8 | 74.1 ± 5.7 | 99 ± 6 | 145 ± 2 |
| Native pfCA | 3.7 ± 0.2 | 10.4 ± 1.2 | 247 ± 14 | 56 ± 4 |
| Recombinant pfCA | 5.6 ± 0.3 | 8.2 ± 1.6 | 315 ± 26 | 84 ±10 |
AZA and SFA are acetazolamide and sulfanilamide (4-aminobenzenesulfonamide), respectively[59].
6. Inhibition of malaria parasite α-carbonic Anhydrase by aromatic/heterocyclic Sulfonamides
As mentioned earlier, the pyrimidine biosynthetic pathway represents a key difference between the parasite and its human host, it constitutes an important feature for the possible targeting of pfCA for the design of novel antimalarials. Indeed, pfCA catalyzes the formation of HCO3−, serving as a substrate for the first enzyme in the pyrimidine biosynthetic pathway, carbamoylphosphate synthetase II. Thus, inhibitors of pfCA may affect the entire biosynthetic pathway, leading to antimalarials which possess a different mechanism of action as compared to the presently known drugs, most of which are rather toxic and led to the emergence of drug-resistance[3],[7],[8].
It is well established that the α-CAs are strongly inhibited by aromatic/heterocyclic sulfonamides, which bind in the deprotonated state to Zn2+ within the enzyme active site[68]–[70]. Of note, some compounds belonging to this class, such as AZA, methazolamide, dichlorophenamide or indisulam among others, are widely used pharmacological agents, mainly as diuretics, antiglaucoma, antiepileptics or anticancer agents[17]–[19],[30],[33],[34],[40]. Indisulam is in advanced clinical trials for the treatment of solid tumors[40],[68].
In our previous studies[24],[25],[71], we investigated the first library of aromatic sulfonamides, most of which were Schiff's bases derived from sulfanilamide/homosulfanilamide/4-aminoethylbenzene-sulfonamide and substituted-aromatic aldehydes, or ureido-substituted sulfonamides, some of which proved to be effective inhibitors of the parasitic enzyme pfCA. In this review, we discuss our extending the previous works for detecting potent sulfonamide CA inhibitors (CAIs) targeting malaria CAs. We focus the in vitro pfCA inhibition studies with the second library of aromatic/heterocyclic sulfonamides possessing a large diversity of scaffolds[72]. The most effective CAIs for pfCA were also assayed in vitro, for the inhibition of the parasite growth in cell cultures, as well as in vivo, in an animal model of drug testing for human malaria, i.e., mice infected with P. berghei.
The chemical structures of the new library of 34 aromatic/heterocyclic sulfonamides and AZA are shown in Figure 3. The sulfonamides 1-34 include both aromatic (benzenesulfonamides as warheads to bind the Zn(II) ion within the CA active site) as well as heterocyclic such compounds, i.e., 1,3,4-thiadiazole- and 1,3,4-thiadiazoline-2-sulfonamides. Various tails are attached to these aromatic/heteroyclic sulfonamide scaffolds, such as the perfluoroaryl/alkyl-sulfonamido-; aryl/diaryl-ureido/thioureido-; arylcarboxamido-; diethyl-dithiocarbamoylamino-; aryomatic Schiff's base, coumarinyl-3-carboxamido- as well as 7-methoxy-coumarin-4-yl-acetamido-, in order to include a large structural variation as well as physico-chemical properties to the test compounds. Indeed, these sulfonamides have been assayed earlier for the inhibition of the human CA I, II, IV and IX isozymes, showing a great variation of potency and different affinities for the various human isozymes.
Figure 3. Chemical structures of aromatic/heterocyclic sulfonamide carbonic anhydrase inhibitors 1-34 and AZA[72]. Coumarine sulfonamide derivatives are 21-32.

In vitro inhibition data of sulfonamides 1-34 against recombinant purified pfCA enzyme, in vitro data for the growth inhibition of P. falciparum in cell cultures as well as in vivo antimalarial data in mice infected with P. berghei, are shown in Table 3. Acetazolamide AZA has been included as standard sulfonamide CAI. Three antimalarial, clinically used drugs, i.e., quinine, qinghausu and chloroquine were also included in these assays as standards.
