

Abstract
Obtaining high yields of membrane proteins necessary to perform detailed structural study is difficult due to poor solubility and variability in yields from heterologous expression systems. To address this issue, an Escherichia coli-based membrane protein overexpression system utilizing an engineered bacterial outer membrane protein F (pOmpF) fusion has been developed. Full-length human receptor activity-modifying protein 1 (RAMP1) was expressed using pOmpF, solubilized in FC15 and purified to homogeneity. Using circular dichroism and fluorescence spectroscopy, purified full-length RAMP1 is composed of approximately 90% α-helix, and retains its solubility and structure in FC15 over a wide range of temperatures (20–60°C). Thus, our approach provides a useful, complementary approach to achieve high-yield, full-length membrane protein overexpression for biophysical studies.
Keywords: membrane protein expression, OmpF, circular dichroism, RAMP1
Introduction
Membrane receptors play a significant role in regulating numerous fundamental biological processes, including membrane trafficking, signal transduction, and cell–cell communication.1 Thus, it is estimated that 20–30% of all predicted proteins in sequenced genomes encode for integral membrane proteins.2 Despite their prevalence and numerous important biological roles, the number of integral membrane protein high-resolution structures available in the protein data bank (PDB) is <1%.3 The challenges in solving membrane protein structures are many, including their insolubility in aqueous solutions and low abundance in their native membrane environment. Thus, obtaining the high yields of homogeneous, soluble membrane protein required for detailed structural studies is a major challenge.4–6
Numerous approaches have been developed to improve the yield and recovery of integral membrane proteins. Among the most popular methods for large-scale, recombinant7 membrane protein expression are E. coli-based strategies utilizing protein fusions to improve yield.8–14 E. coli has several advantages for membrane protein overexpression: defined growth media is relatively inexpensive, numerous genetic tools are available for straightforward cell and target protein manipulation, and several expression conditions are established for large-scale synthesis.10,15 In particular, fusions that direct hydrophobic polypeptides into inclusion bodies have been particularly successful for cell-based membrane protein overexpression by minimizing toxicity associated with disruption of bacterial membranes.11,13,14 Both soluble and insoluble fusions have also been effective in improving membrane protein yield from cell-free expression systems.12 However, in all cases, no single fusion protein was identified that gave consistently high expression independent of the particular target membrane protein. Furthermore, the effectiveness of many fusion protein vectors is limited to expressing fragments of, rather than full-length membrane proteins. Thus, a range of vectors is typically screened initially in order to identify specific fusions that maximize membrane protein production for a new target.
To address these issues, we developed a T7-based expression vector (pOmpF) using an engineered fragment of outer membrane protein F (OmpF) as a fusion protein to direct full-length membrane protein overexpression in E. coli. OmpF is the major outer membrane porin in E. coli, and exhibits high stability due to its β-barrel structure, including eight short periplasmic β-hairpins and eight extracellular antiparallel β-strands.16,17 High-yield expression, purification, and refolding protocols have been established for OmpF, suggesting it may be an effective fusion partner to promote high-yield expression of full-length integral membrane proteins.18–20 OmpF as well as other β-barrel membrane proteins are also considerably more polar than α-helical membrane proteins, and thus the improved solubility of OmpF in aqueous solution enhances its refolding using detergents.19–21 We utilized pOmpF to overexpress human receptor activity-modifying protein 1 (RAMP1), a type I integral membrane protein co-receptor for calcitonin with calcitonin-like receptor (CLR).22 Prior work concerning RAMP1 structure has focused mainly on the extracellular domain, whereas, relatively little is known concerning the structure of the transmembrane (TM) and cytoplasmic domains.23 We demonstrate that pOmpF is able to generate high-yield expression of full-length, human RAMP1, in contrast to other fusion proteins commonly used for membrane protein overexpression, and identified conditions that promote the stability and structure of the purified co-receptor. Thus, pOmpF provides a useful, complementary tool to enable overexpression and purification of full-length integral membrane proteins such as RAMP1 for biophysical studies.
Results and Discussion
Expression plasmid pOmpF was constructed using a T7 promoter to drive overexpression in E. coli. Details of the plasmid design and construction are provided in “Materials and Methods”. The main features of the expression system are illustrated in Supporting Information Figure S1: N-terminal poly-histidine tag, an engineered OmpF fragment (amino acids A23-S164; Uniprot ID P02931), a polyglycine linker, a thrombin cleavage site, and multiple cloning sites (MCS). In the engineered OmpF fusion, the signal peptide of OmpF was removed to prevent protein trafficking to the outer membrane and direct inclusion body formation. We found that the 15.4 kDa fragment (A23-S164) including strand 7 (G157-S163) was most robust to consistent, high-yield expression, and was therefore chosen for further study. The addition of an N-terminal poly-histidine tag in-frame with the expressed protein enables purification via IMAC and immunoblotting to confirm expression from cell lysates. Moreover, the incorporation of a thrombin cleavage site allows the option of cleaving the target protein from aqueous solutions in a detergent-solubilized, native-like state. Previous work has indicated that thrombin is a suitable protease for cleavage of membrane protein fusions, and is largely insensitive to the type of detergent present when solubilizing fusion proteins.24
We generated an in-frame fusion with our engineered OmpF fragment to full-length human RAMP1 (amino acids M1-V148; Uniprot ID O60894), and compared expression to three other commonly-used fusions for membrane protein overexpression: trpLE, BclXL, and glutathione-S-transferase [pET-42a (+) expression plasmid].13,14 Fusions were expressed in LB broth using BL21 (DE3) cells for 12–16 h at a growth temperature of 20°C as described in “Materials and Methods”. As illustrated in Figure 1, RAMP1 expression was only observed as a fusion with pOmpF, with a prominent band visible by PAGE at the expected fusion MW (35.1 kDa) present in the whole-cell extract and confirmed by western blotting using anti-His antibody. Furthermore, high-yield OmpF-RAMP1 expression is observed for several different media formulations (Fig. 2), including autoinduction (ZYP growth media) at 20°C and 37°C, LB media with 1 mM IPTG at 20°C, and M9 minimal media with IPTG at 20°C.25 Thus, the engineered OmpF fragment is able to facilitate overexpression of full-length RAMP1, in contrast to other fusions used previously for membrane protein overexpression. Furthermore, OmpF fusions can be expressed in minimal media at a scale suitable for metabolic labeling and structure determination as well as in rich media to increase protein expression levels for biophysical analysis. We also successfully expressed a 2 TM fragment of the cognate co-receptor for RAMP1, CLR, using pOmpF (Supporting Information Fig. S2) under similar conditions to those described for RAMP1, further demonstrating the potential of pOmpF to produce single- and multi-pass TM proteins at a scale appropriate for biophysical studies.
