

Background: Bacterial ATP synthases are autoinhibited by the subunit ϵ C-terminal domain.
Results: Nucleotide hydrolysis is required to form the ϵ-inhibited state, which also responds dynamically to different ligand conditions.
Conclusion: ϵ inhibition initiates at the catalytic dwell angle, but reversible rotation over ∼40° is probably involved in nucleotide effects on the inhibitory state of ϵ.
Significance: ϵ inhibition may provide a new target for antimicrobial discovery.
Keywords: ATP Synthase, Bacteria, Enzyme Inhibitors, Escherichia coli, Protein-Protein Interactions
Abstract
F1-ATPase is the catalytic complex of rotary nanomotor ATP synthases. Bacterial ATP synthases can be autoinhibited by the C-terminal domain of subunit ϵ, which partially inserts into the enzyme's central rotor cavity to block functional subunit rotation. Using a kinetic, optical assay of F1·ϵ binding and dissociation, we show that formation of the extended, inhibitory conformation of ϵ (ϵX) initiates after ATP hydrolysis at the catalytic dwell step. Prehydrolysis conditions prevent formation of the ϵX state, and post-hydrolysis conditions stabilize it. We also show that ϵ inhibition and ADP inhibition are distinct, competing processes that can follow the catalytic dwell. We show that the N-terminal domain of ϵ is responsible for initial binding to F1 and provides most of the binding energy. Without the C-terminal domain, partial inhibition by the ϵ N-terminal domain is due to enhanced ADP inhibition. The rapid effects of catalytic site ligands on conformational changes of F1-bound ϵ suggest dynamic conformational and rotational mobility in F1 that is paused near the catalytic dwell position.
Introduction
ATP synthases play a key role in energy metabolism in most living organisms and achieve energy coupling as dual engine rotary nanomotors (1–3). The F-type ATP synthase of Escherichia coli (Fig. 1), a bacterial prototype, is composed of core subunits that all have homologs in the ATP synthases of mitochondria and chloroplasts (4). The membrane-embedded FO complex (ab2c10) acts like a turbine to transport protons across the membrane, and the external F1 complex (α3β3γδϵ) contains three cooperative catalytic sites for ATP synthesis or hydrolysis. The ring of c-subunits, with the critical proton transport sites, is the rotor complex of FO and connects to the central rotor stalk of F1, composed of γ and the N-terminal domain (NTD)2 of ϵ. The three catalytic β subunits alternate with three α subunits to surround the upper half of the asymmetric rotor stalk of γ, and the δ-b2 connection forms a peripheral stator stalk anchoring α3β3 to the other stator subunit of FO, a. In vitro, F1 from eukaryotes and bacteria can be dissociated from FO as a soluble, rotary motor ATPase, and these F1-ATPases have been useful for both mechanistic studies and the determination of high resolution structures.
FIGURE 1.

Architecture of bacterial ATP synthase and alternate conformations of subunit ϵ. The smaller image (bottom left) depicts the E. coli ATP synthase, with FO subunits spanning the membrane bilayer (shaded box); the arrow across the bilayer indicates the direction of proton (H+) transport during net ATP synthesis. FO subunits (ribbons, a (dark red), b2 (gray), and c10 (green)) and F1 subunit δ (orange ribbon) are from a homology-modeled assembly (67). All other F1 subunits are from determined structures and are surface-rendered in the FOF1 model but displayed as ribbons in the magnified view of E. coli F1 (3α (green), 3β (shades of blue), γ (yellow), ϵNTD(1–87) (light pink), and ϵCTD(88–138) (dark pink)). The FOF1 model shows ϵ in the ϵC or compact conformation (Protein Data Bank entry 1BSN), docked to γ of EF1-δ (Protein Data Bank entry 3OAA). The magnified ribbon diagram shows the ϵ-inhibited F1-δ structure (Protein Data Bank entry 3OAA) and omits the foremost α subunit to reveal the extended conformation of ϵ (ϵX); for comparison, a ribbon model of the ϵC state is shown offset to the right. The ribbon diagram of each ϵ conformation shows space-filling side-chain atoms (colored by element) predicted in silico for mutations ϵA101C/L121C. Space-filling atoms are also shown for ADP and SO42− on the one occupied catalytic β subunit (chain D). The molecular graphics were prepared with Chimera (81).
Despite general conservation between bacterial and mitochondrial ATP synthases, it has been demonstrated that bacterial ATP synthase can be an effective target for antibacterial treatment. It is the target of a novel class of compounds that are bactericidal for actively replicating and dormant mycobacteria (5, 6) and that show promising effects against multidrug-resistant tuberculosis in phase II clinical trials (7). However, the lead compound is only effective against a narrow spectrum of mycobacteria, and, because it targets the H+-transporting sites of FO, adapting this scaffold to target other pathogenic bacteria introduces a significant risk of cross-reaction with mitochondrial ATP synthase. Recently, our group determined the first crystal structure of a bacterial F1-ATPase that is in an autoinhibited state mediated by the C-terminal domain (CTD) of its ϵ subunit (8). Inhibition by ϵ may serve regulatory roles in ATP synthases of bacteria (2, 9) and chloroplasts (10) but does not occur in mitochondrial ATP synthase, which has a distinct inhibitor protein (11). Recent studies confirmed that the bacterial ϵCTD inhibits ATP synthesis as well as hydrolysis (12, 13), indicating that ϵ inhibition may provide a new target for future development of antimicrobial drugs selective for bacteria. With that in mind, the current study focuses on improving our biochemical understanding of how the catalytic F1 complex of E. coli ATP synthase is inhibited by ϵ.
As shown in Fig. 1, the ϵ subunit has two domains. The ϵNTD, essential for the F1 rotor connection to the c-ring in FO (2, 9), is a β-sandwich fold and exhibits a similar conformation and association with γ in several structures of bacterial F1 (8, 14) and mitochondrial F1 (MF1) (15, 16); essentially the same ϵNTD structure is also seen for isolated bacterial ϵ (17–19). However, the α-helical ϵCTD has been observed in dramatically different conformations (Fig. 1). A compact conformation (the ϵC state) has a coiled-coil between its two α-helices, and the second helix packs against the ϵNTD. The ϵC state has been observed for isolated bacterial ϵ (17–19) and in one bacterial F1 structure (14). In structures of MF1 (15) and MF1·c-ring (20), the homolog of ϵ appears to be locked in the ϵC state by a mitochondria-specific subunit. E. coli ATP synthase can synthesize and hydrolyze ATP when ϵ is restricted to the ϵC state (21), in which the ϵCTD does not contact any F1 subunits (Fig. 1, left). In contrast, in the recently determined structure of E. coli F1 (Fig. 1) (8), an extended conformation of the ϵCTD (ϵX state) contacts five other subunits, and its terminal half is inserted into the central cavity of F1. The position and subunit contacts of the ϵCTD within the E. coli F1 structure correlate well with many biochemical studies of ϵ inhibition and interaction with other F1 subunits (reviewed in Refs. 2 and 9). The extensive buried surface of the ϵCTD within the F1 structure and its interactions with two catalytic β subunits suggest that this form of the enzyme represents an inactive state. This correlates with results of “single-molecule” (SM) studies of F1 from E. coli (22) and other bacteria (23–26), showing that ϵ can induce or extend long “pauses” (seconds) during which γ does not rotate in the presence of substrate MgATP. Some SM studies concluded that ϵ inhibits by stabilizing or extending an ADP-induced inhibitory pause that occurs at the catalytic dwell (22, 24, 27), whereas another recently concluded that ADP- and ϵ-induced inhibitions are separate processes for cyanobacterial F1 (25). Some studies also concluded that ϵ inhibition includes or is dominated by changes to one or more intrinsic kinetic steps along the catalytic pathway (12, 22, 28, 29). In the current study, we adapt an optical assay to directly measure the kinetics of binding and dissociation for E. coli F1·ϵ and correlate these with inhibitory effects for wild type (WT) and mutant forms of ϵ. Our biochemical evidence confirms that inhibition by the CTD of ϵ initiates at the catalytic dwell but also shows that ϵ inhibition competes with formation of the ADP-inhibited state. Further, whereas ϵ inhibition initiates at the catalytic dwell, we also show that the balance between active and ϵ-inhibited states responds dynamically to changing nucleotide conditions.
EXPERIMENTAL PROCEDURES
Plasmid Constructs for Affinity-tagged E. coli ϵ Subunit
A plasmid described previously (30) (noted here as pH6ϵ) encodes a His6-tagged ϵ (H6-ϵ), with a tobacco etch virus protease cleavage site following the N-terminal His6 tag. Site-directed mutagenesis was used to create the following mutations in H6-ϵ. A pair of Cys mutations, ϵA101C/L121C, was created on pH6ϵ using the QuikChange Multi site-directed mutagenesis kit (Stratagene) with primers 5′-CATGGAAGCGA-AACGTAAGTGTGAAGAGCACATTAGGAG-3′ (ϵA101C) and 5′-GCTCAGGCGTCTGCGG-AATGCGCCAAAGCGATC-3′(ϵL121C). H6-ϵ that expresses only the ϵNTD (H6-ϵ88stop; see Ref. 31) was created with the QuikChange-II XL kit (Stratagene) (forward primer, 5′-CAATTCGCGGCCAGTAAGTCGACGAAGCG-3′). Plasmid pBKH2 was created to add a biotin acceptor peptide (Bap) before the N-terminal His6 tag on H6-ϵ. This BapH6-ϵ has 49 residues before the native initial Met of ϵ, and tobacco etch virus cleavage would yield ϵ with three extra N-terminal residues (GAM). It was created with pH6ϵ as a template, using PCR to generate a 514-bp amplicon with restriction sites added before (XhoI) and after (BamHI) the gene for H6-ϵ (primers, 5′-CGACTCGAGCATGTCGTACTA-CCATCACC-3′ and 5′-CTCGGATCCTTACATCGCTTTTTTGGTCAAC-3′). Following cleavage with XhoI and BamHI, this amplicon was cloned into the same sites of pDW363 (32), replacing the malE gene, to create pBKH2. BirA (E. coli biotin holoenzyme synthetase) is co-expressed from pBKH2, allowing in vivo biotinylation of the biotin acceptor peptide on BapH6-ϵ (32). BapH6-ϵ expressed with ϵ88stop or ϵA101C/L121C had poor protein yields, so ϵ was also expressed as a fusion protein following an N-terminal maltose-binding protein (MBP), a cleavage site for PreScission protease (GE Healthcare), and the Bap tag. This MBP-Bap-ϵ has a 31-residue segment between MBP and the initial Met of ϵ, and after cleavage by PreScission protease, Bap-ϵ would retain a 25-residue N-terminal Bap tag. The vector for this construct, pMal-PPase, was derived from pMAL-c2e (New England Biolabs), with a PreScission protease cleavage sequence after malE (33), and an NdeI site was removed by cleavage and polymerase fill-in. The sequence encoding the Bap tag was PCR-amplified from pDW363, with flanking restriction sites before (StyI) and after (NdeI and BamHI) the Bap sequence (primers, 5′-CATCCCAAGGCTGGAGGCCTGAACGATATTTTC-3′ and 5′-CTCGGATCCCATATGGCCACCAGTGTCCTCGTG-3′). After cleavage with StyI and BamHI, this amplicon was inserted into StyI-BamHI sites of pMal-PPase to generate pMAL-PP-Bap. The atpC gene for WT ϵ was extracted from p3U (34) as a 625-bp NdeI-XbaI fragment and ligated into the same sites of pMAL-PP-Bap to create pBKH8, encoding a fusion protein of MBP-Bap-ϵ. Plasmids encoding MBP-Bap-ϵ with mutation ϵ88stop (pBKH9) or ϵA101C/L121C (pBKH10) were produced in the same way, but the NdeI-XbaI inserts were 799 bp (extra sequence downstream of atpC) because those mutant atpC genes had been passed through an intermediate vector.