Table 3. In vitro inhibition data of pfCA (KI, µM), in vitro inhibition of growth of P. falciparum in cell cultures (IC50, µM) and in vivo antimalarial activity in P. berghei infected mice (ID50, mg/kg) of sulfonamides (1-34).
| Compound | KI (µM) | IC50 (µM) | ID50 (mg/kg) |
| 1 | 2.58 | >50 | - |
| 2 | 3.44 | >50 | - |
| 3 | 6.87 | >50 | - |
| 4 | 0.19 | >50 | - |
| 5 | 5.58 | >50 | - |
| 6 | 3.06 | >50 | - |
| 7 | 1.99 | >50 | - |
| 8 | 16.52 | >50 | - |
| 9 | 16.04 | >50 | - |
| 10 | 0.18 | 1.00 | 10.00 |
| 11 | 4.62 | >50 | - |
| 12 | 2.10 | >50 | - |
| 13 | 3.05 | >50 | - |
| 14 | 2.99 | >50 | - |
| 15 | 8.39 | >50 | - |
| 16 | 0.25 | 3.89 | No effect |
| 17 | >25 | >50 | - |
| 18 | >25 | >50 | - |
| 19 | >25 | >50 | - |
| 20 | >25 | >50 | - |
| 21 | 13.17 | >50 | - |
| 22 | 5.05 | >50 | - |
| 23 | 5.82 | >50 | - |
| 24 | 18.51 | >50 | - |
| 25 | 4.06 | >50 | - |
| 26 | >25 | >50 | - |
| 27 | 9.03 | >50 | - |
| 28 | 1.44 | >50 | - |
| 29 | 4.58 | >50 | - |
| 30 | 3.51 | >50 | - |
| 31 | 0.97 | >50 | - |
| 32 | 2.52 | >50 | - |
| 33 | 4.16 | >50 | - |
| 34 | 6.73 | >50 | - |
| AZA | 0.32 | 20.00 | No effect |
| Quinine | No inhibition | - | - |
| Qinghausu | No inhibition | - | - |
| Chloroquine | No inhibition | - | 5.00 |
Acetazolamide AZA and antimalarial drugs quinine, qinghausu and chloroquine, as standards. Compounds 10, 16 and AZA are tested at 25.0, 10.0, 5.0, 2.5 mg/kg body weight for the in vivo study. They showed no cytotoxicity in human KB and BC cells.[72]
The information of Table 3 shows the following structure activity relationship (SAR) for the inhibition of pfCA with sulfonamides 1-34:
Group I (moderate micromolar sulfonamide), several compounds among the tested sulfonamides, such as 8, 9, 17-21, 24 and 26, showed ineffective pfCA inhibitory activity, with inhibition constants (KIs) in the range of 13.17 - > 25.00 µM. They include various scaffolds, such as the N,N-diphenyl-ureas 8 and 9, the Schiff's bases 17-20 or the coumarine-3-carboxamids 21, 24 and 26. SAR is difficult to interpret in these cases also considering the fact that some structurally related derivatives to these compounds showed much better pfCA inhibitory activity.
Group II (low micromolar sulfonamide), another subgroup of derivatives, including 3, 5, 11, 15, 22, 23, 25, 27, 29, 33 and 34 showed a better pfCA inhibitory activity as compounds mentioned above, with KIs in the range of 4.16-9.03 µM. Again the structures of these medium potency inhibitors are very diverse, incorporating basically all classes of the investigated sulfonamides, such as the perfluoroaryl- (3), urea (5 and 34), thiourea (11 and 34), N,N-diethyldithiocarmaboylamino (15) coumarines (22, 23, 25, 27 and 29) as well as Schiff's base (33) derivatives. The sulfonamide heads incorporated in these CAIs include both benezenesulfonamide as well as 1,3,4-thiadiazole/thiadiazolines warheads.