Figure 1.
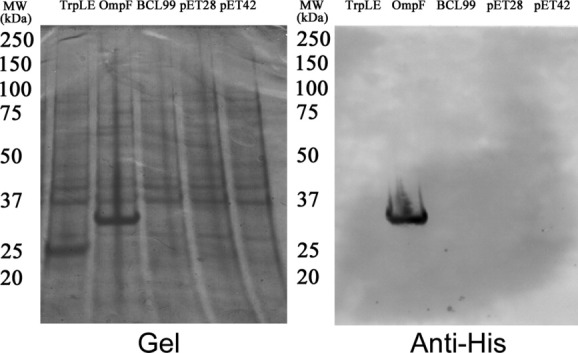
Comparison of RAMP1 expression with different fusion. Full-length RAMP1 CDS was subcloned into three different expression vectors (TrpLE, BCL99, and pET42) containing commonly used fusions for membrane protein overexpression, as well as pOmpF, which contains an engineered OmpF fragment, and pET28, which does not contain a fusion tag. RAMP1 fusions were expressed using IPTG induction from BL21 (DE3) cells for 12–16 h at a growth temperature of 20°C. A prominent band at the expected size (35.1 kDa) for the OmpF-RAMP1 fusion is observed in whole-cell lysates from pOmpF, whereas no prominent bands are observed for any of the other constructs. Immunoblotting using an anti-His antibody confirms the band at the expected size for OmpF-RAMP1 is specific, whereas no bands are observed for any of the other constructs. [TrpLE: pET-TrpLE, OmpF: OmpF, BCL99: pBCL99, pET28: pET-28a(+), and pET42: pET-42a (+)]
Figure 2.
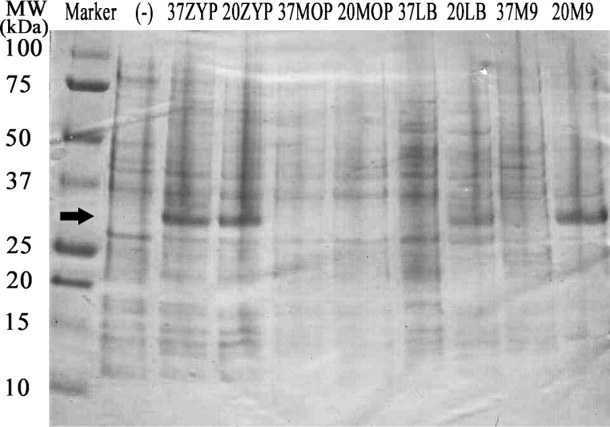
Different induction conditions for pOmpF Expression Vector. pOmpF containing full-length RAMP1 was transformed into BL21 (DE3) cells and expression induced using different growth medias, growth temperatures and inducers. Prominent bands at the expected size for OmpF-RAMP1 were observed from whole-cell lysates expressed using ZYP media at 37°C, ZYP media at 20°C, LB media + IPTG at 20°C, and M9 minimal media + IPTG at 20°C (band between 25kDa and 37kDa). [(-): Uninduced culture, 37ZYP: ZYP media at 37°C, 20ZYP: ZYP media at 20°C, 37MOP: MOPS media at 37°C, 20MOP: MOPS media at 20°C, 37LB: LB media at 37°C, 20LB: LB media at 20°C, 37M9: M9 minimal media at 37°C, and 20M9: M9 minimal media at 20°C].
As expected, the overexpressed OmpF-RAMP1 fusions were retained in the insoluble fraction of the cell after lysis using 1% Triton X-100 and sonication (Figs. 3 and Supporting Information S3). An advantage of insoluble membrane protein fusions is the ability to use differential extraction with detergents, salts and other solutes to selectively enrich for the fusion in the insoluble fraction prior to solubilization.26,27 For OmpF-RAMP1, we were able to achieve >90% pure OmpF-RAMP1 fusion protein as assessed by PAGE (Fig. 4A lane WS2) using a series of washes followed by resolubilizing the protein in zwitterionic detergent fos-choline 15 (FC15). Further details of the wash and solubilization procedure are provided in “Materials and Methods”.
Figure 3.
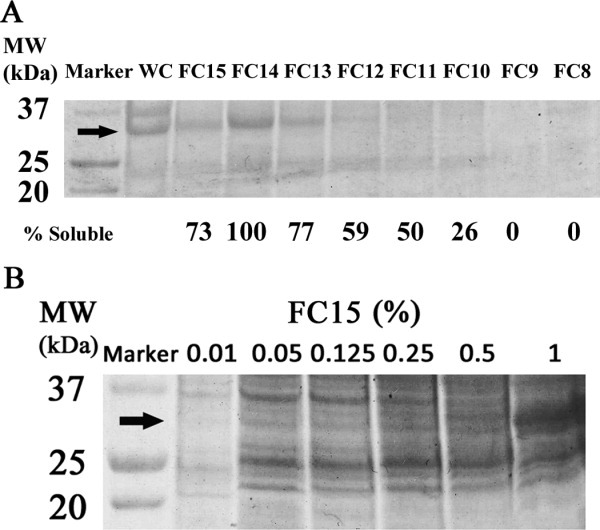
Extraction and solubilization of OmpF-RAMP1 as a function of surfactant type. (A) Induced cultures were lysed, centrifuged and the insoluble fraction mixed with a test detergent solution at 1% (w/v) concentration. After incubation, the soluble fraction was collected by centrifugation and analyzed by SDS-PAGE. OmpF-RAMP1 was highly soluble in FC15, FC14, and FC13 and moderately soluble in FC12 and FC11 (WC: whole cell lysate, FC15: FOS-CHOLINE-15, FC14: FOS-CHOLINE-14, FC13: FOS-CHOLINE-13, FC12: FOS-CHOLINE-12, FC11: FOS-CHOLINE-11, FC10: FOS-CHOLINE-10, FC9: FOS-CHOLINE-9, and FC8: FOS-CHOLINE-8). (B) OmpF-RAMP1 was solubilized in different concentration of FC15, and the minimum FC15 concentration necessary to solubilize OmpF-RAMP1 is 1% w/v. Percent solubilization of RAMP1 by a given test detergent is reported in terms of the relative ratio of RAMP1 present in the whole-cell extract versus that in a given test detergent solution.