Purification of Proteins
SDS-PAGE (35) (Bio-Rad Ready Gels, 12% or 4–15%) was used to analyze the purity of all protein preparations, with staining by SYPRO Orange (Invitrogen) and scanning on a Typhoon 9410 imager (GE Healthcare; 488-nm laser excitation, 526-nm SP emission filter). With gels to test for internal disulfide bonding in Bap-ϵA101C/L121C, concentrated gel sample buffer contained 0.5 mm N-ethylmaleimide instead of 2-mercaptoethanol (βME). Concentration of protein samples was determined by a modified Lowry assay (36). E. coli FOF1 was expressed, and F1 was purified and depleted of subunit δ to form F1(−δ) as described (8). F1(−δ) was depleted of subunit ϵ by immunoaffinity chromatography (37), using three passages of 5–7 mg of F1−δ through an anti-ϵ column (3 ml). At this stage, upon dilution and passage through two sequential centrifuge columns, luciferase assays (8) showed that F1(−δϵ) had the following endogenous nucleotide content (mol/mol F1(−δϵ) ± S.E. from four different samples): noncatalytic sites, 0.89 ± 0.13 ATP and 0.83 ± 0.04 ADP; catalytic sites, 0.1 ± 0.08 ATP and 1.49 ± 0.17 ADP.
Wild-type and mutant forms of H6-ϵ were expressed in E. coli BL21 strain “T7 Express lysY” (New England Biolabs). H6-ϵ was purified by affinity chromatography (column with 10 ml of TALON resin; Clontech) as described (30) but with ϵ-buffer at pH 7.5 (20 mm Tris-HCl, 100 mm NaCl, pH 7.5). After loading, the column was washed with buffer + 10 mm imidazole (10 column volumes) and buffer + 15 mm imidazole (4 column volumes) before elution with buffer + 100 mm imidazole (4 column volumes). Residual impurities were removed from H6-ϵ by gel filtration (HiPrep-16/60, Sephacryl S100 HR, GE Healthcare Life Sciences), and pure H6-ϵ was exchanged into ϵ-buffer + 10% (v/v) glycerol before storage at −80 °C. For some experiments, the His6 tag was removed by treatment with 25 units/ml AcTEVTM protease (Life Technologies) protease for 6 h. The sample was then loaded on the TALON column, and untagged ϵ was collected in the flow-through, concentrated, and frozen in ϵ-buffer + 10% glycerol.
BapH6-ϵ was expressed from pBKH2 in E. coli strain BL21. Cells were grown at 22 °C in a medium containing Luria broth, biotin (12.2 mg/liter), 4.5 m sorbitol, and 1 m betaine. Protein expression was induced by adding isopropyl 1-thio-β-d-galactopyranoside when the A595 reached 0.5. Cells were grown for 4 h after induction until the A595 reached ∼1.0. BapH6-ϵ was partially purified by TALON chromatography (as for H6-ϵ). Fractionation with ammonium sulfate was then used; BapH6-ϵ that precipitated between 25 and 55% saturation was dissolved in ϵ-buffer + 10% glycerol. Finally, BapH6-ϵ was purified by gel filtration (Sephadex G-50 column, 44 cm × 1-cm diameter), concentrated by ultrafiltration (Vivaspin 6 concentrator, 5000 molecular weight cut-off), frozen in liquid N2, and stored at −80 °C.
To express biotinylated MBP-Bap-ϵ mutants, E. coli strain DH5α was co-transformed with pBirAcm (which expresses BirA; Avidity (Aurora, CO)) and either pBKH9 (MBP-Bap-ϵ88stop) or pBKH10 (MBP-Bap-ϵA101C/L121C). Each strain was grown overnight at 37 °C in LB with ampicillin (100 μg/ml) and chloramphenicol (25 μg/ml). Overnight cultures were used to inoculate 2 liters of the same medium plus 0.4% glucose and 0.1 mm biotin. Cells were grown at 37 °C to A595 ∼0.5, 0.4 mm isopropyl 1-thio-β-d-galactopyranoside was added to induce expression of MBP-Bap-ϵ and BirA, and growth continued for ∼3.5 h at 37 °C. Cells were harvested by centrifugation and washed once with column buffer (20 mm Tris-HCl, pH 7.5, 200 mm NaCl, 1 mm EDTA). Cells were lysed by sonication and centrifuged at 11,290 × g for 30 min, and the supernatant was passed through a 0.45-μm filter. This sample was mixed with 5 ml of amylose-agarose resin (New England Biolabs; pre-equilibrated with column buffer) and incubated at 4 °C, with rocking, for 2 h. The amylose resin was then sedimented (1000 × g, 3 min), the supernatant was discarded, and the resin was washed five times by centrifugation with 40 ml of column buffer + 5 mm βME. For the ϵA101C/L121C mutant, 5 mm βME was present throughout purification. The resin was incubated with 0.1 mg of PreScission Protease (4 °C, 3 h, in 15 ml of column buffer + 5 mm βME) to release Bap-ϵ, which was collected in the supernatant and in a subsequent wash of the resin with 10 ml of column buffer + 5 mm βME. Ultrafiltration (Vivaspin-20, 5000 molecular weight cut-off) was used to concentrate the Bap-ϵ from 25 to 1 ml and exchange it into ϵ buffer + 10% glycerol, including 1 mm βME for Bap-ϵA101C/L121C. Concentrated Bap-ϵ was frozen in liquid N2 and stored at −80 °C. For some experiments, His6- or Bap-tagged ϵA101C/L121C was treated with 5,5′-dithiobis(2-nitrobenzoate) (DTNB) to induce disulfide bonding between closely approaching cysteines (38). The sample was first passed through a Biogel P6 centrifuge column (39) (pre-equilibrated with 20 mm MOPS-Tris, 50 mm KCl, pH 8 (MTK8)) to remove βME. Tagged ϵA101C/L121C (∼30 μm) was then incubated with 50 μm DTNB for 15 min or less at room temperature and passed through a second centrifuge column to remove excess DTNB.
ATPase Assays
A coupled enzyme assay (40) was used for continuous monitoring of ATP hydrolysis, and assays were done at 30 °C. Decrease in NADH concentration was monitored at 340 nm in a Hewlett-Packard 8453 spectrophotometer. The standard assay buffer was MTK8 supplemented with 1 mm phosphoenolpyruvate and 0.3 mm NADH. MgATP substrate was added from stock solutions of Mg acetate and Na2ATP; concentrations and ratios of Mg2+/ATP are noted for specific experiments. Pyruvate kinase (rabbit muscle, Roche Applied Science catalog number 109045) and lactate dehydrogenase (Porcine heart, Calbiochem catalog number 427211) were each present at 0.1 mg/ml in assays with excess Mg2+ versus ATP; for assays with excess ATP versus Mg2+, pyruvate kinase was 0.2 mg/ml. In some assays, hydrolysis of GTP was measured rather than ATP. For assays measuring inhibition by ϵ, BSA (fatty-acid free) was added at 0.5 mg/ml, EDTA was added to 0.1 mm, and F1(−δϵ), ϵ, and ATP were added to final concentrations and preincubated at 30 °C for 10 min; the assay was initiated by adding magnesium acetate from a concentrated stock and mixing. Hydrolysis rates were measured at steady state, typically 12–15 min after adding Mg2+. For each data set varying ϵ concentration, a fixed concentration of F1(−δϵ) was used (FT = 0.6 or 1.2 nm), and hydrolysis rates were fit by nonlinear regression (Prism, GraphPad, Inc.) to the following equation,
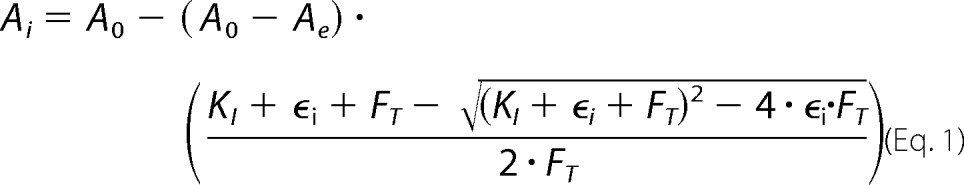 |
where A0 is the rate measured for F1(−δϵ) alone, Ai is the rate measured at each ϵ concentration, ϵi, Ae is the rate fitted for ϵ-saturated F1(−δϵ), and KI is the apparent dissociation constant fitted for F1·ϵ binding. For assays of inhibition by azide, magnesium acetate, ATP, sodium azide, and ϵ (if any) were added first, and the assay was initiated by adding F1(−δϵ) from a concentrated stock; steady-state rates were measured as above.
Kinetic Assays of Binding and Dissociation between F1(−δϵ) and Biotinylated ϵ, Using Biolayer Interferometry (BLI)
An Octet-RED system and streptavidin-coated sensors (FortéBio, SA biosensors, catalog number 18-5019) were used to monitor BLI kinetics of protein-protein binding and dissociation, analogous to surface plasmon resonance techniques (41). MTK8 buffer included 0.5 mg/ml BSA to minimize nonspecific binding of proteins to the sensors and as carrier protein for nanomolar dilutions of F1(−δϵ) or ϵ. All steps were done at 30 °C, with each sensor stirred in 0.2 ml of sample at 1000 rpm and a standard measurement rate of 5 s−1. Wild-type or mutant forms of biotinylated ϵ (BapH6-ϵ, or Bap-ϵ after cleavage from MBP-Bap-ϵ) were immobilized on SA biosensors. Levels of in vivo biotinylation varied between ϵ samples, so preliminary BLI titrations were done for each biotinylated ϵ to determine the Bap-ϵ concentration and loading time needed for optimal BLI kinetic responses in subsequent binding of F1(−δϵ) to the ϵ-loaded sensors. Minimal loading of ϵ was found to be favorable for kinetic responses to F1(−δϵ) binding, so most subsequent experiments loaded biotinylated ϵ to yield 0.2–0.4 nm of BLI signal per sensor. Use of reference sensors with immobilized biotinylated ϵ but without added F1(−δϵ) confirmed that added ligands (nucleotides, Mg2+, EDTA, and azide) did not alter the BLI signal for immobilized ϵ. To correct for BLI base-line drift and minimal nonspecific binding of F1(−δϵ) to sensors, all BLI experiments included one or more reference sensors in parallel for which biotinylated ϵ was omitted from the ϵ-loading step, but F1(−δϵ) was included in the association step, usually at the highest concentration of F1(−δϵ) used for each experiment. FortéBio's analysis software (version 6.4) was used for reference subtraction, Savitsky-Golay filtering, and global fitting of kinetic rates for F1·ϵ binding and dissociation.