Group III (submicromolar sulfonamide), the remaining derivatives, including 1, 2, 4, 6, 7, 10, 12-14, 16, 28, 30-32 and AZA were better pfCA inhibitors, showing low micromolar or submicromolar affinities for the enzyme, with KIs in the range of 0.18 - 3.51 µM. It may be observed the large variation of inhibitory activity of these compounds, when small structural variations are incorporated. For example, incorporation of an extra-methyl group in 2, as in the thiadiazoline 3, led to a 2-fold decrease of the inhibitory activity of 3 compared to 2. Replacement of the perfluorophenyl-sulfonyl moiety of 1 with the corresponding perfluoro-octylsulfonyl moiety present in 4, led to a 13.5 times gain in inhibitory activity of the later compound compared to the former one. In fact 4, together with the ureido-thiourea 10, are among the most potent pfCA inhibitors ever detected up until now. Indeed, compound 10 possess the same 3,4-dichlorophenyl-urea moiety also found in 5, but 10 has an additional thiourea fragment in its molecule, and it is 31 times more effective a pfCA inhibitor as compared to 5. Probably the more elongated shape of 10 compared to the compact 5, lead to better interactions with the enzyme active site and to this very good in vitro inhibition of pfCA. However, compound 34 possess a similar elongated shape with 10, and also the phenylureido-thiourea fragment, but 34 is a 37.4 times less effective inhibitor as compared to 10. Probably the two chlorine atoms present in 10 are crucial for the effective binding to the enzyme active site. It is also interesting to note the very effective pfCA inhibition with the Schiff's base 16, incorporating again a chlorophenyl moiety in its molecule, which is at least 100-times more effective as an enzyme inhibitor as compared to the structurally related derivatives 17-20, all quite ineffective pfCA inhibitors.
The large variations of activity were also observed for the coumarines 21-32, with only one compound 31 possessing submicromolar pfCA inhibitory activity, the vast majority of these sulfonamides being medium potency inhibitors. The standard CAI acetazolamide, AZA, behaves as a strong pfCA inhibitor too, with a KI of 0.32 µM. However, it is difficult to rationalize these data in the absence of an X-ray crystal structure of this enzyme, but we can state that small structural variations in the scaffold of our tested sulfonamides lead to very different inhibition profiles, which is noteworthy, meaning that it is possible to detect much more effective pfCA inhibitors by an intense screening effort of various libraries of structurally diverse compounds. In addition, our data also demonstrate that antimalarials such as quinine, chloroquine or qinghausu, which do not possess moieties present in CAIs (sulfonamides and their bioisosteres), are not at all pfCA inhibitors up to millimolar concentrations (Table 3). Thus, the sulfonamides investigated in our study are specifically interacting with the pfCA active site, similarly to all sulfonamides targeting α-CAs investigated earlier[17]–[19],[40].
7. In vitro and In vivo antimalarial properties of α-carbonic anhydrase inhibitors
By using [3H]hypoxanthine incorporation for monitoring growth of P. falciparum in in vitro cell culture[45], the 50% inhibitory concentration (IC50) for AZA is 20 µM. Based on the morphology examination of treated parasites in in vitro culture, the effect of AZA was more pronounced in the ring/trophozoite forms than the schizont stage of P. falciparum, as shown by clumping of nucleus and collapsing cytoplasm (Figure 4). This is consistent with the stage-dependent activity of the enzyme in that more maturing parasites contain higher activity (Table 1). Pretreatment of the human red cell with AZA, which totally abolished the host enzyme activity, show no pronounced effect on the parasite invasion. It is then concluded that the CAI directly affect the parasite carbonic anhydrase and lead to eventually death of the parasite in the host red cell.
Figure 4. Antimalarial effect of acetazolamide AZA on P. falciparum morphology during an intra-erythrocytic cycle (ring, trophozoite and schizont stages of development)[59].

The morphological abnormalities were examined in the absence (panels A, B and C; control) or in the presence of 100 µM AZA (panels D, E and F; AZA-treated culture) at various times of the parasite culture starting with ring stage development.
The most effective in vitro pfCA inhibitors, i.e, 10, 16 and AZA also showed interesting growth inhibition of in vitro culture of P. falciparum parasite (Table 3). Indeed, AZA was a rather weak antimalarial compound, with an IC50 of 20 µM, but two of the 34 newly investigated derivatives, i.e., 10 and 16, showed appreciable activity, with IC50s in the low micromolar range, of 1.00-3.89 µM. All other derivatives, irrespective of their KI values against the purified pfCA enzyme, were ineffective inhibitors for the in vitro growth of the parasite (Table 3), with IC50s > 50 µM.
Acetazolamide (as standard), chloroquine (as clinically used antimalarial drug) and the two active sulfonamides, 10 and 16, were also tested in vivo, in an animal model of human malaria, i.e., mice infected with P. berghei, for their antimalarial activity (Table 3). It may be observed that AZA and compound 16 were ineffective antimalarials in this animal model (at doses of 25.0, 10.0, 5.0 and 2.5 mg/kg body weight daily for four days, respectively) whereas chloroquine and compound 10 showed appreciable antimalarial activity, with ID50s (amount of compound protecting 50% of the test animals against malaria infection) in the range of 5-10 mg/kg. Thus, the newly investigated sulfonamide 10 has a comparable activity with chloroquine, being slightly less in vivo effective as compared to this widely used drug, but its probable mechanism of action is very different compared to that of chloroquine, involving inhibition of the malaria parasite CA.