Figure 4.

Purification of RAM1 from OmpF-RAMP1 fusion. (A) Cellular proteins from whole-cell extracts are removed by successive washes of Insoluble fraction using water (WW) and FC15 (WS) (WW1: water wash 1, WW2: water wash 2, WW3: water wash 3, WW4, water wash 4, WS1: FOS-15 buffer solubilization 1, WS2: FOS-15 buffer solubilization 2, and WS3: FOS-15 buffer solubilization 3). (B) Immobilized metal ion affinity chromatography (IMAC) was used to isolate His-tagged OmpF-RAMP1 fusion protein using a series of low imidazole washes (IMW; 10–30 mM) to remove impurities followed by high imidazole wash (IME; 100 mM) to elute bound OmpF-RAMP1 (UB: unbound, IMW1: IMAC wash 1, IMW2: IMAC wash 2, IMW3: IMAC wash 3, IMW4: IMAC wash 4, IMW5: IMAC wash 5, IME1: IMAC elution 1, and IME2: IMAC elution 2). (C) Purified protein samples were transferred to a nitrocellulose membrane, transferred proteins were visualized using Ponceau S stain and RAMP1 identified in visualized bands by immunoblotting with anti-RAMP1 antibody (Anti-RAMP1). The anti-RAMP1 immunoblot confirms RAMP1 expression in the OmpF-RAMP1 fusion and in the thrombin-cleaved sample to remove OmpF (PSUC: Ponceau S stained uncleaved OmpF-RAMP1 fusion, PSC: Ponceau S stained thrombin cleaved OmpF-RAMP1 fusion, WBUC: anti-RAMP1 blotted uncleaved OmpF-RAMP1 fusion, and WBC: anti-RAMP1 blotted thrombin cleaved OmpF-RAMP1 fusion). (D) Anion exchange chromatography (AEC) at pH 8.3 was used to isolate RAMP1 from OmpF and other proteins present in cleavage solution (UC: uncleaved, C: cleaved, UB: unbound, EXW1: AEX wash 1, EXW2: AEX wash 2, EXW3: AEX wash 3, EXW4: AEX wash 4, EXE1: AEX elution 1, and EXE2: AEX elution 2). (E) MALDI MS of purified RAMP1 from lane EXE1 in Figure 4D indicates a single peak at 17 kDa, which corresponds to MW of RAMP1. (Theoretical pI and MW: OmpF-RAMP1 [6.59 and 35.1kDa), OmpF (5.73 and 17.4kDa), RAMP1 (8.56 and 17.8kDa), Bovine Thrombin (7.05)]
A major challenge in working with membrane proteins is identifying conditions that promote both solubility and stability of the fusion and purified target membrane protein. In general, membrane proteins are only soluble in strong denaturants such as urea and SDS or organic solvents such as formic acid due to their hydrophobicity. However, harsh conditions typically used for solubilization, such 70% formic acid, 1–5% SDS or 8M urea, not only unfold the membrane protein, but may also lead to irreversible denaturation and aggregation.26,28 Thus, choice of a polar fusion protein, such as OmpF, can often enhance the overall solubility of the fusion, thereby reducing the amount of denaturant required or in some cases, enable solubilization using non-denaturing detergents. We tested the ability of a series of non-ionic, ionic, and zwitterionic detergents to effectively solubilize OmpF-RAMP1, with the expectation that mild detergents will be effective at solubilizing the fusion. Of those tested, OmpF-RAMP1 was highly soluble in the denaturing detergent SDS as well as the zwitterionic FC detergents FC13 (77%), FC14 (100%) and FC15 (73%), and moderately soluble in FC11 and FC12 (Fig. 3); percent solubilization for each sample is reported as the ratio of RAMP1 present in the detergent extract relative to the whole-cell lysate. FC detergents have been used successfully for solubilization, chromatographic purification, and biophysical characterization of a wide range of integral membrane proteins overexpressed in E. coli, including G-protein coupled receptors and OmpF.16,29 For FC15, the minimum detergent concentration required to solubilize the fusion protein was determined to be 1% (w/v) (Fig. 3B). Therefore the mild zwitterionic detergent FC15 is a suitable choice to solubilize OmpF-RAMP1 directly, without a need for harsh denaturants or high detergent concentrations.5,30
For purification, IMAC affinity chromatography was used as an initial capture step for the OmpF-RAMP1 fusion protein, followed by ion exchange chromatography to separate OmpF from RAMP1 after treatment with thrombin. Having pre-washed the OmpF-RAMP1 fusion present in the insoluble fraction to improve purity to >90% (Fig. 4A), the fusion was immobilized onto IMAC, washed with low concentrations of imidazole to remove residual impurities and eluted using 100 mM imidazole (Fig. 4B). After IMAC, the OmpF-RAMP1 fusion was essentially pure as assessed by PAGE. Thrombin treatment of the eluate was robust to the presence of FC15 and imidazole, and >90% of RAMP1 was released from OmpF after a 40-min digestion. Importantly, OmpF-RAMP1 and RAMP1 both remained soluble and stable in FC15 throughout the digest. Due to the similar sizes of RAMP1 (17.8 kDa) and OmpF (17.4 kDa), an anti-RAMP1 western blot was used to confirm the presence of full-length RAMP1 before and after thrombin treatment. As observed on nitrocellulose membranes visualized by Ponceau S stain (Fig. 4C), two prominent species between 15 and 20 kDa are present in the thrombin-treated sample. However, only one of the two species present in the thrombin-treated sample were reactive against anti-RAMP1, confirming RAMP1 was successfully removed from the OmpF fusion in a soluble, stable form. Interestingly, RAMP1 appeared to migrate faster than OmpF on a PAGE gel, which may be a result of altered detergent binding to TM helices resistant to unfolding by SDS.31 Thus, FC15 is compatible with chromatographic capture of OmpF-RAMP1 using IMAC, thrombin cleavage and stabilization of full-length RAMP1 in solution.