RESULTS
Inhibition of E. coli F1 by ϵ with and without the ϵCTD
Upon in vitro dissociation of E. coli F1 from the membrane, ϵ becomes more inhibitory but can dissociate upon dilution of F1, relieving inhibition of F1-ATPase activity (2). For most experiments in this study, we used F1 that was depleted of δ and ϵ subunits, or F1(−δϵ). The stator subunit δ does not significantly affect F1-ATPase activity (42) but was removed because its dissociation from F1 could interfere with assays below for F1·ϵ binding and dissociation. Fig. 2 compares inhibition of F1(-δϵ) by WT and mutant forms of H6-ϵ, and Table 1 summarizes the inhibition parameters from regression curves of Fig. 2 and an additional data set. As noted before (30), the N-terminal His6 tag on WT ϵ did not significantly alter inhibition compared with WT ϵ that had the tag removed (Table 1). Also, inhibition was not altered by the N-terminal Bap tag added to ϵ (WT and mutants) for kinetic assays of F1·ϵ binding and dissociation (not shown). For WT ϵ, values for the inhibitory constant KI and residual activity of ϵ-saturated F1 agree with earlier estimates (29, 43). We obtained nearly the same parameters for ϵ inhibition in assays with ATP < Km (not shown), consistent with noncompetitive inhibition by ϵ versus ATP (29, 44). We also show that the >90% inhibition by saturating WT ϵ was unaffected by excess Mg2+ (Fig. 2A), although F1(−δϵ) alone was inhibited >50% by 1 mm excess Mg2+ (Table 1).
FIGURE 2.

Effects of truncating or cross-linking the ϵCTD on inhibition of F1(−δϵ). A, F1(−δϵ) was preincubated for 10 min with 0.1 mm EDTA and ATP (black and orange symbols) or GTP (green symbols) and the indicated concentrations of wild-type H6-ϵ (♦) or H6-ϵ88stop (●). Hydrolysis was initiated by adding magnesium acetate. Final concentrations of added ligands were as follows: 2:1 mm ATP/Mg2+ (black); 1:2 mm ATP/Mg2+ (orange); 1:2 mm GTP/Mg2+ (green). B, ATPase assays as for A, but H6-ϵA101C/L121C was used with 1 mm DTT present (▴) or with an ϵA101C–L121C disulfide bond (■). See “Experimental Procedures” for assay details and regression analysis. With H6-ϵ88stop, data points are averages from three (ATP) or two (GTP) experiments, and standard error bars are included but are smaller than the symbols for most points. Results of regression analyses for these and for a data set with untagged WT ϵ are summarized in Table 1.
TABLE 1.
Inhibition of F1(−δϵ) by variants of subunit ϵ
Results of nonlinear regression for data shown in Fig. 2 and a data set with untagged WT ϵ. Equation 1 was used (see “Experimental Procedures”).
| ϵ | NTP/Mg2+ ratio | Activity of F1(−δϵ) (A0)a | Activity of ϵ-saturated F1 (Ae/A0)b ± S.E.c | KI ± S.E.c |
|---|---|---|---|---|
| mm | μmol min−1 mg−1 | % | nm | |
| Wild type | ATP 2:1 | 78 | 7.9 ± 0.6 | 0.49 ± 0.02 |
| Wild-type H6-ϵ | ATP 2:1 | 78 | 6.8 ± 0.8 | 0.67 ± 0.04 |
| ATP 1:2 | 37 | 7.9 ± 0.5 | 0.46 ± 0.02 | |
| GTP 1:2 | 94 | 7.3 ± 0.7 | 0.87 ± 0.04 | |
| H6-ϵ88stop | ATP 2:1 | 73 | 76.3 ± 1.2 | 12.4 ± 1.9 |
| ATP 1:2 | 36 | 54.6 ± 1.4 | 7.2 ± 0.8 | |
| GTP 1:2 | 95 | 84 ± 2.0 | 13 ± 7 | |
| H6-ϵ101C/121C, + 1 mm DTT | ATP 2:1 | 80 | 31 ± 1 | 1.2 ± 0.1 |
| ATP 1:2 | 41 | 27 ± 1 | 0.98 ± 0.06 | |
| H6-ϵ101C/121C, disulfide-bonded | ATP 2:1 | 80 | 69 ± 2 | 23 ± 3.7 |
| ATP 1:2 | 38 | 58 ± 1 | 31 ± 2 |
a Hydrolysis activity units are μmol·min−1·mg−1 F1(−δϵ).
b Activity of ϵ-saturated F1(−δϵ), Ae, is listed as a percentage of A0, the measured activity of F1(−δϵ) alone.
c S.E., standard error from nonlinear regression.
To test for inhibition by ϵ lacking its CTD, we used ϵ88stop, one of the largest C-terminal deletions that still allows assembly of FOF1 that is functionally coupled, both in vivo and in vitro (31). In Fig. 1, both conformations of ϵ are colored darker for the C-terminal region that is absent in ϵ88stop. As shown in Fig. 2A, H6-ϵ88stop caused much less inhibition than WT H6-ϵ and had a >15-fold larger KI, confirming that the ϵCTD is responsible for the majority of inhibition. However, unlike WT H6-ϵ, the maximal extent of inhibition by H6-ϵ88stop almost doubled to ∼45% in the presence of excess free Mg2+. Inhibitory effects of free Mg2+ are linked to inhibitory MgADP bound at a catalytic site on F1 from E. coli (45, 46), from other bacteria (47), from mitochondria (48, 49) and chloroplasts (50, 51), and hydrolysis of GTP is less sensitive to this type of inhibition (45, 48, 52). For example, with 1 mm excess Mg2+, GTPase turnover by F1(−δϵ) is ∼2.5-fold faster than ATPase (Table 1). We show that WT H6-ϵ exhibits similar high affinity inhibition for GTPase and ATPase (Fig. 2A and Table 1). However, H6-ϵ88stop inhibited GTPase much less, ∼16% maximal, both with excess free Mg2+ present (Fig. 2A) and without it (not shown). Thus, observed partial inhibition of ATPase by the ϵNTD is largely due to increased MgADP inhibition in the absence of the ϵCTD. This can also explain why ϵ truncated after ϵ94 (with only about half of the first helix remaining) inhibited E. coli F1 ∼50% because the assays contained 2 mm excess free Mg2+ (53). The effects of the ϵNTD are distinct from the >90% inhibition caused by the ϵCTD of intact WT ϵ, which is not sensitive to the effects of excess Mg2+.
As an alternative to removing the ϵCTD, we also used the ϵA101C/L121C mutant (21).3 This cysteine pair can form a disulfide bond in nearly 100% yield (Fig. 3) that cross-links the two α-helices of the ϵCTD in the ϵC conformation, preventing ϵ from switching to the ϵX conformation (see Fig. 1). As shown in Fig. 2B, this disulfide linkage prevented high affinity inhibition of F1(−δϵ) as effectively as removing the ϵCTD. However, the partial inhibition observed was less sensitive to excess free Mg2+ than with H6-ϵ88stop; this suggests that ϵCTD/ϵNTD interactions in the ϵC state can influence interactions of ϵNTD with γ that alter catalytic behavior. With DTT present to prevent the disulfide bond, H6-ϵA101C/L121C could access the ϵX state and showed high affinity inhibition (KI ∼1 nm), similar to that with WT H6-ϵ (KI ∼0.5 nm). However, the activity of F1 saturated with reduced H6-ϵA101C/L121C was 4-fold greater than with WT H6-ϵ. In the structure of ϵ-inhibited F1 (8), ϵLeu-121 is in a coiled-coil interface with the γ N-terminal helix, and the ϵL121C mutation probably perturbs this interface, favoring more F1 complexes in the active state on average. This supports the concept that, with ϵ-saturated F1, the residual ATPase activity (7–8% with WT ϵ) is due to the time-averaged fraction of F1 complexes in which ϵ is not in the inhibitory ϵX conformation.
FIGURE 3.
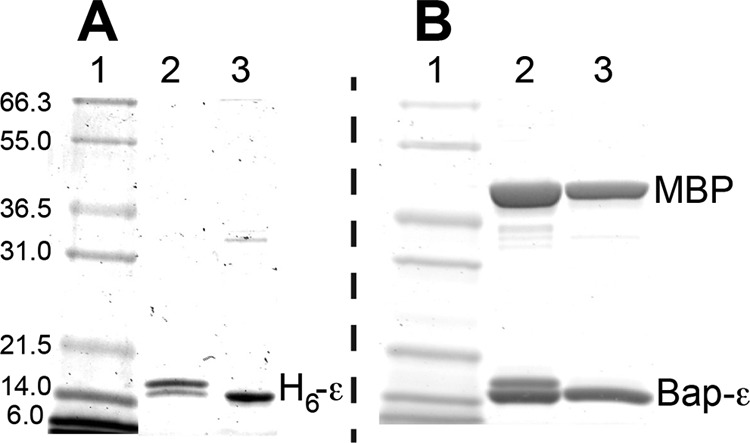
SDS-PAGE analysis of disulfide bond formation with ϵA101C/L121C. Samples were applied to 12% SDS-PAGE under non-reducing conditions. Lane 1 of each gel, molecular mass markers (approximate kDa noted for gel A). Gel A, samples of H6ϵA101C/L121C (1 μg/lane) were taken immediately after DTT was removed from the sample by centrifuge column (lane 2) or after 2 min of reaction with 50 μm DTNB (lane 3). Gel B, samples of (MBP-cleaved) Bap-ϵ (5 μg/lane) were taken after removing DTT as above (lane 2) or after reaction with 50 μm DTNB for 15 min (lane 3). With 2-mercaptoethanol in the gel sample buffer (not shown), each ϵA101C/L121C migrated only at the upper band position (above 14 kDa standard), confirming that the lower band seen here was a faster migrating band due to the internal ϵA101C–L121C disulfide bond.