In conclusion, a library of aromatic/heterocyclic sulfonamides possessing a large diversity of scaffolds has been assayed for inhibition of CA from the malaria parasite P. falciparum. Moderate, low micromolar and submicromolar in vitro inhibitors, were detected, whereas several compounds showed anti-P. falciparum growth in in vtro cultures. One derivative 10, 4-(3,4-dichlorophenylureido)thioureido-benzenesulfonamide was an effective in vitro pfCA inhibitor (KI of 0.18 µM), inhibited the in vitro growth of P. falciparum with an IC50 of 1 µM, and was also effective as an antimalarial agent in mice infected with P. berghei, an animal model of human malaria infection, with an ID50 of 10 mg/kg (chloroquine as standard, showed an ID50 of 5 mg/kg).
8. Concluding remarks and future directions
In malaria parasites, there are at least four species known to contain putative genes encoding the α-CAs family. One P. falciparum gene has been cloned and functionally expressed in E. coli. The recombinant enzyme is catalytically active and has authentic properties similar to the wild type enzyme purified directly from in vitro P. falciparum cultures. Our studies on the pfCA inhibitor affecting the in vitro P. falciparum growth and in vivo P. berghei infection in mouse indicate therapeutic potential use of sulfonamide CA inhibitors targeting the malaria parasite CA enzyme. This also provides that antimalarial drugs possessing a novel mechanism of action can be obtained, by inhibiting a critical enzyme in a metabolism for the life cycle of the malaria parasite, e.g., the first step of pyrimidine biosynthesis, which is the CA-mediated carbamoylphosphate synthesis. Thus, the sulfonamide inhibitors of the parasite CA may have potential for the development of novel therapies of human malaria and thus shortcut the drug resistance to the clinically used antimalarials.
Moreover, the evolutionary relationship of the parasite enzymes to other organisms is not understood. Finally, the functional roles of the enzyme in parasite metabolisms need to be further investigated. Our ultimate goal is the elucidation of the 3 D structure of the parasite CA for rationale drug design lending further insights into its differences from the equivalent enzyme in human.
Acknowledgments
The research in our laboratory was supported by the UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases, the National Science and Technology Development Agency of Thailand (Career Development Award), the Thailand Research Fund (Basic Research), and the Office of Higher Education Commission (University Staff Development Consortium), Thailand. We thank Professor CT Supuran (Florence University) for the 3 D structure of human CA II active site.
Footnotes
Foundation Project: Supported by a grant from UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (No.900142, 930143, 960103, 970074, 990490), the National Science and Technology Development Agency of Thailand (Career Development Award ID no.01-38-007), the Thailand Research Fund (Basic Research Grants ID No. BRG/13/2543, BRG4580020, BRG 4880006).
Conflict of interest statement: We declare that we have no conflict of interest.
References
- 1.Cox FEG. Modern parasitology. Oxford: Blackwell Scientific Publications; 1982. pp. p.1–346. [Google Scholar]
- 2.Attaran A, Barnes KI, Curtis C, d'Alessandro U, Fanello CI, Galinski MR, et al. WHO, the global fund, and medical malpractice in malaria treatment. Lancet. 2004;36:237–240. doi: 10.1016/S0140-6736(03)15330-5. [DOI] [PubMed] [Google Scholar]
- 3.White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hay SI, Okiro EA, Gething PW, Patil AP, Tatem AJ, Guerra CA, et al. Estimating the global clinical burden of Plasmodium falciparum malaria in 2007. Plos Medicine. 2010;7:e1000290. doi: 10.1371/journal.pmed.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pain A, Bohme U, Berry AE, Mungall K, Finn RD, Jackson AP, et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455:799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pink R, Hudson A, Mouries MA, Bendig M. Opportunities and challenges in antiparasitic drug discovery. Nature Rev Drug Discos. 2005;4:727–740. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- 8.Hopkins AL, Witty MJ, Nwaka S. Mission possible. Nature. 2007;449:166–169. doi: 10.1038/449166a. [DOI] [PubMed] [Google Scholar]