Anion exchange chromatography (AEC) was used to remove residual OmpF and thrombin from FC15-solubilized RAMP1. AEC was preferred based on the predicted isoelectric point (pI) of RAMP1 (8.6), which is considerably higher than that of OmpF-RAMP1 (6.6), OmpF (5.7) and thrombin (7.0). After loading the thrombin digestion mixture onto the AEC at pH 8.3, an initial isocratic wash with low NaCl (50 mM) at pH 8.3 was used to remove OmpF-RAMP1, OmpF, and thrombin. A subsequent isocratic wash at high NaCl concentration (0.5M) then enabled purification of bound, FC15-solubilized RAMP1 (Fig. 4D). Purity was assessed MALDI MS (Fig. 4E), which indicates a single species at the expected MW for full-length, human RAMP1. Additionally, using dynamic light scattering (DLS), we observe a single, monodisperse population for FC-solubilized, purified RAMP1 with an estimated hydrodynamic radius (3 nm) consistent with a RAMP1-FC15 complex (Fig. 5). From an initial culture volume of 25 mL, we are able to purify approximately 1 mg of RAMP1 solubilized in FC15. Thus, using RAMP1 as a representative integral membrane protein, we anticipate yields of purified, target membrane protein on the order of 1–10 mg purified receptor per liter culture from pOmpF, which enables membrane protein purification on a scale necessary for biophysical and structural studies.
Figure 5.
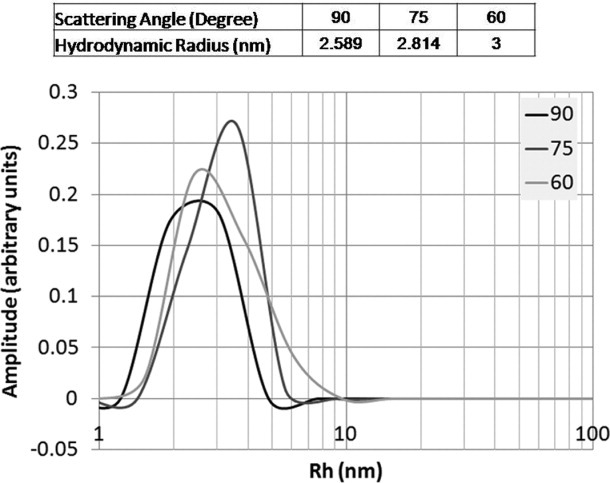
Dynamic light scattering analysis of RAMP1-FC15 complex. A homogeneous population of RAMP1 solubilized in FC15 is observed, with an estimated hydrodynamic radius of about 3 nm, which is consistent with a RAMP1 monomer solubilized in FC15.
In order to confirm purified RAMP1 is soluble and stable after purification from OmpF, we used circular dichroism (CD) to measure the secondary structure of purified RAMP1 solubilized in FC15. At 20°C, the two local minima at 208 nm and 222 nm and maximum between 190 and 195 nm indicative of α-helical secondary structure (Fig. 6).32 We also examined the thermal stability of RAMP1 secondary structure between 20 and 60°C in FC15, and observed only a modest (5%) decrease in the molar ellipticity at 195 nm and increase in the molar ellipticity at 208 nm at 60°C which was reversible upon cooling to 20°C. Importantly, no precipitation of RAMP1 was observed over the entire 20–60°C temperature range using either FC15. Thus, we conclude RAMP1 is stable in FC15 at the level of secondary structure, and the observed secondary structure is insensitive to temperature. We also measured the secondary structure and thermal stability of purified RAMP1 in 1:1 dodecyl β-D-maltoside:FC15 (Supporting Information Fig. S4) and found essentially identical results to FC15 alone, indicating the secondary structure and thermal stability of purified RAMP1 is independent of detergent type.
Figure 6.
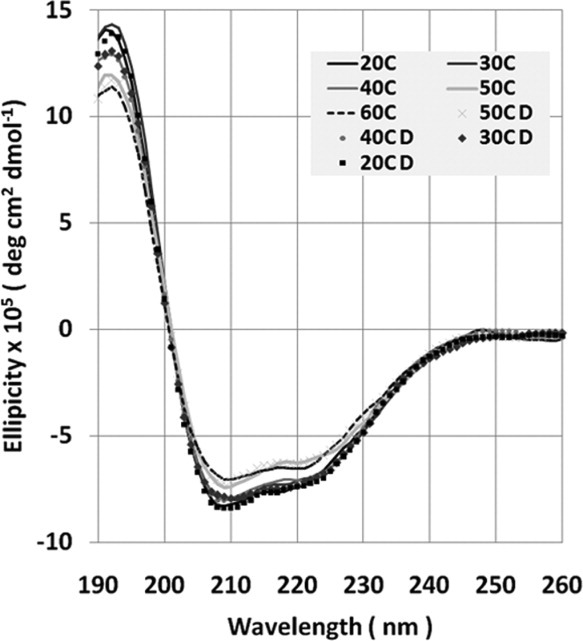
Circular dichroism spectra of purified RAMP1. All spectra represent an average of three scans. As temperature was increased to 60°C, the minima at 208 nm and 222 nm increased slightly (5%), but the overall spectra remained α-helical. The change in minima was completely reversible upon cooling samples to 20°C. (D: reducing temperature).