Kinetics of F1·ϵ Binding and Dissociation, Assayed by BLI
In preliminary assays, H6-ϵ was loaded on BLI sensors coated with Ni2+-nitrilotriacetic acid, but slow dissociation of WT H6-ϵ from the sensors prevented accurate measures of the slow dissociation rate of F1 from WT H6-ϵ. To achieve more stable and specific attachment of ϵ to the sensor surface, ϵ was engineered with an N-terminal Bap tag, so that a specific lysine could be biotinylated in vivo (32). Biotinylated Bap-ϵ could be stably bound to streptavidin-coated sensors (supplemental Fig. S1), and BLI was then used to measure binding and dissociation kinetics of F1(−δϵ). For each Bap-ϵ variant, 4–7 sensors were used in parallel, with F1(−δϵ) concentrations varied ≥10-fold in the association samples, and association/dissociation kinetics were fit globally to determine the rate constants (Table 2). KD values derived from the rate constants correlate well with inhibitory KI values (Table 1) for WT ϵ, ϵ88stop, and disulfide-bonded ϵA101C/L121C. Representative kinetics for binding/dissociation of F1(−δϵ) with sensors containing WT ϵ or ϵ88stop are shown in Fig. 4. The ϵCTD did not significantly alter the association rate, indicating that only ϵNTD/γ interactions are involved in initial F1·ϵ binding. In contrast, removing the ϵCTD (Fig. 4) or preventing it from adopting the ϵX state (disulfide-bonded ϵA101C/L121C) increased the dissociation rate by ≥80-fold (Table 2). For sensors loaded with biotinylated WT BapH6-ϵ, only a small fraction of bound F1(−δϵ) could be observed to dissociate in buffer only (Fig. 4), but results presented below show that essentially all F1(−δϵ) on the sensor is reversibly bound. Additional assays (not shown) included excess, non-biotinylated WT H6-ϵ in the dissociation phase and confirmed that the observed, slow dissociation rate was not due to rebinding of F1(−δϵ) to the sensor. Thus, the much slower dissociation of F1(−δϵ)/WT-ϵ is probably due to strong bias of bound WT ϵ to reside in the ϵX state, with the ϵCTD buried within F1. However, from the KD values (Table 2), note that the ϵCTD contributes only ∼20% to the net free energy for F1·ϵ binding (ΔΔG, −10 or −12 kJ/mol for WT ϵ versus ϵ88stop or disulfide-bonded ϵA101C–L121C, respectively). This does not mean the ϵX state of ϵCTD has only weak interactions with other F1 subunits; rather, the small contribution to net binding energy is probably due to the loss of favorable interactions between γ and α3β3 that are blocked by insertion of the ϵCTD.
TABLE 2.
Binding/dissociation rates and KD values for F1(−δϵ) with variants of subunit ϵ
| Biotinylated ϵ | ka ± S.E.a | kd ± S.E.a | KD |
|---|---|---|---|
| m−1 s−1 | s−1 | nm | |
| Wild type | 2.0 × 105 ± 0.1% | 4.8 × 10−5 ± 0.1% | 0.24 |
| Wild type, +1 mm EDTA/ATP | 2.3 × 105 ± 0.1% | 4.1 × 10−3 ± 0.1% | 17 |
| ϵ88stop | 3.1 × 105 ± 0.6% | 3.8 × 10−3 ± 0.4% | 12.2 |
| ϵΑ101C/L121C, disulfide-bonded | 2.1 × 105 ± 0.3% | 6.6 × 10−3 ± 0.2% | 32 |
a S.E., standard error from global fitting analysis (presented as a percentage of the parameter's value).
FIGURE 4.

Binding and dissociation kinetics for F1(−δϵ) and sensor-bound biotinylated ϵ, with or without ϵCTD. Biotinylated Bap-ϵ (wild type or ϵ88stop, as noted) was loaded onto streptavidin biosensors. At time 0, each sensor was transferred from buffer alone to a sample containing 15 nm F1(−δϵ). After 900 s (vertical dashed line), each sensor was moved into buffer without F1(−δϵ). Black lines, experimental data; red lines, the kinetic fits for each ϵ, from global regression of kinetic data at varied concentrations of F1(−δϵ). Kinetic parameters and KD values derived from the fittings are summarized in Table 2.
Effects of F1 Ligands (Mg2+, Nucleotides, Pi) on Conformational Bias of Bound, Full-length ϵ
From the crystal structure of ϵ-inhibited F1 (8), the extensive surface area of ϵCTD that is buried within the central cavity of F1 suggests that the ϵX state of ϵ does not directly dissociate from F1; the slow dissociation observed in Fig. 4 probably occurs due to dynamic transition of ϵ between ϵX and conformations like ϵC in which the ϵCTD is outside of the central rotor cavity. Thus, factors that influence the fraction of F1 complexes with ϵ in the ϵX state should alter the kinetics of F1·ϵ dissociation. To show that the conformation of ϵ on E. coli F1 and FOF1 can be influenced by nucleotides and other ligands that interact with catalytic sites, early studies used static assays, such as the capacity to form a β-ϵ cross-link; cross-linking of β-ϵ was minimized by non-hydrolysis conditions, such as ATP/EDTA or MgAMPPNP but maximized by post-hydrolysis conditions (MgADP/Pi) (54, 55). The β-ϵ cross-linking residues (56) are within hydrogen-bonding distance in the structure of ϵ-inhibited F1 but should be at least 35 Å apart with ϵ in the ϵC state (8). Here, we use the BLI assay for F1·ϵ binding/dissociation for more dynamic analyses of how different ligands may shift the conformation of F1-bound ϵ between the ϵX state and other conformations of the ϵCTD that allow faster F1·ϵ dissociation. In control assays (not shown), the various ligands tested did not alter the rate at which F1(-δϵ) dissociated from biotinylated ϵ88stop, confirming that the ligand effects are specific to the ϵCTD of WT ϵ. F1(−δϵ) was bound in parallel to multiple sensors with biotinylated WT ϵ, and Fig. 5 shows dissociation of F1(−δϵ) when sensors were exposed to different ligands. For Fig. 5A, F1(−δϵ) was bound to all sensors in MTK8 buffer + BSA, and dissociation in this buffer was slow (Fig. 5A, curve 4, ∼5.4 × 10−5 s−1 ± 0.2%). This is consistent with access to the ϵX state when F1 is in a post-hydrolysis conformation because isolated F1(−δϵ) retained ∼1.5 ADP (mol/mol) but negligible ATP at catalytic sites (see “Experimental Procedures”). Added MgADP/Pi (Fig. 5A, curve 5) appeared to stabilize the ϵX state, consistent with prior β-ϵ cross-linking results (55). However, a similar effect was achieved by adding only Mg2+ and Pi (Fig. 5A, curve 6), suggesting that the endogenous ADP in isolated F1(−δϵ) was sufficient to stabilize the ϵX state upon the addition of Mg2+ and Pi. The importance of Pi in stabilizing this state (55) was also observed here because MgADP alone (Fig. 5A, curve 2) allowed a significant fraction of F1 to dissociate faster.
FIGURE 5.

Effects of other ligands on dissociation of F1 from sensor-bound wild-type ϵ. F1(−δϵ) (50 nm) was incubated for ∼10 min in MTK8 + BSA buffer only (A) or plus 1 mm ATP/EDTA (B) and then incubated with BLI sensors containing WT BapH6-ϵ to equilibrate F1·ϵ binding. Signals for bound F1(−δϵ) varied slightly between sensors (6–9%), so data were normalized for display. Results show the kinetics of F1(−δϵ) dissociation upon moving sensors into buffer with different ligands. C and D, selected results from A and B, respectively, for the initial 60 s of dissociation. When present, Mg2+ was at 2 mm; all other ligands were 1 mm. Curve 1, ATP/EDTA; curve 2, MgADP; curve 3, MgATP; curve 4, buffer only; curve 5, MgADP/Pi; curve 6, Mg2+/Pi.
In contrast to slow F1·ϵ dissociation under post-hydrolysis conditions, the addition of 1 mm ATP/EDTA caused ∼94% of F1·ϵ to dissociate ∼80-fold faster (Fig. 5A, curve 1, 4.2 × 10−3 s−1 ± 0.1%) and with <3-s transition to faster dissociation (Fig. 5C). This effect is due to ATP because EDTA alone had a minimal effect on F1 dissociation from immobilized WT ϵ (not shown). Further, by including 1 mm ATP/EDTA during association and dissociation phases, global analysis of F1·ϵ binding/dissociation shows that ATP/EDTA did not alter the F1·ϵ binding rate but gave a dissociation rate and KD similar to values for ϵ88stop (Table 2). The presence of nonhydrolyzable MgAMPPNP (2:1 mm) during the dissociation phase (not shown) also accelerated F1·ϵ dissociation, indicating that nucleotide binding alone is sufficient to shift F1 to a conformation that does not allow the ϵCTD to insert into F1 and form the ϵX state. Also, the ability of MgADP/Pi to stabilize the inhibitory state of ϵ was readily reversible; even when F1(-δϵ) was bound to WT ϵ/sensors for 45 min with MgADP/Pi present, switching the sensors to buffer with MgAMPPNP immediately caused >90% of F1(−δϵ) to dissociate at the faster rate (not shown; 3.7 × 10−3 s−1 ± 0.1%).
For the experiment in Fig. 5B, F1(−δϵ) was bound to immobilized WT ϵ in the presence of 1 mm ATP/EDTA, so that most F1·ϵ complexes would not have ϵ in the slowly dissociating ϵX state at the time the sensors were moved to dissociation wells. As expected, F1 dissociation was fast with ATP/EDTA present (Fig. 5B, curve 1). With buffer only (Fig. 5B, curve 4), most F1 still dissociated fast. This could indicate that ATP bound during the F1·ϵ association phase dissociated slowly or that endogenous ADP had dissociated from F1 during the association phase due to the ATP/EDTA present. MgADP alone (Fig. 5B, curve 2) slowed dissociation of most F1, but MgADP/Pi (Fig. 5B, curve 5) or Mg2+/Pi (Fig. 5B, curve 6) effectively reversed the ATP/EDTA effect so that almost all F1 dissociated very slowly. Mg2+ was essential for this effect; without it, F1 dissociation in the presence of 1 mm Pi (not shown) was nearly identical to that in buffer alone (Fig. 5B, curve 4). The effect of Mg2+ was enhanced by submillimolar Pi (not shown, K½ ∼0.2 mm Pi), similar to how Pi enhanced MgADP protection of F1-bound ϵ from trypsin (55).
With Mg2+/Pi, there was no apparent lag in reverting F1·ϵ to slow dissociation (Fig. 5D, curve 6). This suggested that, after F1·ϵ association with ATP/EDTA present, rapid reversion by Mg2+/Pi to slow F1·ϵ dissociation required hydrolysis of ATP that remained bound at a catalytic site. In the dissociation step, added Mg2+ could complex with the bound ATP, and hydrolysis would return F1 to the catalytic dwell step, at which insertion of the ϵCTD into the central cavity appears to occur. The bound MgADP/Pi present would then stabilize F1 with ϵ in the ϵX state. The experiment shown in Fig. 6 tested this possibility. With non-hydrolyzable MgAMPPNP present during F1·ϵ association, dissociation of most F1·ϵ was fast in the presence of MgAMPPNP (Fig. 6, curve 1) or in buffer alone (Fig. 6, curve 2), similar to the effects of ATP/EDTA in Fig. 5B. However, with MgAMPPNP present during association, inclusion of Mg2+/Pi during dissociation failed to prevent fast dissociation of most F1 (Fig. 6, curve 3), in contrast to the parallel control with ATP/EDTA in association (Fig. 6, curve 4). These results indicate that, with catalytic nucleotide bound in a prehydrolysis state, the rotary conformation of F1 does not allow insertion of the ϵCTD into the central rotor cavity, but hydrolysis at the catalytic dwell allows the ϵCTD access to insert and form the ϵX state.