- 9.Krungkrai J, Imprasittichai W, Otjungreed S, Pongsabut S, Krungkrai SR. Artemisinin resistance or tolerance in human malaria patients. Asian Pac J Trop Med. 2010;3:748–753. [Google Scholar]
- 10.Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415:686–693. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- 11.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meldrum NU, Roughton FJW. Carbonic anhydrase: its preparation and properties. J Physiol. 1933;80:113–142. doi: 10.1113/jphysiol.1933.sp003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tashian RE. Genetic variation and evolution of the carboxylic esterases and carbonic anhydrases of primate erythrocytes. Am J Hum Genet. 1965;17:257–272. [PMC free article] [PubMed] [Google Scholar]
- 14.Smith KS, Jakubzick C, Whittam TS, Ferry JG. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc Natl Acad Sci USA. 1999;96:15184–15189. doi: 10.1073/pnas.96.26.15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujikawa AK, Nishimori I, Taguchi T, Onishi S. Human carbonic anhydrase XIV (CA14): cDNA cloning, mRNA expression, and mapping to chromosome 1. Genomics. 1999;61:74–81. doi: 10.1006/geno.1999.5938. [DOI] [PubMed] [Google Scholar]
- 16.Winum JY, Scozzafava A, Montero JL, Supuran CT. Sulfamates and their therapeutic potential. Med Res Rev. 2005;25:186–228. doi: 10.1002/med.20021. [DOI] [PubMed] [Google Scholar]
- 17.Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem. 2007;15:4336–4350. doi: 10.1016/j.bmc.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 18.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 19.Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett. 2010;15:3467–74. doi: 10.1016/j.bmcl.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Hunt JA, Ahmed M, Fierke CA. Metal binding specificity in carbonic anhydrase is influenced by conserved hydrophobic core residues. Biochemistry. 1999;38:9054–62. doi: 10.1021/bi9900166. [DOI] [PubMed] [Google Scholar]
- 21.Fisher Z, Prada JAH, Tu C, Duda D, Yoshioka C, An H, et al. Structural and kinetic characterization of active-site histidine as a proton shuttle in catalysis by human carbonic anhydrase II. Biochemistry. 2005;44:1097–1105. doi: 10.1021/bi0480279. [DOI] [PubMed] [Google Scholar]
- 22.Altschul SF, Madden TL, Schaffer AA, Zang J, Zang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krungkrai J, Krungkrai SR, Supuran CT. Malarial parasite carbonic anhydrase and its inhibitors. Curr Top Med Chem. 2007;7:909–917. doi: 10.2174/156802607780636744. [DOI] [PubMed] [Google Scholar]
- 25.Krungkrai J, Supuran CT. The alpha-carbonic anhydrase from the malaria parasite and its inhibition. Curr Pharm Des. 2008;14:631–640. doi: 10.2174/138161208783877901. [DOI] [PubMed] [Google Scholar]
- 26.Hall RA, Vullo D, Innocenti A, Scozzafava A, Supuran CT, Klappa P, et al. External pH influences the transcriptional profile of carbonic anhydrase, CAH-4b in Caenorhabditis elegans. Mol Biochem Parasitol. 2008;161:140–149. doi: 10.1016/j.molbiopara.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Sein KK, Aikawa M. The pivotal role of carbonic anhydrase in malaria infection. Med Hypotheses. 1998;5:9–23. doi: 10.1016/s0306-9877(98)90172-4. [DOI] [PubMed] [Google Scholar]
- 28.Krungkrai SR, Suraveratum N, Rochanakij S, Krungkrai J. Characterisation of carbonic anhydrase in Plasmodium falciparum. Int J Parasitol. 2001;31:661–668. doi: 10.1016/s0020-7519(01)00172-2. [DOI] [PubMed] [Google Scholar]
- 29.Tripp BC, Smith K, Ferry JG. Carbonic anhydrase: new insights for an ancient enzyme. J Biol Chem. 2001;276:48615–48618. doi: 10.1074/jbc.R100045200. [DOI] [PubMed] [Google Scholar]