Additionally, we used K2D3 to extract secondary structure from the experimental spectra of purified RAMP1.33 We find full-length RAMP1 is composed of 90% α-helix, with negligible β-strand. For comparison, the previously published RAMP1 extracellular domain crystal structure (PDB 2YX8) is comprised of a three-helix bundle with approximately 81% α-helix, which is less than the estimated 90% helical content by CD for full-length RAMP1.23 Thus, we conclude the RAMP1 TM and likely cytosolic domains are also contributing to the additional α-helical secondary structure estimated by CD for full-length, TM RAMP1. Given that RAMP1 is a type I integral membrane protein with a predicted single, TM α-helix, the experimentally measured increase in helicity for full-length, TM RAMP1 versus soluble, extracellular RAMP1 is not surprising. Thus, we conclude that FC15 is an appropriate detergent for stabilization of purified, full-length RAMP1, with the measured secondary structure (90% α-helix) consistent with the expected result based on the previous crystal structure for the RAMP1 extracellular domain.23
As further confirmation that the FC15-RAMP1 complex forms a stable tertiary structure, we compared tryptophan emissions spectra for wild-type RAMP1 with and without reducing agent dithiothreitol (DTT). Compared to free tryptophan, which has an emissions maximum at 359 nm, wild-type RAMP1 is significantly blue-shifted, with an emissions maximum at 340 nm (Fig. 7), consistent with burial of extracellular tryptophan residues. Addition of DTT to wild-type RAMP1 causes a nearly 25% increase in the intensity of the tryptophan emission spectrum within 10 min; the increase in fluorescence under reducing conditions is indicative of tryptophan fluorescence quenching in the non-reduced, purified RAMP1-FC15 sample due to disulfide bond formation.34 RAMP1 is predicted to have three extracellular disulfide bonds that contribute to tertiary structure, and multiple studies of the effects of disulfide bond formation on protein tertiary structure have demonstrated that an increase in the intensity of tryptophan fluorescence upon disulfide bond reduction occurs concomitant with an increase in the partial molar volume due to loss of tertiary structure.34,35 RAMP1 has also been shown to regulate the glycosylation of its cognate co-receptor CLR, which could potentially influence the tertiary structure of RAMP1 in a RAMP1-CLR heterodimeric complex.36 However, previous studies of E. coli-expressed RAMP1-CLR extracellular domain fusions have demonstrated comparable ligand binding affinity to full-length receptors in mammalian membranes despite not being glycosylated.37 Thus, we conclude that RAMP1 assumes a tertiary structure consistent with a stable, folded state, particularly when considered along with the expected secondary structure (Figs. 6 and Supporting Information S4), monodispersity (Fig. 5), and thermal stability (Figs. 6 and Supporting Information S4) measured for RAMP1-FC15.
Figure 7.
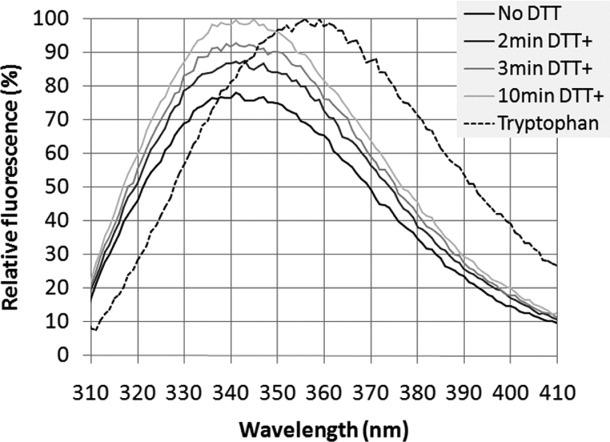
Fluorescence spectrum of wild-type and reduced RAMP1. An excitation wavelength of 295 nm was used to selectively observe the effect of tryptophan residues. Compared to the fully solvent exposed tryptophan sample (maximum at 359 nm), the fluorescence spectrum of unreduced RAMP1 is blue shifted (maximum at 341 nm), indicating burial of the extracellular tryptophan residues. Furthermore, the 25% increase in spectrum maximum in the reduced RAMP1 sample relative to unreduced RAMP1 is consistent with the presence of disulfide bonds, which when reduced, lead to increased tryptophan exposure (No DTT: no DTT added, 2min DTT+: 2 min after DTT was added, 3min DTT+: 3 min after DTT was added, 10min DTT+: 10 min after DTT was added, and Tryptophan: free tryptophan).
Conclusions
The engineered OmpF fusion (pOmpF) provides a novel, complementary vector to others used for high-yield expression and purification of full-length, integral membrane proteins from E. coli. In particular, we find that the engineered OmpF truncation enables overexpression of full-length integral membrane proteins such as the 17 kDa human RAMP1 in a form suitable for isolation and enrichment from whole-cell lysates, yet provides adequate solubility to enable extraction directly into zwitterionic detergents such as FC15 without a need for harsh denaturants. High-yield expression of human RAMP1 using pOmpF is observed from a variety of expression conditions, including minimal media suitable for metabolic labeling and enriched autoinduction broth for high-density protein expression. The ability to extract soluble, stable membrane protein fusion directly from cell lysates into FC15 simplifies subsequent purification, with a single IMAC capture step capable of purifying the fusion to near homogeneity. Furthermore, the solubility and stability of the FC15-solubilised OmpF-RAMP1 fusion enables removal of the OmpF fusion and recovery of the purified, full-length target membrane protein in yields sufficient for biophysical and structural characterization. In the case of purified, full-length RAMP1, the experimentally measured secondary structure by CD was consistent with the previous structure of full-length human RAMP1 extracellular domain, exhibited high thermal stability and tryptophan fluorescence emissions spectra consistent with a folded, stable RAMP1 tertiary structure.23 Overall, the engineered OmpF provides a useful method for robust, high-yield membrane protein expression and purification, particularly in instances where other commonly-used fusions tags are not effective at improving expression yield or conferring solubility.
Materials and Methods
Subcloning and plasmid design
The engineered OmpF sequence (amino acids A23-S164; Uniprot ID P02931) was designed to include strands 1–7 and remove the N-terminal signal peptide sequence. An optimized nucleotide sequence (Genscript) corresponding to the polyhistidine tag, fusion protein sequence, polyglycine linker, thrombin cleavage site, and MCS were subcloned into the kanamycin-resistant pET-28a(+) vector as a NcoI/XhoI fragment to construct plasmid pOmpF (Supporting Information Fig. S1). Plasmid pTrpLE containing an optimized TrpLE fragment was kindly provided by Dr. Jebrell Glover (Department of Chemistry, Lehigh University).13 Plasmid pBCL99 was generated by amplifying the corresponding 99-amino acid BclXL fragment described previously for high-level membrane protein expression from plasmid pEF6-BclXL using specific primers, and subcloned as an NcoI-XhoI fragment into plasmid pET28a(+).14,38 The human RAMP1 CDS (amino acids M1-V148; Uniprot ID O60894) was generated using overlap extension PCR with synthetic, E. coli codon-optimized oligonucleotides. The gene was inserted as a BamHI/XhoI fragment in pOmpF.