FIGURE 6.

Contrasting effects of Mg2+/Pi on F1·ϵ dissociation after preincubating F1 with ATP/EDTA or MgAMPPNP. F1(−δϵ) (50 nm) was incubated ∼10 min in MTK8 buffer containing 2:1 mm MgAMPPNP (curves 1–3) or 1 mm ATP/EDTA (curve 4) and incubated with BLI sensors containing WT BapH6-ϵ. BLI signals for bound F1(−δϵ) were normalized as in Fig. 5. Kinetics of F1 dissociation were monitored in the presence of 2:1 mm MgAMPPNP (curve 1), buffer only (curve 2), or 2:1 mm Mg2+/Pi (curves 3 and 4).
In the experiments of Fig. 4, hydrolysis conditions had complex effects on F1·ϵ dissociation. With or without ATP/EDTA during F1·ϵ binding, MgATP in the dissociation phase (curve 3) induced a small or negligible rate of F1·ϵ dissociation during the initial 60 s (Fig. 5, C and D). Comparable with conditions for Fig. 5B, assays for ϵ inhibition of F1-ATPase (Fig. 2) included an ATP/EDTA preincubation, and ϵ-saturated F1 had initial ATPase activity (1–2 min, not shown) that was ∼85% of the steady-state, inhibited rate. Thus, compared with fast F1 dissociation in the continued presence of ATP/EDTA, hydrolysis of MgATP initially reverted F1·ϵ complexes to slow dissociation, rapidly re-establishing the bias toward the inhibitory ϵX state (Fig. 5D, curves 1 and 3). This is consistent with noncompetitive inhibition of ϵ versus MgATP and, combined with other results above, indicates that ϵ accesses the inhibitory ϵX state following hydrolysis at the catalytic dwell step. On the longer time scale of Fig. 5 (A and B), hydrolysis conditions increased the dissociation rate for a fraction of F1·ϵ complexes. As indicated by other experiments below, this slow effect on F1·ϵ dissociation is probably due to gradual competitive transition of some active complexes to the ADP-inhibited state, which favors faster dissociation of ϵ. The fast dissociating fraction was substantially smaller for F1·ϵ complexes formed in the presence of ATP/EDTA (Fig. 5B), probably because the ATP/EDTA preincubation minimizes initial inhibition (not shown) due to ADP-inhibited complexes.
Inhibition of F1(−δϵ) by Azide and the ϵCTD Are Competing Processes
The above results support prior SM mechanics studies (22–24, 26) that concluded that ϵ inhibition pauses rotation at the catalytic dwell position. In the absence of ϵ, long pauses at the catalytic dwell have been documented and attributed to inhibitory MgADP (57, 58). However, there have been conflicting conclusions about the relationship between inhibitory MgADP and ϵ inhibition for F1 of different bacterial species (22, 24, 25, 27). We investigated this by testing interactions between ϵ inhibition and inhibition by sodium azide, which acts by stabilizing the MgADP-inhibited state (45, 59–61). We first tested inhibition of F1(−δϵ) by azide, with or without excess WT or mutant forms of H6-ϵ present. Inhibition of F1(−δϵ) alone showed a KI of ∼5 μm azide, whether assays were done with 2:1 mm Mg/ATP (Fig. 7) or with 1:2 mm Mg/ATP (not shown). Thus, for E. coli F1, azide inhibition is separated from the step that confers sensitivity to inhibition by excess free Mg2+. The KI for azide was not altered by bound H6-ϵ mutants that could not access the ϵX state due to truncation (ϵ88stop) or disulfide bonding (ϵA101C–L121C). The presence of 100 nm WT ϵ reduced the activity of F1(−δϵ) ∼10-fold, but the residual activity was still inhibited by excess azide. However, bound WT H6-ϵ increased the KI for azide ∼5-fold (Fig. 7). Also, saturating F1(-δϵ) with reduced H6-ϵA101C/L121C, which was less inhibitory than WT H6-ϵ, yielded an intermediate KI value for azide inhibition. These results indicate that forming the inhibitory ϵX state competes with azide's capacity to inhibit F1-ATPase. To test whether azide also competed with formation of the ϵ-inhibited state, F1(−δϵ) was bound to immobilized WT BapH6-ϵ in buffer alone, and F1·ϵ dissociation was measured under hydrolysis conditions with varied concentrations of azide (Fig. 8A). Increasing azide concentrations caused greater fractions of F1·ϵ to dissociate at a faster rate, and the hyperbolic dependence on azide (Fig. 8B) yielded a K½ of ∼14 μm, comparable with the mid-range of KI values for azide inhibition with or without WT H6-ϵ (Fig. 6). Taken together, the results of Figs. 7 and 8 show competition between (i) the ability of the ϵCTD to insert, forming the inhibitory ϵX state, and (ii) the ability of azide to bind to and stabilize the MgADP-inhibited state of F1.
FIGURE 7.

Effects of excess ϵ variants on inhibition of F1-ATPase by azide. Steady-state ATPase rates were measured for F1(−δϵ) in the presence of varied concentrations of sodium azide. Assays were done in the absence of ϵ or in the presence of excess variants of H6-ϵ. Each data set was fit to the equation, v = V/(1 + [azide]/KI), and then ATPase rates were normalized to V (μmol·min−1·mg−1 F1(−δϵ), without azide) = 100%. Without ϵ, similar KI values (μm) were obtained for assays done with 2:1 mm ATP/Mg2+ (V = 78 ± 3, KI = 4.7 ± 0.7, not shown) or 1:2 mm ATP/Mg2+ (○; dashed line, V = 29.3 ± 0.5, KI = 5.3 ± 0.3). Assays were done with 1:2 mm ATP/Mg2+ in the presence of 100 nm wild-type ϵ (●; V = 3.8 ± 0.04, KI = 24.5 ± 0.9) or 100 nm ϵ88stop (▵; V = 17.9 ± 0.3, KI = 4.6 ± 0.2) or with 2:1 mm ATP/Mg2+ in the presence of 100 nm ϵA101C/L121C + 5 mm DTT (▾; V = 30.9 ± 0.3, KI = 13.5 ± 0.4) or in the presence of 300 nm disulfide-bonded ϵA101C/L121C (▿; V = 75.6 ± 0.7, KI = 6.2 ± 0.2). In control assays (not shown), 5 mm DTT had no direct effect on ATPase activity of F1(−δϵ).
FIGURE 8.

Effects of azide on dissociation of F1 from sensor-bound, wild-type ϵ. A, kinetics for F1·ϵ dissociation measured by BLI. Prior to the F1 dissociation phase shown, BLI sensors had been loaded with biotinylated wild-type ϵ and then incubated for 900 s in buffer with 20 nm F1(−δϵ); the normalized BLI signal at time 0 represents bound F1(−δϵ). Dissociation of F1(−δϵ) from immobilized ϵ was done in hydrolysis conditions (1 mm ATP, 2 mm Mg2+) with increasing concentrations of azide, as indicated. Dissociation kinetics were biphasic, with “fast” (1–2.5 × 10−3 s−1) and “slow” (0.8–1.6 × 10−4 s−1) fractions (nonlinear regression fits not shown but essentially superimpose with data at scale shown; R2 > 0.9995 for all fits). B, the azide dependence of F1·ϵ dissociation. The fraction of complexes that dissociated fast versus slow shows hyperbolic dependence on azide concentration, with K½ = 14 ± 2.3 μm; R2 = 0.9935.
DISCUSSION
Correlating Results with Functional and Rotational States of the Enzyme
Hydrolysis by the three alternating catalytic sites of F1 drives a full 360° rotation of the γ central rotary shaft, so hydrolysis of each ATP molecule involves a 120° rotation of γ relative to α3β3. As depicted schematically in Fig. 9, SM microscopy studies of bacterial F1-ATPases (reviewed in Refs. 62 and 63) have shown that each 120° rotation is comprised of two observable rotary substeps (solid arrows); MgATP binding at one alternating catalytic site drives an ∼80° rotation of γ to the catalytic dwell angle, whereas the subsequent ∼40° rotation is limited by the intrinsic rates of hydrolysis and Pi dissociation at the other alternating catalytic sites. At 120°, Mg2+ and ADP dissociate from one site before or in concert with binding of MgATP at another site to drive the next 80° substep. Fig. 9 also includes bars along the rotational arc that depict the observed angular position of γ (relative to α3β3) in crystal structures of F1 or F1·c-ring complexes. This is based on alignment of structures by a structurally conserved, apparently stiff core of the γ subunit that we identified previously (8). A recent analysis of MF1 structures concluded that the position of γ could not be correlated with rotational angle during the catalytic cycle, arguing that the part of γ protruding below α3β3 is variably displaced by lattice contacts in different crystals (64). However, biophysical characterization of the stiffness of γ indicates that only the lowest portion of γ, near its interface with the c-ring of FO, is extremely pliable (65). Also, the 99 residues of γ that we identified as a structurally conserved “γ-core” include (i) most of the γ coiled-coil that is inside α3β3, (ii) the first ∼15 residues of the γ C-terminal helix that protrude below α3β3, and (iii) 41 residues of the γ Rossmann fold domain that pack beside the protruding part of the γ C-terminal helix (see supplemental Figs. 4 and 5 of Ref. 8). We have updated our analysis of the rotational angle of γ to include 34 F1 or F1·c-ring structures and find that all γ subunits superimpose well with the γ-core (supplemental Table S-I). Thus, the distribution of F1 structures along the rotary arc of γ in Fig. 9 should be useful for comparing structural states with functional and rotational data.
FIGURE 9.

Proposed paths for inhibition by ϵCTD or ADP/azide, relative to rotary catalytic substeps and angular positions of γ in F1 structures. A 120° segment is shown for the γ rotational cycle, corresponding to net hydrolysis of 1 ATP. Two rotary substeps, observed by SM microscopy, are depicted by solid arrows (direction is for rotation of γ during hydrolysis). Bars along the inner edge of the 80–120° arc indicate the rotary angle of γ in 34 F1 crystal structures (see supplemental Table S-I). Bars are spaced every 5°, and the height represents the number of F1 structures aligned near each angle ± 2.5°; shortest bar, one structure; longest bar, 12 structures. Bars are shaded for structures of bovine F1 (black), yeast F1 (gray), and E. coli F1 (checkered). The dotted arrows below the bars indicate the proposed paths to and from the ADP-inhibited state, as stabilized by azide at ∼95° (61). The dashed arrow shows the ∼40° rotary shift proposed to occur during transition into (counterclockwise) or out of (clockwise) the ϵ-inhibited state. The two F1 aligned past 120° are also shown near 0° because this position should be the starting point for the next 120° turn.