- 30.Scozzafava A, Mastrolorenzo A, Supuran CT. Modulation of carbonic anhydrase activity and its applications in therapy. Expert Opin Ther Patents. 2004;14:667–702. [Google Scholar]
- 31.Tripp BC, Bell III CB, Cruz F, Krebs C, Ferry JG. A role of iron in an ancient carbonic anhydrase. J Biol Chem. 2004;279:6883–6887. doi: 10.1074/jbc.M311648200. [DOI] [PubMed] [Google Scholar]
- 32.Liljas A, Laurberg M. A wheel invented three times: the molecular structures of the three carbonic anhydrases. EMBO Reports. 2000;1:16–17. doi: 10.1093/embo-reports/kvd016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Supuran CT. Carbonic anhydrase as drug targets-an overview. Curr Top Med Chem. 2007;7:825–833. doi: 10.2174/156802607780636690. [DOI] [PubMed] [Google Scholar]
- 34.Cupuran CT, Mastrolorenzo A, Scozzafava A. Carbonic anhydrase inhibitors and activators and their use in therapy. Expert Opin Ther Patents. 2006;16:1627–1664. [Google Scholar]
- 35.Mizumori M, Meyerowitz J, Takeuchi T, Lim S, Lee P, Supuran CT, et al. Epithelial carbonic anhydrases facilitate pCO2 and pH regulation in rat duodenal mucosa. J Physiol. 2006;573:827–842. doi: 10.1113/jphysiol.2006.107581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah GN, Ulmasov B, Waheed A, Becker T, Makani S, Svichar N, et al. Carbonic anhydrase IV and XIV knockout mice: roles of the respective carbonic anhydrases in buffering the extracellular space in brain. Proc Natl Acad Sci USA. 2005;102:16771–16776. doi: 10.1073/pnas.0508449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kivela A, Kivela J, Saarnio J, Parkkila S. Carbonic anhydrases in normal gastrointestinal tract and gastrointestinal tumors. World J Gastroenterol. 2005;11:155–163. doi: 10.3748/wjg.v11.i2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther. 1997;74:1–20. doi: 10.1016/s0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 39.Chakravarty S, Kannan KK. Drug-protein interactions: refined structures of three sulfonamide drug complexes of human carbonic anhydrase I enzyme. J Mol Biol. 1994;243:298–309. doi: 10.1006/jmbi.1994.1655. [DOI] [PubMed] [Google Scholar]
- 40.Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- 41.Hyde JE. Drug-resistant malaria. FEBS J. 2007;274:4688–4698. doi: 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krungkrai J, Kanchanaphum P, Pongsabut S, Krungkrai SR. Putative metabolic roles of the mitochondria in asexual blood stages and gametocytes of Plasmodium falciparum. Asian Pac J Trop Med. 2008;1:31–49. [Google Scholar]
- 43.Krungkrai J, Cerami A, Henderson GB. Pyrimidine biosynthesis in parasitic protozoa: purification of a monofunctional dihydroorotase from Plasmodium berghei and Crithidia fasciculata. Biochemistry. 1990;29:6270–6275. doi: 10.1021/bi00478a023. [DOI] [PubMed] [Google Scholar]
- 44.Krungkrai J, Cerami A, Henderson GB. Purification and characterization of dihydroorotate dehydrogenase from the rodent malaria parasite Plasmodium berghei. Biochemistry. 1991;30:1934–1939. doi: 10.1021/bi00221a029. [DOI] [PubMed] [Google Scholar]
- 45.Krungkrai J, Krungkrai SR, Phakanont K. Antimalarial activity of orotate analogs that inhibit dihydroorotase and dihydroorotate dehydrogenase. Biochem Pharmacol. 1992;43:1295–1301. doi: 10.1016/0006-2952(92)90506-e. [DOI] [PubMed] [Google Scholar]
- 46.Krungkrai J. Dihydroorotase and dihydroorotate dehydrogenase as a target for antimalarial drugs. Drugs Fut. 1993;18:441–450. [Google Scholar]
- 47.Krungkrai J. Purification, characterization and localization of mitochondrial dihydroorotate dehydrogenase in Plasmodium falciparum, human malaria parasite. Biochim Biophys Acta. 1995;1243:351–360. doi: 10.1016/0304-4165(94)00158-t. [DOI] [PubMed] [Google Scholar]
- 48.Krungkrai J, Prapunwattana P, Wichitkul C, Reungprapavut S, Krungkrai SR, Horii T. Molecular biology and biochemistry of malarial parasite pyrimidine biosynthetic pathway. Southeast Asian J Trop Med Public Health. 2003;34(S2):32–43. [PubMed] [Google Scholar]
