

Abstract
Rationale
Failing cardiomyocytes exhibit decreased efficiency of excitation-contraction (E-C) coupling. The down-regulation of junctophilin-2 (JP2), a protein anchoring the sarcoplasmic reticulum (SR) to T-tubules (TTs), has been identified as a major mechanism underlying the defective E-C coupling. However, the regulatory mechanism of JP2 remains unknown.
Objective
To determine whether microRNAs regulate JP2 expression.
Methods and Results
Bioinformatic analysis predicted two potential binding sites of miR-24 in the 3′-untranslated regions of JP2 mRNA. Luciferase assays confirmed that miR-24 suppressed JP2 expression by binding to either of these sites. In the aortic stenosis model, miR-24 was up-regulated in failing cardiomyocytes. Adenovirus-directed over-expression of miR-24 in cardiomyocytes decreased JP2 expression and reduced Ca2+ transient amplitude and E-C coupling gain.
Conclusions
MiR-24-mediated suppression of JP2 expression provides a novel molecular mechanism for E-C coupling regulation in heart cells, and suggests a new target against heart failure.
Keywords: myocardial contractility, excitation-contraction coupling, heart failure, calcium signaling, heart failure
INTRODUCTION
The contractile strength of the heart is controlled by the excitation-contraction (E-C) coupling process, in which the voltage-gated L-type Ca2+ current (LCC) through the plasma membrane, including transverse tubules (TTs), activates Ca2+ release from ryanodine receptors (RyRs) in the sarcoplasmic reticulum (SR) and initiates cell contraction.1-3 TT-SR structural coupling across a ~15 nm junctional cleft4 relies on junctophilin-2 (JP2).5 During heart failure, the defective E-C coupling3,6 is accompanied by decreased expression of JP2.7-9 Knockdown of JP2 compromises E-C coupling.9-12 Therefore, deciphering the mechanisms of JP2 down-regulation is important in understanding the pathogenesis of E-C coupling.
MicroRNAs are ~22-nt non-coding RNAs that suppress gene expression by binding to the 3′ untranslated region (3′UTR) of target mRNAs.13 Here we report that miR-24, a microRNA up-regulated in heart failure,14 is an immediate upstream suppressor of JP2.
METHODS
Putative miRNA binding sites on the 3′ UTR of JP2 mRNA was identified by TargetScan and verified by luciferase assays (online supplemental materials). Rat ventricular cardiomyocytes were cultured and infected15 with adenoviral vectors containing GFP or miR-24 precursor sequences. After 48 hours of culture, miR-24 and JP2 expression analysis, TEM analysis, whole-cell patch clamp, and confocal Ca2+ imaging were performed as reported12,15.
RESULTS
Bioinformatic analysis identified two putative miR-24 binding sites within the 3′UTR of human, rat and mouse JP2 mRNAs (Fig. 1A-C). In rat aortic stenosis models of compensated hypertrophy (CHT) and decompensated heart failure (DHT) (Fig. 1D and E), miR-24 was up-regulated in isolated ventricular cardiomyocytes (Fig. 1F), accompanied by JP2 down-regulation (Fig. 1G), as reported previously7-9.
Figure 1. MiR-24 was predicted to bind to the 3′UTR of JP2 mRNA, and was up-regulated in heart failure.

A-C, Predicted miR-24 binding sites on the 3′UTRs of human, rat and mouse JP2 mRNA sequences. D, Representative m-node echocardiograph from the decompensated hypertrophy (DHT) group. E, Posterior end-diastolic wall thickness (PWD, left) and fractional shortening (FS, right) were used to identify the stages of compensated hypertrophy (CHT) and DHT. F-G, Real-time PCR assessments of miR-24 and JP2 expression. n = 5/group. *P <0.05 and **P <0.01 vs sham-operated animals.
To determine whether JP2 is a bona fide target of miR-24, we constructed luciferase expression plasmids containing the JP2 3′ UTR segments with putative miR-24 binding sites. Co-transfection of miR-24 mimics and the constructed plasmids in HEK293 cells led to significant suppression of luciferase expression (Fig. 2). Mutating either site I or site II alone by replacing the GAG (Fig. 1, bold font) with CUC did not compromise the effect of miR-24. Only double mutation of both binding sites eliminated the suppression (Fig. 2). Similar results were shared by the rat, mouse and human sequences, indicating that the miR-24 targeting of JP2 expression was doubly-secured through evolution.
Figure 2. Luciferase assays of miR-24 binding sites on human, rat and mouse JP2 3′UTRs.
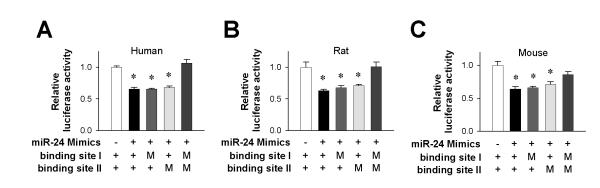
Transfection of luciferase reporters with wild-type 3′UTR alone was used as control (white). In other groups, miR-24 Mimics was co-transfected with luciferase reporters containing wild-type 3′UTR (black), 3′UTR with mutant binding site I (grey), with mutant binding site II (light grey) and with both mutations (dark grey). n = 4/group. *P <0.05 and **P <0.01 vs control.