Protein expression
Auto-induction
The pOmpF-RAMP1 plasmid was transformed into BL21 cells and allowed to grow for 1 h in SOC media before 10 μL of cells were plated on a LB agar plate containing kanamycin (50 μg/mL) and grown overnight at 37 °C. The following day, individual colonies were isolated and grown in selective LB media (50 μg/mL Kan) overnight at 37 °C. The next day, saturated cell culture was diluted 100-fold into fresh, selective ZYP media and grown at 37 °C for 24 h.25 Cell pellets from induced cultures was harvested by centrifugation and stored at –20°C until further use.
IPTG induction
The pOmpF-RAMP1 plasmid was transformed into BL21 cells and grown for 1 h in SOC media. Ten mirolitre of transformed cells were then plated on a kanamycin selective (50 μg/mL) LB agar plate and grown overnight at 37°C. Colonies were then isolated and allowed to grow in selective LB media (50 μg/mL Kan) overnight at 37°C. The next day, saturated cell culture was centrifuged, spent media decanted, and the cell pellet resuspended in fresh M9 minimal media. The cell suspension was then diluted to OD600 of 0.4 in fresh M9 minimal media containing 0.5% v/v glucose and 50 μg/mL kanamycin, and grown at 20°C for 16 h to equilibrate growth temperature. Equilibrated cells were harvested by centrifugation, spent media decanted, then resuspended in fresh M9 minimal media. Equilibrated cultures were then diluted to OD600 of 0.4 in M9 minimal media containing 0.5% v/v glycerol and 50 μg/mL kanamycin, grown at 20 °C for an hour, then 1 mM IPTG added to induce protein expression. After 16–24 h, cells were collected from induced cultures by centrifugation and stored at –20 °C until further use.
Protein purification
Surfactant extraction and solubility test
Induced cultures were centrifuged (12,000 x g) to remove spent media, the pellet resuspended in a 1% (w/v) test surfactant solution, then mixed for 20 min. Samples were then centrifuged and the supernatant analyzed by PAGE gel electrophoresis using 12% acrylamide gels with 1x MES running buffer. To measure the percentage recovery of RAMP1 from the whole-cell extract (% soluble), the intensity of the band corresponding to RAMP1 in the whole-cell extract was compared to that of RAMP1 solubilized using a given test surfactant solution using the densiometry function in ImageJ. The total volume for each sample was kept constant, and the result reported as the ratio of test detergent band intensity relative to the whole-cell extract.
Inclusion body preparation
Fifty milliliter of induced culture was centrifuged (12,000 x g) for 15 min and the supernatant removed. The resulting pellet was resuspended in 50 mL of deionized water and sonicated (15 W) for 20 min to disrupt cell membranes. Next, cultures were mixed using a stir plate with agitation (200 rpm) for 20 min to release soluble proteins, and then centrifuged (12,000 x g) for 15 min. The wash procedure using deionized water was repeated a minimum of three times. After three washes, the resulting pellet containing primarily OmpF-RAMP1 was resuspended in 5 mL of FC15 buffer (1% w/v FC15, 20 mM Tris, pH 8.3), mixed using a stir plate with agitation (200 rpm) for 20 min, and centrifuged (12,000 x g) for 15 min. The supernatant, containing primarily residual contaminating proteins, was decanted, the pellet collected, and the entire FC15 wash procedure repeated at least once. To solubilize OmpF-RAMP1, the FC15-washed pellet was then resuspended in an equal volume of FC15 buffer supernatant.
Immobilized metal ion affinity chromatography (IMAC)
Five-hundred microliter of chelating sepharose Ni-NTA resin (GE Healthcare) was charged using 0.2 M NiCl2 and equilibrated with 1 mL of FC15 buffer prior to use. An equal volume of FC15-solubilized OmpF-RAMP1 solution and settled Ni-NTA resin was mixed in a 2 mL microcentrifuge tube for 20 min on a rotator mixer. The sample was centrifuged briefly to sediment the resin, and the supernatant containing unbound impurities was removed. The resin was then sequentially washed with 1 bed volume of FC15 buffer, 4 bed volumes of FC15 IMAC wash buffer 1 (FC15 buffer + 10 mM imidazole), 1 bed volume of FC15 IMAC wash buffer 2 (FC15 buffer + 20 mM imidazole), 1 bed volume of FC15 IMAC wash buffer 3 (FC15 buffer + 30 mM imidazole), and 1 bed volume of FC15 IMAC wash buffer 4 (FC15 buffer + 50 mM imidazole) to remove bound impurities. Finally, the target protein was eluted with 1 bed volume of FC15 IMAC elution buffer 1 (FC15 buffer + 100 mM imidazole) and 1 bed volume of FC15 IMAC elution buffer 2 (FC15 buffer + 300 mM imidazole).
Thrombin cleavage
One milliliter of IMAC eluate was cleaved with bovine thrombin (BioPharm Laboratories) by incubation for 40 min (5 U/mL thrombin, 2.5 mM CaCl2) with mixing at room temperature on a rotisserie.
Anion exchange chromatography (AEC)
Five-hundred microliter of SP Sepharose (GE Healthcare) was equilibrated with 1 mL of FC15 buffer, then an equal volume of thrombin-cleaved OmpF-RAMP1 solution and settled SP Sepharose was mixed for 10 min. The mixture was centrifuged briefly to sediment resin and the supernatant containing unbound protein removed. The resin was then sequentially washed with 4 bed volumes of FC15 buffer, 1 bed volume of FC15 IEX wash buffer 1 (FC15 buffer + 10 mM NaCl), 1 bed volume of FC15 IEX wash buffer 2 (FC15 buffer + 20 mM NaCl), 1 bed volume of FC15 IEX wash buffer 3 (FC15 buffer + 30 mM NaCl), and 1 bed volume of FC15 IEX wash buffer 4 (FC15 buffer + 50 mM NaCl) to remove any weakly bound material. Solublilized RAMP1 was eluted with 1 bed volume of FC15 IEX elution buffer 2 (FC15 buffer + 500 mM NaCl). NaCl was removed by dialysis against a 1000-fold excess of low-salt FC15 buffer using a 3500 MWCO dialysis membrane (Pierce).