Although there is no current consensus for correlating the rotary position of γ in known structures with the dwell states observed in SM assays after 80 and 40° substeps (63, 66, 67), we assign γ to be at 80° for one MF1 structure, with all three catalytic sites filled with nucleotide (Protein Data Bank entry 1H8E), because structural considerations (68) and molecular dynamics simulations (69) suggest that it is closest to the catalytic dwell state. Most MF1 structures have no nucleotide in the βE site due to an open conformation that distorts the nucleotide-binding pocket, but 1H8E has a “half-closed” conformation of βE with bound MgADP and SO42− (thought to mimic Pi binding). Another recent MF1 structure (Protein Data Bank entry 4ASU) also has a nucleotide in all three catalytic sites, but its βE is much closer to the open conformation and has ADP but no bound SO42− (or Pi) or Mg2+ (64). The 4ASU structure was proposed to represent the catalytic intermediate from which final products Mg2+ and ADP dissociate, and its rotary position at ∼123° (Fig. 9) correlates with SM studies indicating that ADP dissociates after ∼40° rotation to one of the 120° dwell positions (70–72). The structural indication that Mg2+ dissociates before ADP (64) suggests that the inhibitory effect of free Mg2+ occurs at the 120° position. This can explain results showing that excess free Mg2+ does not affect inhibitory transitions that occur at the catalytic dwell step at 80°; free Mg2+ does not affect the rate at which actively rotating E. coli F1(−ϵ) switches to the ADP-inhibited (paused) state (22), and, as shown here, free Mg2+ does not alter inhibition of E. coli F1 by ϵCTD or azide.
Although our results indicate that ADP and ϵ inhibition begin as competing processes after hydrolysis at the catalytic dwell, F1 structures of these inhibited states have γ positioned at angles that are distinct from the catalytic dwell; in Fig. 9, azide-inhibited MF1 (61) has γ at 93°, and ϵ-inhibited E. coli F1 (8) has γ rotated much further to 123° (checkered bar). In contrast, SM studies concluded that both ADP and ϵ inhibition cause long paused/inactive states at the catalytic dwell at 80° (22–24, 57). With a bead attached to γ or ϵ, it is possible that SM microscopy could have overlooked dynamic oscillations between 80 and 120° because those assays can exhibit broad angular distributions of events. For example, during long paused periods (up to 1–2 s) without net rotation, a γ-attached bead on E. coli F1(−ϵ) showed rapid, ongoing angular fluctuations spanning at least ±30° (Fig. 3A in Ref. 22). This could represent dynamic rotational oscillations in E. coli F1 or could be a technical limitation because the bead was attached to γ by a single cysteine, allowing for significant flexibility in the linkage. On the other hand, there is prior evidence that functional rotation is needed for transition to and from the ϵ-inhibited state; with E. coli FOF1 in liposomes, a chemical treatment that blocks rotation of FO also prevented nucleotide-dependent changes in the conformation of ϵ (54). With F1 exposed to MgADP/Pi, conditions that stabilize the ϵ-inhibited state (Fig. 5), cryoelectron microscopy of E. coli F1 showed a unique, ϵ-dependent position of γ and a unique position of ϵ relative to α3β3 (73). Also, with FOF1-liposomes, SM fluorescence assays showed a shift in the position of the ϵ NTD relative to the FO stator for active versus inactive complexes (74). Thus, we propose that the ϵCTD begins inserting into bacterial F1 near the catalytic dwell (80°) but then induces partial rotation to ∼120° to achieve the final ϵ-inhibited state, with the last half of the ϵCTD buried in the central cavity of F1 (8). This is similar to a proposal that insertion of the mitochondrial inhibitor protein (IF1) into MF1 involves rotational steps (75), and IF1-inhibited MF1 has γ rotated ∼27° past the catalytic dwell in Fig. 9. The transitions of azide-inhibited MF1 and ϵ-inhibited E. coli F1 to distinct rotary angles may help explain the competition between these inhibitory paths. Azide-inhibited MF1 has azide bound with MgADP in the closed, high affinity βD site, but azide inhibition is unlikely to occur in the ϵ-inhibited state of E. coli F1 because insertion of the ϵCTD and rotation of γ shift βD to a distinct “half-closed” conformation. Conversely, if E. coli F1 first shifted to the ADP-inhibited state, azide would probably stabilize the closed βD state and so prevent opening of the αEβD interface with γ that is necessary to allow insertion of the ϵCTD. In Fig. 9, the broken lines below the arc indicate the proposed rotational paths leading to and from ADP- or ϵ-inhibited states. In SM tests of forced rotation, TF1 was preferentially activated from the ADP-inhibited state (paused at ∼93°; Fig. 9) by >40° rotation forward, and, consistent with proposed release of ADP near 120° (Fig. 9), added ADP suppressed or reversed rotational activation (76). In contrast, forced rotation of up to 120° in either direction failed to reactivate ϵ-paused TF1 (26). Based on the asymmetric insertion of ϵCTD within F1 (8), we suspect that reactivation from the ϵX-inhibited state (Fig. 9, near 120°) occurs with the lowest activation barrier by reverse rotation of γ/ϵNTD toward the catalytic dwell angle at 80° (Fig. 9, dashed arrow). Such reactivation by rotation in the direction of ATP synthesis may be indicated by a study with E. coli FOF1-liposomes; with MgADP and Pi present, prior exposure to proton motive force activated the initial rate of subsequent ATPase activity up to 9-fold (77).
Dynamics of ϵ Conformational Changes and the Influence of Nucleotides/Ligands
Under conditions for ATP hydrolysis, SM assays with a 60-nm gold bead attached to γ showed that E. coli F1 complexes switch back and forth between actively rotating and paused states every few seconds, with or without ϵ bound to F1 (22). Our results on F1·ϵ dissociation kinetics are consistent with such rapid exchange between active and inactive states. As shown in Fig. 5 (C and D), there were no more than a few seconds lag in ligand-dependent switching between the slowest and fastest modes of F1·ϵ dissociation. Exposure to saturating nucleotide without hydrolysis (ATP/EDTA or MgAMPPNP) induced the fastest and essentially monophasic F1·ϵ dissociation (Figs. 5 and 6), indicating that few F1·ϵ complexes remained in or could regain access to the ϵX-inhibited state. However, most F1·ϵ complexes still dissociated slowly upon exposure to hydrolysis conditions (Fig. 5), consistent with noncompetitive inhibition by ϵ versus ATP for E. coli F1 (29, 44). Also, the KI for ϵ inhibition of steady-state hydrolysis (Table 1, ∼0.5 nm) is similar to the KD for F1·ϵ binding (Table 2, 0.24 nm), which was measured without added nucleotides. Thus, it is unlikely that catalytic binding of ATP or MgAMPPNP directly increases the rate at which the ϵX-paused state returns to an active form. Rather, we propose that the intrinsic rates to and from the ϵX-inhibited state are fast enough (seconds or less) to allow ligands to influence subsequent conformational changes once F1 exits from the ϵ-inhibited state. Once the ϵCTD escapes from the central cavity of F1 near 80°, F1 would be most likely to rotate forward, completing an active 40° step (Fig. 9, solid arrow). At that point, binding of nucleotide would drive an ∼80° step toward the next catalytic dwell but, without hydrolysis, would trap F1 in a conformation and rotary position that would not allow the ϵCTD to reinsert; thus, F1·ϵ complexes would dissociate at the faster rate observed with only γ/ϵNTD interactions. With hydrolysis conditions, each return to a catalytic dwell step would provide the same low probability for insertion of the ϵCTD, and, as indicated by SM studies (22), the long durations of the paused/inhibited states (1 to 3.5 s) relative to the limiting catalytic steps (1–2 ms) would result in a large fraction of inhibited complexes during steady-state hydrolysis.
In an SM study with E. coli F1 (22), the presence of ϵ did not alter the duration time of active complexes (0.5–1 s), but ϵ-paused states had longer duration times (up to 3.5 s) than ADP-paused states (∼1 s). This is consistent with other indications that ϵ inhibition predominates over ADP inhibition. Oxyanions, which are thought to activate F1 by promoting release of inhibitory ADP (78), activate E. coli F1 more if ϵ is absent (28), and recently it was shown that the oxyanion selenite optimally activates ϵ-depleted E. coli F1 ∼10-fold4 with excess free Mg2+ present but only activates ϵ-saturated F1 2–2.5-fold (79). Also, because our results show that inhibition by the ϵCTD is distinct from the ADP-inhibited transition, the unaffected duration of the active state (22) suggests that a prior common step is rate-limiting for transitions to either ADP- or ϵ-inhibited states. This common step is probably the end of hydrolysis that precedes Pi release because ϵ reduces the rate of Pi release ∼15-fold following “unisite” hydrolysis (28). Pi stabilizes F1 with ϵ in the ϵX state, which could mean that Pi rebinds to βD (with bound MgADP) and delays an active 40° rotation, allowing more time for a possible transition to the ϵX-inhibited state. However, we cannot rule out that the Pi effect could be due in part to binding to other β(s), which show SO43− bound at the “P-loop” in ϵ-inhibited F1 (8).
Conclusions
This study sheds further light on how the CTD of subunit ϵ inhibits the catalytic F1 complex of a bacterial ATP synthase. Most significantly, results reveal that ATP hydrolysis is required for insertion of the inhibitory ϵCTD into F1 at the catalytic dwell step and that ϵ inhibition competes with conversion to an ADP-inhibited state of the enzyme. With insertion of the ϵCTD starting at the catalytic dwell (∼80°), the dynamic response of the conformation of ϵ to catalytic site ligands and the structurally observed γ angle of ∼123° in ϵ-inhibited F1 suggest dynamic, reversible rotation over the 40° substep. Our results also show that the ϵCTD has a small energetic contribution to net binding of ϵ to F1. Thus, there is potential for antibiotic development by discovering or designing compounds that enhance or mimic ϵ inhibition of bacterial ATP synthases. The BLI assays established here for kinetics of F1·ϵ binding and dissociation should be valuable in further analyzing which ϵCTD residues and interactions are critical for ϵ inhibition in F1-ATPase from E. coli and other bacteria. Of course, the BLI assay cannot be used to study ϵ inhibition in membrane-bound ATP synthase because ϵ does not dissociate from intact FOF1. The extent of ϵ inhibition can vary widely for membranes isolated from different bacteria. For example, E. coli membranes exhibit substantial ATPase activity, whereas mycobacterial membranes are devoid of ATPase activity but can be activated by treatment that probably damages the ϵ subunit (80). Thus, additional approaches will be needed to determine what other factors influence ϵ inhibition in ATP synthases of different bacterial species.
Acknowledgments
We thank Y. M. Milgrom for stimulating discussions and for sharing unpublished results. We thank Profs. Richard L. Cross and Stephan Wilkens for critical reading of the manuscript. We thank Prof. Robert K. Nakamoto (University of Virginia) for plasmid pH6ϵ.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM083088 (to T. M. D.).

This article contains supplemental Fig. S1 and Table S-I.