To test whether miR-24 interferes with endogenous JP2 expression, we constructed adenovirus to over-express miR-24 in rat ventricular myocytes (Fig. 3A). Real-time PCR (Fig. 3B) and Western blot assay (Fig. 3C) showed that miR-24 over-expression by 150% down-regulated the JP2 protein level by 50-60% compared with cells either without infection (control) or infected with adenovirus containing GFP only. This result agreed well with the effect of miR-24 over-expression in neonatal cardiomyocytes (Online Figure I)
Figure 3. The effect of miR-24 over-expression on JP2 expression and TT-SR junctions in rat cardiomyocytes.

A, Real-time PCR assay of miR-24 expression in cultured rat cardiomyocytes without infection (control), infected with adenovirus containing GFP (GFP) and infected with adenovirus containing miR-24 precursors (miR-24). n = 4/group. B-C, Real-time PCR (B) and Western blot (C) analysis of JP2 expression. D-E, Stereological measurements of the volume density (D) and the surface area density (E) of TTs. Data from 56 (GFP) and 99 (miR-24) TEM images in 3 independent experiments/group. F, Representative TEM images demonstrating the apparent junction length (yellow) between the TT (red) and SR (green) membranes. G-H, The apparent length of TT-SR junction (G) and its distribution (H). Data from 67 (GFP) and 108 (miR-24) TEM images in 3 independent experiments/group. *P <0.05 and **P <0.01 vs GFP.
To assess the structural consequences of miR-24-induced JP2 down-regulation, ultrathin sections of isolated ventricular myocytes were imaged with transmission electron microscopy (TEM). Stereological analysis19 (Online Figure II) showed that the volume density (Fig. 3D) and the surface area (Fig. 3E) of SR-coupled TTs were reduced by 30.7% and 24.0%, respectively, in the miR-24 group. In contrast, those of TTs apparently not coupled to SRs were increased by 63.8% and 65.2%, respectively. The surface area of junctional SRs was reduced by 28.6% accordingly. Moreover, the spatial span of individual TT-SR junctions, measured by the apparent curvilinear length of parallel TT and SR membranes (the yellow line in Fig. 3F), was curtailed by 26.3% (Fig. 3G), with its logarithmic normal distribution shifted to shorter lengths (Fig. 3H).
As decreased density and size of TT-SR junctions leads to asynchronous and inhomogeneous Ca2+ release12, we next recorded the strength and timing of local Ca2+ release spikes at individual sarcomeres16 (Fig. 4A) when 10 mM EGTA was included in the intracellular electrode solution. Compared with those in the control and GFP groups, the Ca2+ spike amplitude in the miR-24 group was decreased (Fig. 4B), and the delay from depolarization to the spike peak of individual sarcomeres (Dspike) was prolonged and dispersed (Fig. 4C). Due to the reduction and desynchronization of local Ca2+ release, the global Ca2+ transient evoked by whole-cell depolarization in the absence of EGTA (Fig. 4D) was significantly lower in the miR-24 group than in the control and GFP groups (Fig. 4E). As the LCC Ca2+ current was not altered, the gain of E-C coupling, indexed by the amplitude of the Ca2+ transient per unit ICa, was curtailed by nearly half in the miR-24 group (Fig. 4F), leading to a ~40% reduction in the fractional shortening of cardiomyocytes (Fig. 4G).
Figure 4. The effect of miR-24 over-expression on E-C coupling in rat cardiomyocytes.

A, Typical local Ca2+ release spikes evoked by the depolarization from −70 to 0 mV in GFP (left) and miR-24 (right) groups. The black/white strip beside each color image is a positioning reference of Z-lines derived from the background fluorescence prior to depolarization. The time-courses of Ca2+ spikes at positions 1-6 are plotted in the lower panels. B, Amplitude of Ca2+ spikes in control, GFP and miR-24 groups (n = 10, 9 and 11, respectively). C, Delay of Ca2+ spikes (Dspike) measured as the time from depolarization to the highest spike peak as illustrated in A. The standard deviations (SD) of Dspike are compared in the right panel. D, Typical recordings of LCC Ca2+ currents (ICa, upper plots), Ca2+ transients (middle images and plots) and cell length (lower plots) in response to the depolarization from −70 to 0 mV. E, ICa density (upper) and Ca2+ transient amplitude (lower) compared among control (n = 13), GFP (n = 17) and miR-24 (n = 19) groups. Two-way ANOVA with repeated measures identified a significant difference between miR-24 and other two groups. F, The Gain of E-C coupling was calculated as the Ca2+ transient amplitude per unit ICa density at 0 mV. G, Fractional shortening of cardiomyocytes measured by cell edge-detection of Ca2+ transients at 0 mV. *P <0.05 and **P <0.01 vs GFP group.