- 49.Krungkrai SR, Aoki S, Palacpac NMQ, Sato D, Mitamura T, Krungkrai J, et al. Human malaria parasite orotate phosphoribosyltransferase: functional expression, characterization of kinetic reaction mechanism and inhibition profile. Mol Biochem Parasitol. 2004;134:245–255. doi: 10.1016/j.molbiopara.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Krungkrai SR, Prapunwattana P, Horii T, Krungkrai J. Orotate phosphoribosyl transferase and orotidine 5′-monophosphate decarboxylase exist as multienzyme complex in human malaria parasite Plasmodium falciparum. Biochem Biophys Res Commun. 2004;318:1012–1018. doi: 10.1016/j.bbrc.2004.04.124. [DOI] [PubMed] [Google Scholar]
- 51.Krungkrai SR, DelFraino BJ, Smiley JA, Prapunwattana P, Mitamura M, Horii T, et al. A novel enzyme complex of orotate phosphoribosyltransferase and orotidine 5′-monophosphate decarboxylase in human malaria parasite Plasmodium falciparum: physical association, kinetics and inhibition characterization. Biochemistry. 2005;44:1643–1652. doi: 10.1021/bi048439h. [DOI] [PubMed] [Google Scholar]
- 52.Krungkrai SR, Kusakari Y, Tokuoka K, Inoue T, Adachi H, Matsumura H, et al. Crystallization and preliminary crystallographic analysis of orotidine 5′-monophosphate decarboxylase from human malaria parasite Plasmodium falciparum. Acta Cryst. 2006;F62:542–545. doi: 10.1107/S1744309106015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tokuoka K, Kusakari Y, Krungkrai SR, Matsumura H, Krungkrai J, Horii T, et al. Structural basis for the decarboxylation of orotidine 5′-monophosphate (OMP) by Plasmodium falciparum OMP decarboxylase. J Biochem. 2008;143:69–78. doi: 10.1093/jb/mvm193. [DOI] [PubMed] [Google Scholar]
- 54.Kanchanaphum P, Krungkrai J. Kinetic benefits and thermal stability of orotate phosphoribosyltransferase and orotidine 5′-monophosphate decarboxylase enzyme complex in human malaria parasite Plasmodium falciparum. Biochem Biophys Res Commun. 2009;390:337–341. doi: 10.1016/j.bbrc.2009.09.128. [DOI] [PubMed] [Google Scholar]
- 55.Kanchanaphum P, Krungkrai J. Co-expression of human malaria parasite Plasmodium falciparum orotate phosphoribosyltransferase and orotidine 5′-monophosphate decarboxylase as enzyme complex in Escherichia coli: a novel strategy for drug development. Asian Biomed. 2010;4:297–306. [Google Scholar]
- 56.Krungkrai SR, Wutipraditkul N, Krungkrai J. Dihydroorotase of human malarial parasite Plasmodium falciparum differs from host enzyme. Biochem Biophys Res Commun. 2008;366:821–826. doi: 10.1016/j.bbrc.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 57.Sherman IW. Biochemistry of Plasmodium (malaria parasites) Microbiol Rev. 1979;43:453–495. doi: 10.1128/mr.43.4.453-495.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheibel LW. Plasmodial metabolism: carbohydrate. In: Wernsdorfer WH, McGregor I, editors. Malaria vol. 1. New York: Churchill Livingstone; 1988. pp. 171–217. [Google Scholar]
- 59.Reungprapavut S, Krungkrai SR, Krungkrai J. Plasmodium falciparum carbonic anhydrase is a possible target for malaria chemotherapy. J Enz Inhib Med Chem. 2004;19:249–256. doi: 10.1080/14756360410001689577. [DOI] [PubMed] [Google Scholar]
- 60.Adler L, Brundell J, Falkbring SO, Nyman PO. Carbonic anhydrase from Neisseria sicca, strain 6021 I. Bacterial growth and purification of the enzyme. Biochim Biophys Acta. 1972;284:298–310. doi: 10.1016/0005-2744(72)90068-x. [DOI] [PubMed] [Google Scholar]
- 61.Brundell J, Falkbring SO, Nyman PO. Carbonic anhydrase from Neisseria sicca, strain 6021 II. Properties of the purified enzyme. Biochim Biophys Acta. 1972;284:311–323. doi: 10.1016/0005-2744(72)90069-1. [DOI] [PubMed] [Google Scholar]
- 62.Garg LC. Catalytic activity and inhibition of carbonic anhydrase of rat tissues. Biochem Pharmacol. 1974;23:3153–3161. doi: 10.1016/0006-2952(74)90601-7. [DOI] [PubMed] [Google Scholar]