The atomic coordinates and structure factors (code 3OAA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The original study with ϵA101C/L121C (21) showed that an ϵA101C–L121C disulfide bond could be formed in membrane-bound FOF1 but did not appear to alter ATPase activity. We tested membranes expressing FOF1 with ϵA101C/L121C and showed that removing DTT from the sample activated ATPase 2-fold; this is the same activation seen originally (21) after inducing a disulfide between the ϵCTD and ϵNTD (ϵM49C-A126C). We believe that their ATPase assays did not include reductant for the non-oxidized sample of FOF1 + ϵA101C/L121C and that the disulfide formed quickly and spontaneously, as we have observed (Fig. 3). Thus, they would not have observed activation of ATPase by oxidation of ϵA101C–L121C if the disulfide had also formed in the non-oxidized sample.
Y. M. Milgrom, personal communication.
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- ϵC and ϵX
- compact and extended conformations of ϵ (Fig. 1)
- BLI
- biolayer interferometry
- Bap
- biotin acceptor peptide
- βME
- 2-mercaptoethanol
- DTNB
- 5,5′-dithiobis(2-nitrobenzoate)
- MF1
- mitochondrial F1-ATPase
- TF1
- F1-ATPase of thermophilic Bacillus sp. PS3
- SM
- single-molecule
- H6-ϵ
- His6-tagged ϵ
- MBP
- maltose-binding protein
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate.
REFERENCES
- 1. Boyer P. D. (1997) The ATP synthase. A splendid molecular machine. Annu. Rev. Biochem. 66, 717–749 [DOI] [PubMed] [Google Scholar]
- 2. Duncan T. M. (2004) The ATP Synthase. Parts and Properties of a Rotary Motor. in The Enzymes, Vol. XXIII, 3rd Ed (Hackney D. D., Tamanoi F., eds) pp. 203–275, Elsevier Academic Press, New York [Google Scholar]
- 3. Spetzler D., Ishmukhametov R., Hornung T., Martin J., York J., Jin-Day L., Frasch W. D. (2012) Energy Transduction by the Two Molecular Motors of the F0F1 ATP Synthase (Eaton-Rye J. J., Tripathy B. C., Sharkey T. D., eds) pp. 561–590, Springer, Dordrecht, Netherlands [Google Scholar]
- 4. Cox G. B., Devenish R. J., Gibson F., Howitt S. M., Nagley P. (1992) The structure and assembly of ATP synthase. in Molecular Mechanisms in Bioenergetics (Ernster L., ed) pp. 283–315, Elsevier Science, New York [Google Scholar]
- 5. Andries K., Verhasselt P., Guillemont J., Göhlmann H. W., Neefs J. M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E., Williams P., de Chaffoy D., Huitric E., Hoffner S., Cambau E., Truffot-Pernot C., Lounis N., Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 [DOI] [PubMed] [Google Scholar]
- 6. Koul A., Vranckx L., Dendouga N., Balemans W., Van den Wyngaert I., Vergauwen K., Göhlmann H. W., Willebrords R., Poncelet A., Guillemont J., Bald D., Andries K. (2008) Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 283, 25273–25280 [DOI] [PubMed] [Google Scholar]
- 7. Diacon A. H., Pym A., Grobusch M., Patientia R., Rustomjee R., Page-Shipp L., Pistorius C., Krause R., Bogoshi M., Churchyard G., Venter A., Allen J., Palomino J. C., De Marez T., van Heeswijk R. P., Lounis N., Meyvisch P., Verbeeck J., Parys W., de Beule K., Andries K., Mc Neeley D. F. (2009) The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 360, 2397–2405 [DOI] [PubMed] [Google Scholar]
- 8. Cingolani G., Duncan T. M. (2011) Structure of the ATP synthase catalytic complex (F1) from Escherichia coli in an autoinhibited conformation. Nat. Struct. Mol. Biol. 18, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feniouk B. A., Suzuki T., Yoshida M. (2006) The role of subunit ϵ in the catalysis and regulation of FOF1-ATP synthase. Biochim. Biophys. Acta 1757, 326–338 [DOI] [PubMed] [Google Scholar]
- 10. Richter M. L. (2004) γ-ϵ interactions regulate the chloroplast ATP synthase. Photosynth. Res. 79, 319–329 [DOI] [PubMed] [Google Scholar]
- 11. Campanella M., Casswell E., Chong S., Farah Z., Wieckowski M. R., Abramov A. Y., Tinker A., Duchen M. R. (2008) Regulation of mitochondrial structure and function by the F1FO-ATPase inhibitor protein, IF1. Cell Metab. 8, 13–25 [DOI] [PubMed] [Google Scholar]
- 12. Iino R., Hasegawa R., Tabata K. V., Noji H. (2009) Mechanism of inhibition by C-terminal α-helices of the ϵ subunit of Escherichia coli FOF1-ATP synthase. J. Biol. Chem. 284, 17457–17464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masaike T., Suzuki T., Tsunoda S. P., Konno H., Yoshida M. (2006) Probing conformations of the β subunit of FOF1-ATP synthase in catalysis. Biochem. Biophys. Res. Commun. 342, 800–807 [DOI] [PubMed] [Google Scholar]
- 14. Stocker A., Keis S., Vonck J., Cook G. M., Dimroth P. (2007) The Structural basis for unidirectional rotation of thermoalkaliphilic F1-ATPase. Structure 15, 904–914 [DOI] [PubMed] [Google Scholar]
- 15. Gibbons C., Montgomery M. G., Leslie A. G., Walker J. E. (2000) The structure of the central stalk in bovine F1-ATPase at 2.4 Å resolution. Nat. Struct. Biol. 7, 1055–1061 [DOI] [PubMed] [Google Scholar]
- 16. Kabaleeswaran V., Puri N., Walker J. E., Leslie A. G., Mueller D. M. (2006) Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J. 25, 5433–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uhlin U., Cox G. B., Guss J. M. (1997) Crystal structure of the ϵ subunit of the proton-translocating ATP synthase from Escherichia coli. Structure 5, 1219–1230 [DOI] [PubMed] [Google Scholar]
- 18. Wilkens S., Capaldi R. A. (1998) Solution structure of the ϵ subunit of the F1-ATPase from Escherichia coli and interactions of this subunit with β subunits in the complex. J. Biol. Chem. 273, 26645–26651 [DOI] [PubMed] [Google Scholar]
- 19. Yagi H., Kajiwara N., Tanaka H., Tsukihara T., Kato-Yamada Y., Yoshida M., Akutsu H. (2007) Structures of the thermophilic F1-ATPase ϵ subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1. Proc. Natl. Acad. Sci. U.S.A. 104, 11233–11238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watt I. N., Montgomery M. G., Runswick M. J., Leslie A. G., Walker J. E. (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. U.S.A. 107, 16823–16827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulenberg B., Capaldi R. A. (1999) The ϵ subunit of the F1FO complex of Escherichia coli. Cross-linking studies show the same structure in situ as when isolated. J. Biol. Chem. 274, 28351–28355 [DOI] [PubMed] [Google Scholar]
- 22. Sekiya M., Hosokawa H., Nakanishi-Matsui M., Al-Shawi M. K., Nakamoto R. K., Futai M. (2010) Single molecule behavior of inhibited and active states of Escherichia coli ATP synthase F1 rotation. J. Biol. Chem. 285, 42058–42067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konno H., Murakami-Fuse T., Fujii F., Koyama F., Ueoka-Nakanishi H., Pack C. G., Kinjo M., Hisabori T. (2006) The regulator of the F1 motor. Inhibition of rotation of cyanobacterial F1-ATPase by the ϵ subunit. EMBO J. 25, 4596–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsumuraya M., Furuike S., Adachi K., Kinosita K., Jr., Yoshida M. (2009) Effect of ϵ subunit on the rotation of thermophilic Bacillus F1-ATPase. FEBS Lett. 583, 1121–1126 [DOI] [PubMed] [Google Scholar]
- 25. Konno H., Isu A., Kim Y., Murakami-Fuse T., Sugano Y., Hisabori T. (2011) Characterization of the relationship between ADP- and ϵ-induced inhibition in cyanobacterial F1-ATPase. J. Biol. Chem. 286, 13423–13429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saita E. I., Iino R., Suzuki T., Feniouk B. A., Kinosita K., Jr., Yoshida M. (2010) Activation and stiffness of the inhibited states of F1-ATPase probed by single-molecule manipulation. J. Biol. Chem. 285, 11411–11417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feniouk B. A., Suzuki T., Yoshida M. (2007) Regulatory interplay between proton motive force, ADP, phosphate, and subunit ϵ in bacterial ATP synthase. J. Biol. Chem. 282, 764–772 [DOI] [PubMed] [Google Scholar]
- 28. Dunn S. D., Zadorozny V. D., Tozer R. G., Orr L. E. (1987) ϵ subunit of Escherichia coli F1-ATPase. Effects on affinity for aurovertin and inhibition of product release in unisite ATP hydrolysis. Biochemistry 26, 4488–4493 [DOI] [PubMed] [Google Scholar]
- 29. Weber J., Dunn S. D., Senior A. E. (1999) Effect of the ϵ-subunit on nucleotide binding to Escherichia coli F1-ATPase catalytic sites. J. Biol. Chem. 274, 19124–19128 [DOI] [PubMed] [Google Scholar]
- 30. Andrews S. H., Peskova Y. B., Polar M. K., Herlihy V. B., Nakamoto R. K. (2001) Conformation of the γ subunit at the γ-ϵ-c interface in the complete Escherichia coli F1-ATPase complex by site-directed spin labeling. Biochemistry 40, 10664–10770 [DOI] [PubMed] [Google Scholar]
- 31. Cipriano D. J., Dunn S. D. (2006) The role of the ϵ subunit in the Escherichia coli ATP synthase. The C-terminal domain is required for efficient energy coupling. J. Biol. Chem. 281, 501–507 [DOI] [PubMed] [Google Scholar]
- 32. Tsao K.-L., DeBarbieri B., Michel H., Waugh D. S. (1996) A versatile plasmid expression vector for the production of biotinylated proteins by site-specific, enzymatic modification in Escherichia coli. Gene 169, 59–64 [DOI] [PubMed] [Google Scholar]
- 33. Bhardwaj A., Walker-Kopp N., Wilkens S., Cingolani G. (2008) Foldon-guided self-assembly of ultra-stable protein fibers. Protein Sci. 17, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duncan T. M., Zhou Y., Bulygin V. V., Hutcheon M. L., Cross R. L. (1995) Probing interactions of the Escherichia coli FOF1 ATP synthase β and γ subunits with disulphide cross-links. Biochem. Soc. Trans. 23, 736–741 [DOI] [PubMed] [Google Scholar]
- 35. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 36. Peterson G. L. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 83, 346–356 [DOI] [PubMed] [Google Scholar]
- 37. Dunn S. D. (1986) Removal of the ϵ subunit from Escherichia coli F1-ATPase using monoclonal anti-ϵ antibody affinity chromatography. Anal. Biochem. 159, 35–42 [DOI] [PubMed] [Google Scholar]