Immunoblotting
Twenty microliter of protein sample was mixed with 5 μL of 5x Lammeli sample buffer, heated briefly at 90°C, then loaded onto a 12% acrylamide gel with MES running buffer. Samples were run for 1 h at 200 V using Lammeli running buffer, then transferred to a nitrocellulose membrane (Amersham Hybond ECL) for 90 min, blocked for 1 h at room temperature using 5% milk in TBST, then incubated with a 1000x dilution of anti-RAMP1 rabbit polyclonal antibody (ab96125, abcam) for 16 h at 4°C or a 3000x dilution of anti-His antibody (Cell Signaling) for 1 h at room temperature. The membrane was washed with TBST, exchanging solution every 5 min, followed by incubation with a 3000x dilution of goat polyclonal anti-rabbit IgG antibody (ab6721, abcam) for 1 h at room temperature or a 15,000x dilution of anti-mouse IgG antibody for 1 h at room temperature. Membranes were developed using a chemiluminescent substrate (GE) and imaged using a Typhoon imager.
Mass spectrometry
The MW of purified human RAMP1 protein was verified by MALDI-TOF mass spectrometry. Protein samples in FC15 buffer were mixed with sinapinic acid matrix (acetonitrile/water/TFA: 50/50/0.1) at a 1:1 ratio, spotted on a MSP 96 target ground steel plate (Bruker) and allowed to air dry before analysis using a Microflex mass spectrometer (Bruker Daltonics).
Circular dichroism (CD)
Spectra were collected using 400 μL of dialyzed protein samples (0.14 mg/mL) in 0.1 cm quartz cuvettes (Starna) using a J-815 CD spectrometer (JASCO) with heated sample chamber. The scanning speed was set to 500 nm/min and measurements collected from 180 to 260 nm. All CD spectra were collected in nanopure water (Millipore) containing 1% w/v FC15; this solution was also used for background subtraction. For thermal stability measurements, samples were first heated to a specific temperature (in 10° increments) from 20 to 60°C and then cooled down to 20°C. The spectrum of buffer containing 1% w/v FC15 at each temperature was used for background subtraction. Raw spectra (in mdeg) were converted to molar ellipticity (in degree cm2/dmol), and percent secondary structure estimated from the converted spectra using the K2D3 online server.33
Fluorescence measurements
Fluorescence emission spectrums (310–410 nm) were measured at 25°C using a Cary Eclipse Spectrophotometer (Agilent Technologies) with an excitation wavelength of 295 nm. Buffer conditions were identical to those used for CD measurements. For free tryptophan, a concentration of 0.01 mg/mL was used. For the disulfide bond reducing experiment, a DTT concentration of 10 mM was used.
Dynamic light scattering (DLS)
Measurements were performed using a Brookhaven Instruments spectrometer and the intensity–intensity time correlation function was measured by means of a BIO-9000 AT multichannel digital correlator. The scattering angle 90°, 75°, and 60° were used. The CONTIN method was used to analyze the normalized electric field time correlation function to determine the particle's apparent diffusion coefficient (D).39 The mean hydrodynamic radius (Rh) was calculated from the Stokes–Einstein equation:
where kb is the Boltzmann constant, T the temperature, and η the shear viscosity of the solvent. Buffer conditions were identical to those used for CD measurements.
Acknowledgments
This work was supported by a grant from the National Science Foundation (CBET-1227924). Partial support was also provided through a Faculty Research Grant (FRG) from Lehigh University.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Klemm JD, Schreiber SL, Crabtree GR. Dimerization as a regulartory mechanism in signal transduction. Annu Rev Immunol. 1998;16:569–592. doi: 10.1146/annurev.immunol.16.1.569. [DOI] [PubMed] [Google Scholar]
- 2.Wallin E, Von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heijne AE. Membrane protein structure: prediction versus reality. Annu Rev Biochem. 2007;76:125–140. doi: 10.1146/annurev.biochem.76.052705.163539. [DOI] [PubMed] [Google Scholar]
- 4.Seddon AM, Curnow P, Booth PJ. Membrane proteins, lipids and detergents: not just a soap opera. Biochim Biophys Acta. 2004;1666:105–117. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Loll PJ. Membrane protein structural biology: the high throughput challenge. J Struct Biol. 2003;142:144–153. doi: 10.1016/s1047-8477(03)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Wiener MC. A pedestrian guide to membrane protein crystallization. Methods. 2004;34:364–372. doi: 10.1016/j.ymeth.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Zapun A, Bardwell JCA, Creighton TE. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993;32:5083–5092. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
- 8.Kuliopulos A, Walsh CT. Production, purification, and cleavage of tandem repeats of recombinant peptides. J Am Chem Soc. 1994;116:4599–4607. [Google Scholar]
- 9.Leviatan S, Sawada K, Moriyama Y, Nelson N. Combinatorial method for overexpression of membrane proteins in Escherichia coli. J Biol Chem. 2010;285:23548–23556. doi: 10.1074/jbc.M110.125492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernaudat F, Frelet-Barrand A, Pochon N, Dementin S, Hivin P, Boutigny S, Rioux JB, Salvi D, Seigneurin-Berny D, Richaud P, Joyard J, Pignol D, Sabaty M, Desnos T, Pebay-Peyroula E, Darrouzet E, Vernet T, Rolland N. Heterologous expression of membrane proteins: choosing the appropriate host. PLoS ONE. 2011;6:e29191. doi: 10.1371/journal.pone.0029191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laage R, Langosch D. Strategies for prokaryotic expression of eukaryotic membrane proteins. Traffic. 2001;2:99–104. doi: 10.1034/j.1600-0854.2001.020204.x. [DOI] [PubMed] [Google Scholar]