- 63.Graham D, Reed ML, Patterson BD, Hockley DG, Dwyer MR. Chemical properties, distribution, and physiology of plant and algal carbonic anhydrases. Ann N Y Acad Sci. 1984;429:222–237. doi: 10.1111/j.1749-6632.1984.tb12340.x. [DOI] [PubMed] [Google Scholar]
- 64.King RW, Garg LC, Huckson J, Maren TH. The isolation and partial characterization of sulfonamide-resistant carbonic anhydrases from the liver of the male rat. Mol Pharmacol. 1974;10:335–343. [PubMed] [Google Scholar]
- 65.Shoaf WT, Jones ME. Carbonic anhydrase of microorganisms I: an enzyme from baker's yeast which catalyzes the formation of carbamate from ammonium bicarbonate solutions. Arch Biochem Biophys. 1970;139:130–142. doi: 10.1016/0003-9861(70)90054-8. [DOI] [PubMed] [Google Scholar]
- 66.Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Slow-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- 67.Soltes RE, Mulligan ME, Coleman JR. Identification and characterization of a gene encoding a vertebrate-type carbonic anhydrase in cyanobacteria. J Bacteriol. 1997;179:769–774. doi: 10.1128/jb.179.3.769-774.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enz Inhib Med Chem. 2004;19:199–229. doi: 10.1080/14756360410001689540. [DOI] [PubMed] [Google Scholar]
- 69.Lehtonen J, Shen B, Vihinen M, Casini A, Scozzafava A, Supuran CT, et al. Characterization of CA XIII, a novel member of the carbonic anhydrase isozyme family. J Biol Chem. 2004;279:2719–2727. doi: 10.1074/jbc.M308984200. [DOI] [PubMed] [Google Scholar]
- 70.Wistrand PJ, Lindqvist A. Design of carbonic anhydrase inhibitors and the relationship between the pharmacodymanics and pharmacokinetics of acetazolamide. In: Botrè F, Gros G, Storey BT, editors. Carbonic anhydrase-from biochemistry and genetics to physiology and clinical medicine. Weinheim: Willey-VCH; 1991. pp. p.352–378. [Google Scholar]
- 71.Krungkrai J, Scozzafava A, Reungprapavut S, Krungkrai SR, Rattanajak R, Kamchonwongpaisan S, et al. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: towards antimalarials with a novel mechanism of action? Bioorg Med Chem. 2005;13:483–489. doi: 10.1016/j.bmc.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 72.Krungkrai J, Krungkrai SR, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic/heterocyclic sulfonamides: in vitro and in vivo studies. Bioorg Med Chem Lett. 2008;18:5466–5471. doi: 10.1016/j.bmcl.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 73.Fan ZG, Zhang LM, Yan GG, Wu Q, Gan XF, Zhong SF, et al. Bioinformatics analysis for structure and function of CPR of Plasmodium falciparum. Asian Pac J Trop Med. 2011;4(2):85–87. doi: 10.1016/S1995-7645(11)60042-4. [DOI] [PubMed] [Google Scholar]
- 74.Fan ZG, Gang LV, Zhang LM, Gan XF, Wu Q, Zhong SF, et al. Bioinformatics analysis and prediction for structure and function of nitric oxide synthase and similar proteins from Plasmodium berghei. Asian Pac J Trop Med. 2011;4(1):1–4. doi: 10.1016/S1995-7645(11)60021-7. [DOI] [PubMed] [Google Scholar]
- 75.Tangpukdee N, Wai KM, Muangnoicharoen S, Kano S, Phophak N, Tiemprasert J, et al. Indicators of fatal outcome in severe Plasmodium falciparum malaria: a study in a tertiary-care hospital in Thailand. Asian Pac J Trop Med. 2010;3(11):855–859. [Google Scholar]
- 76.Wisedpanichkij R, Chaicharoenkul W, Mahamad P, Prompradit P, Na-Bangchang K. Polymorphisms of the oxidant enzymes glutathione S-transferase and glutathione reductase and their association with resistance of Plasmodium falciparum isolates to antimalarial drugs. Asian Pac J Trop Med. 2010;3(9):673–677. [Google Scholar]
- 77.Zhong NT, Huang FY, Tang GH, Jiao JG, Lin YZ, Wang CC, et al. Effect of hepatocyte growth factor signaling pathway activation on Plasmodium berghei infection. Asian Pac J Trop Med. 2010;3(3):169–172. [Google Scholar]
- 78.Nmorsi OPG, Isaac C, Ukwandu NCD, Ohaneme BA. Pro-and anti-inflammatory cytokines profiles among Nigerian children infected with Plasmodium falciparum malaria. Asian Pac J Trop Med. 2010;3(1):41–44. [Google Scholar]