- 38. Duncan T. M., Bulygin V. V., Zhou Y., Hutcheon M. L., Cross R. L. (1995) Rotation of subunits during catalysis by Escherichia coli F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 92, 10964–10968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Penefsky H. S. (1977) Reversible binding of Pi by beef heart mitochondrial adenosine triphosphatase. J. Biol. Chem. 252, 2891–2899 [PubMed] [Google Scholar]
- 40. Pullman M. E., Penefsky H. S., Datta A., Racker E. (1960) Partial resolution of the enzymes catalysing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenyl-stimulated adonosine triphophatase. J. Biol. Chem. 235, 3322–3329 [PubMed] [Google Scholar]
- 41. Abdiche Y., Malashock D., Pinkerton A., Pons J. (2008) Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 377, 209–217 [DOI] [PubMed] [Google Scholar]
- 42. Tozer R. G., Dunn S. D. (1986) Column centrifugation generates an intersubunit disulfide bridge in Escherichia coli F1-ATPase. Eur. J. Biochem. 161, 513–518 [DOI] [PubMed] [Google Scholar]
- 43. Dunn S. D. (1982) The isolated γ subunit of Escherichia coli F1 ATPase binds the ϵ subunit. J. Biol. Chem. 257, 7354–7359 [PubMed] [Google Scholar]
- 44. Sternweis P. C., Smith J. B. (1980) Characterization of the inhibitory ϵ subunit of the proton-translocating adenosine triphosphatase from Escherichia coli. Biochemistry 19, 526–531 [DOI] [PubMed] [Google Scholar]
- 45. Hyndman D. J., Milgrom Y. M., Bramhall E. A., Cross R. L. (1994) Nucleotide-binding sites on Escherichia coli F1-ATPase. Specificity of noncatalytic sites and inhibition at catalytic sites by MgADP. J. Biol. Chem. 269, 28871–28877 [PubMed] [Google Scholar]
- 46. Kato Y., Sasayama T., Muneyuki E., Yoshida M. (1995) Analysis of time-dependent change of Escherichia coli F1-ATPase activity and its relationship with apparent negative cooperativity. Biochim. Biophys. Acta 1231, 275–281 [DOI] [PubMed] [Google Scholar]
- 47. Jault J. M., Matsui T., Jault F. M., Kaibara C., Muneyuki E., Yoshida M., Kagawa Y., Allison W. S. (1995) The α3β3γ complex of the F1-ATPase from thermophilic Bacillus PS3 containing the α D261N substitution fails to dissociate inhibitory MgADP from a catalytic site when ATP binds to noncatalytic sites. Biochemistry 34, 16412–16418 [DOI] [PubMed] [Google Scholar]
- 48. Drobinskaya I. Y., Kozlov I. A., Murataliev M. B., Vulfson E. N. (1985) Tightly bound adenosine diphosphate, which inhibits the activity of mitochondrial F1-ATPase, is located at the catalytic site of the enzyme. FEBS Lett. 182, 419–424 [DOI] [PubMed] [Google Scholar]
- 49. Milgrom Y. M., Boyer P. D. (1990) The ADP that binds tightly to nucleotide-depleted mitochondrial F1-ATPase and inhibits catalysis is bound at a catalytic site. Biochim. Biophys. Acta 1020, 43–48 [DOI] [PubMed] [Google Scholar]
- 50. Murataliev M. B., Milgrom Y. M., Boyer P. D. (1991) Characteristics of the combination of inhibitory Mg2+ and azide with the F1 ATPase from chloroplasts. Biochemistry 30, 8305–8310 [DOI] [PubMed] [Google Scholar]
- 51. Guerrero K. J., Xue Z. X., Boyer P. D. (1990) Active/Inactive state transitions of the chloroplast F1 ATPase are induced by a slow binding and release of Mg2+. Relationship to catalysis and control of F1 ATPases. J. Biol. Chem. 265, 16280–16287 [PubMed] [Google Scholar]
- 52. Jault J. M., Divita G., Allison W. S., Di Pietro A. (1993) Glutamine 170 to tyrosine substitution in yeast mitochondrial F1 β-subunit increases catalytic site interaction with GDP and IDP and produces negative cooperativity of GTP and ITP hydrolysis. J. Biol. Chem. 268, 20762–20767 [PubMed] [Google Scholar]
- 53. Xiong H., Zhang D., Vik S. B. (1998) Subunit ϵ of the Escherichia coli ATP synthase. Novel insights into structure and function by analysis of thirteen mutant forms. Biochemistry 37, 16423–16429 [DOI] [PubMed] [Google Scholar]
- 54. Mendel-Hartvig J., Capaldi R. A. (1991) Nucleotide-dependent and dicyclohexylcarbodiimide-sensitive conformational changes in the ϵ subunit of Escherichia coli ATP synthase. Biochemistry 30, 10987–10991 [DOI] [PubMed] [Google Scholar]
- 55. Mendel-Hartvig J., Capaldi R. A. (1991) Catalytic site nucleotide and inorganic phosphate dependence of the conformation of the ϵ subunit in Escherichia coli adenosinetriphosphatase. Biochemistry 30, 1278–1284 [DOI] [PubMed] [Google Scholar]
- 56. Dallmann H. G., Flynn T. G., Dunn S. D. (1992) Determination of the 1-ethyl-3-[(3-dimethylamino)propyl]-carbodiimide-induced cross-link between the β and ϵ subunits of Escherichia coli F1-ATPase. J. Biol. Chem. 267, 18953–18960 [PubMed] [Google Scholar]
- 57. Hirono-Hara Y., Noji H., Nishiura M., Muneyuki E., Hara K. Y., Yasuda R., Kinosita K., Jr., Yoshida M. (2001) Pause and rotation of F1-ATPase during catalysis. Proc. Natl. Acad. Sci. U.S.A. 98, 13649–13654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakanishi-Matsui M., Kashiwagi S., Ubukata T., Iwamoto-Kihara A., Wada Y., Futai M. (2007) Rotational catalysis of Escherichia coli ATP synthase F1 sector. Stochastic fluctuation and a key domain of the β subunit. J. Biol. Chem. 282, 20698–20704 [DOI] [PubMed] [Google Scholar]
- 59. Vasilyeva E. A., Minkov I. B., Fitin A. F., Vinogradov A. D. (1982) Kinetic mechanism of mitochondrial adenosine triphosphatase. Inhibition by azide and activation by sulphite. Biochem. J. 202, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murataliev M. B., Boyer P. D. (1992) The mechanism of stimulation of MgATPase activity of chloroplast F1-ATPase by non-catalytic adenine-nucleotide binding. Acceleration of the ATP-dependent release of inhibitory ADP from a catalytic site. Eur. J. Biochem. 209, 681–687 [DOI] [PubMed] [Google Scholar]
- 61. Bowler M. W., Montgomery M. G., Leslie A. G., Walker J. E. (2006) How azide inhibits ATP hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. U.S.A. 103, 8646–8649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Noji H., Okuno D., Ikeda T. (2011) Mechanochemistry of F1 motor protein. Chem. Sci. 2, 2086–2093 [Google Scholar]
- 63. Kinosita K., Jr. (2012) F1-ATPase. A prototypical rotary molecular motor. Adv. Exp. Med. Biol. 726, 5–16 [DOI] [PubMed] [Google Scholar]
- 64. Rees D. M., Montgomery M. G., Leslie A. G., Walker J. E. (2012) Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 109, 11139–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sielaff H., Rennekamp H., Wächter A., Xie H., Hilbers F., Feldbauer K., Dunn S. D., Engelbrecht S., Junge W. (2008) Domain compliance and elastic power transmission in rotary FOF1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 105, 17760–17765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Okuno D., Fujisawa R., Iino R., Hirono-Hara Y., Imamura H., Noji H. (2008) Correlation between the conformational states of F1-ATPase as determined from its crystal structure and single-molecule rotation. Proc. Natl. Acad. Sci. U.S.A. 105, 20722–20727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sielaff H., Rennekamp H., Engelbrecht S., Junge W. (2008) Functional halt positions of rotary FOF1-ATPase correlated with crystal structures. Biophys. J. 95, 4979–4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Menz R. I., Walker J. E., Leslie A. G. (2001) Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites. Implications for the mechanism of rotary catalysis. Cell 106, 331–341 [DOI] [PubMed] [Google Scholar]
- 69. Pu J., Karplus M. (2008) How subunit coupling produces the γ-subunit rotary motion in F1-ATPase. Proc. Natl. Acad. Sci. U.S.A. 105, 1192–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nishizaka T., Oiwa K., Noji H., Kimura S., Muneyuki E., Yoshida M., Kinosita K., Jr. (2004) Chemomechanical coupling in F1-ATPase revealed by simultaneous observation of nucleotide kinetics and rotation. Nat. Struct. Mol. Biol. 11, 142–148 [DOI] [PubMed] [Google Scholar]
- 71. Adachi K., Oiwa K., Nishizaka T., Furuike S., Noji H., Itoh H., Yoshida M., Kinosita K., Jr. (2007) Coupling of rotation and catalysis in F1-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321 [DOI] [PubMed] [Google Scholar]
- 72. Shimo-Kon R., Muneyuki E., Sakai H., Adachi K., Yoshida M., Kinosita K., Jr. (2010) Chemo-mechanical coupling in F1-ATPase revealed by catalytic site occupancy during catalysis. Biophys. J. 98, 1227–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilkens S., Capaldi R. A. (1994) Asymmetry and structural changes in ECF1 examined by cryoelectronmicroscopy. Biol. Chem. Hoppe-Seyler 375, 43–51 [DOI] [PubMed] [Google Scholar]
- 74. Zimmermann B., Diez M., Zarrabi N., Gräber P., Börsch M. (2005) Movements of the ϵ-subunit during catalysis and activation in single membrane-bound H+-ATP synthase. EMBO J. 24, 2053–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gledhill J. R., Montgomery M. G., Leslie A. G., Walker J. E. (2007) How the regulatory protein, IF1, inhibits F1-ATPase from bovine mitochondria. Proc. Natl. Acad. Sci. U.S.A. 104, 15671–15676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hirono-Hara Y., Ishizuka K., Kinosita K., Jr., Yoshida M., Noji H. (2005) Activation of pausing F1 motor by external force. Proc. Natl. Acad. Sci. U.S.A. 102, 4288–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fischer S., Graber P., Turina P. (2000) The activity of the ATP synthase from Escherichia coli is regulated by the transmembrane proton motive force. J. Biol. Chem. 275, 30157–30162 [DOI] [PubMed] [Google Scholar]
- 78. Milgrom Y. M., Cross R. L. (1993) Nucleotide binding sites on beef heart mitochondrial F1-ATPase. Cooperative interactions between sites and specificity of noncatalytic sites. J. Biol. Chem. 268, 23179–23185 [PubMed] [Google Scholar]
- 79. Bulygin V. V., Milgrom Y. M. (2009) A bi-site mechanism for Escherichia coli F1-ATPase accounts for the observed positive catalytic cooperativity. Biochim. Biophys. Acta 1787, 1016–1023 [DOI] [PubMed] [Google Scholar]
- 80. Haagsma A. C., Driessen N. N., Hahn M. M., Lill H., Bald D. (2010) ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 313, 68–74 [DOI] [PubMed] [Google Scholar]
- 81. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]