- 12.Ishihara G, Goto M, Saeki M, Ito K, Hori T, Kigawa T, Shirouzu M, Yokoyama S. Expression of G protein coupled receptors in a cell-free translational system using detergents and thioredoxin-fusion vectors. Protein Expr Purif. 2005;41:27–37. doi: 10.1016/j.pep.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Diefenderfer C, Lee J, Mlyanarski S, Guo Y, Glover KJ. Reliable expression and purification of highly insoluble transmembrane domains. Anal Biochem. 2009;384:274–278. doi: 10.1016/j.ab.2008.09.038. [DOI] [PubMed] [Google Scholar]
- 14.Thai K, Choi J, Franzin CM, Marassi FM. Bcl-XL as a fusion protein for the high-level expression of membrane-associated proteins. Protein Sci. 2005;14:948–955. doi: 10.1110/ps.041244305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner S, Klepsch MM, Schlegel S, Appel A, Draheim R, Tarry M, Högbom M, van Wijk KJ, Slotboom DJ, Persson JO, de Gier J-W. Tuning Escherichia coli for membrane protein overexpression. Proc Natl Acad Sci USA. 2008;105:14371–14376. doi: 10.1073/pnas.0804090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kefala G, Ahn C, Krupa M, Esquivies L, Maslennikov I, Kwiatkowski W, Choe S. Structures of the OmpF porin crystallized in the presence of foscholine-12. Protein Sci. 2010;19:1117–1125. doi: 10.1002/pro.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit RA, Jansonius JN, Rosenbusch JP. Crystal structures explain functional properties of two E.coli porins. Nature. 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 18.Eisele JLJ, Rosenbusch JPJ. In vitro folding and oligomerization of a membrane protein. Transition of bacterial porin from random coil to native conformation. J Biol Chem. 1990;265:10217–10220. [PubMed] [Google Scholar]
- 19.Visudtiphole V, Thomas MB, Chalton DA, Lakey JH. Refolding of Escherichia coli outer membrane protein F in detergent creates LPS-free trimers and asymmetric dimers. Biochem J. 2005;392:375. doi: 10.1042/BJ20051257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe Y. Characterization of the refolding and reassembly of an integral membrane protein OmpF porin by low-angle laser light scattering photometry coupled with high-performance gel chromatography. J Chromat A. 2002;961:137–146. doi: 10.1016/s0021-9673(02)00540-x. [DOI] [PubMed] [Google Scholar]
- 21.Surrey T, Schmid A, Jahnig F. Folding and membrane insertion of the trimeric β-barrel protein OmpF. Biochemistry. 1996;35:2283–2288. doi: 10.1021/bi951216u. [DOI] [PubMed] [Google Scholar]
- 22.McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- 23.Kusano S, Kukimoto-Niino M, Akasaka R, Toyama M, Terada T, Shirouzu M, Shindo T, Yokoyama S. Crystal structure of the human receptor activity-modifying protein 1 extracellular domain. Protein Sci. 2008;17:1907–1914. doi: 10.1110/ps.036012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vergus JM, Wiener MC. The variable detergent sensitivity of proteases that are utilized for recombinant protein affinity tag removal. Protein Expr Purif. 2011;78:139–142. doi: 10.1016/j.pep.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Rogl H. Refolding of Escherichia coli produced membrane protein inclusion bodies immobilised by nickel chelating chromatography. FEBS Lett. 1998;432:21–26. doi: 10.1016/s0014-5793(98)00825-4. [DOI] [PubMed] [Google Scholar]
- 27.Mouillac BBJ. Mammalian membrane receptors expression as inclusion bodies in Escherichia coli. Methods Mol Biol. 2010;601:39–48. doi: 10.1007/978-1-60761-344-2_3. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Zhang L, Zhang Y, Zhang T, Feng Y, Lu X, Lan W, Wang J, Wu H, Cao C, Wang X. Highly efficient production of soluble proteins from insoluble inclusion bodies by a two-step-denaturing and refolding method. PLoS One. 2011;6:e22981. doi: 10.1371/journal.pone.0022981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren H, Yu D, Ge B, Cook B, Xu Z, Zhang S. High-level production, solubilization and purification of synthetic human GPCR chemokine receptors CCR5, CCR3, CXCR4 and CX3CR1. PLoS One. 2009;4:e4509. doi: 10.1371/journal.pone.0004509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders CR, Kuhn Hoffmann A, Gray DN, Keyes MH, Ellis CD. French swimwear for membrane proteins. ChemBioChem. 2004;5:423–426. doi: 10.1002/cbic.200300830. [DOI] [PubMed] [Google Scholar]
- 31.Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci USA. 2009;106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norden B, Rodger A, Dafforn T. Linear dichroism and circular dichroism : a textbook on polarized-light spectroscopy. Cambridge: Royal Society of Chemistry; 2010. p. 293. [Google Scholar]
- 33.Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2012;80:374–381. doi: 10.1002/prot.23188. [DOI] [PubMed] [Google Scholar]
- 34.Zapun A, Bardwell JCA, Creighton TE. The reactive and destabilizing disulfide bond of DsbA,a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993;32:5083–5083. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
- 35.Gekko K, Kimoto A, Kamiyama T. Effects of disulfide bonds on compactness of protein molecules revealed by volume, compressibility, and expansibility changes during reduction. Biochemistry. 2003;42:13746–13753. doi: 10.1021/bi030115q. [DOI] [PubMed] [Google Scholar]
- 36.Hilairet S, Foord SM, Marshall FH, Bouvier M. Protein-protein interaction and not glycosylation determines the binding selectivity of heterodimers between the calcitonin receptor-like receptor and the receptor activity-modifying proteins. J Biol Chem. 2001;276:29575–29581. doi: 10.1074/jbc.M102722200. [DOI] [PubMed] [Google Scholar]
- 37.ter Haar E, Koth CM, Abdul-Manan N, Swenson L, Coll JT, Lippke JA, Lepre CA, Garcia-Guzman M, Moore JM. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 38.Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 39.Provencher SW. A Fourier method for the analysis of exponential decay curves. Biophys J. 1976;16:27–41. doi: 10.1016/S0006-3495(76)85660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.