DISCUSSION
JP2 is a structural protein linking the SR to cell membrane/TTs,5 and plays a key role in the nanoscopic signaling between LCCs and RyRs during E-C coupling. The healthy operation of E-C coupling requires JP2 levels finely tuned within the homeostatic range. In the present study, we demonstrated that miR-24 regulates JP2 expression by binding to at least one of the two sites within the 3′UTR of JP2 mRNA. Over-expression of miR-24 leads to TT-SR ultrastructural remodeling and defective E-C coupling, reproducing those observed in failing heart cells.
Previous reports have shown that miR-24 is a ubiquitously-expressed microRNA regulating cell proliferation and cancer genesis.17-19 In the heart, miR-24 regulate cardiomyocyte apoptosis20 and endothelial vascularity.15 However, whether or not miR-24 is expressed in cardiomyocytes is controversial.15,20 Here, we measured miR-24 expression in isolated cardiomyocytes (Figs. 1F and 3A), confirming robust expression of miR-24 in cardiomyocytes.
A microRNA usually has multiple targets. Using the TargetScan software, we performed genome-wide scanning of miR-24 targets among human, rat and mouse annotated genes, and identified no E-C coupling components other than JP2. Experimental data showed that miR-24 over-expression did not alter the function or expression of LCC, RyR, SR Ca2+ pumps and the SR Ca2+ buffer calsequestrin (Online Figure III), the miR-24-induced defective E-C coupling cannot be attributed to direct or indirect regulation of major E-C coupling proteins other than JP2. Rather, the E-C coupling effects of miR-24 were fully reproduced by knocking down JP210,12 (Online Figure IV). The luciferase experiments (Fig. 2), together with the highly comparable effects of JP2 knockdown and miR-24 over-expression, doubly confirmed that miR-24 regulates E-C coupling by targeting JP2.
The pathogenesis of heart failure involves a variety of intracellular signaling cascades, including the calcineurin-NFAT pathway, the calmodulin-dependent protein kinase pathway, and pathways involving other protein kinases.21-23 How these pathogenic signals link to the down-regulation of JP2 and SERCA during heart failure is still unclear. As miR-24 is a member of the miR-23a-27a-24-2 cluster up-regulated by calcineurin-NFATc3 signaling,23 the identification of JP2 as a miR-24 target suggests a potential link between the upstream hypertrophy/heart failure signals and defective E-C coupling (Online Figure V).
Supplementary Material
Novelty and Significance.
What Is Known?
In cardiomyocytes, junctophilin-2 (JP2) links the sarcoplasmic reticulum (SR) to the cell membrane, including T-tubules (TT), forming structural units for excitation-contraction (EC) coupling.
During heart failure, the expression of JP2 is decreased, leading to decreased number and size of TT-SR junctions and a decrease in the efficiency of E-C coupling.
Knockdown of JP2 introduces structural and functional defects in TT-SR junctions, indicating that the expression of JP2 expression is one of the determinants of E-C coupling efficiency.
What New Information Does This Article Contribute?
The 3′ untranslated region of JP2 mRNA contains 2 binding sites for miR-24.
Over-expression of miR-24 decreases JP2 expression
MiR-24-induced JP2 down-regulation induces structural and functional defects inTT-SR junctions, indicating that homeostatic levels of miR-24 are important for the physiological E-C coupling.
Myocardial contractility is controlled by the Ca2+ signaling between the cell membrane/TTs and the SR. JP2, anchors the SR to TTs in heart cells and; thereby, determines the efficiency of Ca2+ signaling. In heart failure, JP2 expression is down-regulated, but the regulatory mechanism is not known. In the present study, we found that miR-24, a microRNA up-regulated in heart failure, is a suppressor of JP2 expression. Delivering miR-24 to cardiomyocytes fully reproduced the defective Ca2+ signaling in failing heart cells. These finding reveals a novel mechanism of JP2 regulation, and suggest new therapeutic options for the treatment of heart failure.
ACKNOWLEDGEMENTS
We thank Dr. Xue-Mei Hao for technical support, Drs. Heping Cheng and Haihong Ye for helpful comments, and Dr. IC Bruce for editing.
SOURCES OF FUNDING The 973 Program of China (2011CB809101, 2011CBA01101), the National Natural Science Foundation of China (30800475, 81070196, 81030001 and 30730013), the 863 Program of China (2011AA020108), the Program for New Century Excellent Talents in University, the Beijing Talents Foundation, and the NIH, USA (R01TW007269).
Non-standard Abbreviations
- 3′UTR
3′ untranslated region
- CHT
compensated hypertrophy
- CICR
Ca2+-induced
- Ca2+
release
- CSQ
calsequestrin
- DHT
decompensated hypertrophy
- Dspike
time delay from depolarization to the peak of the Ca2+ spike
- E-C
excitation-contraction
- ICa
whole-cell Ca2+ current through L-type Ca2+ channels
- JP2
junctophilin-2
- LCC
L-type Ca2+ channel
- RyR
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- TAC
transverse aortic constriction
- TT
transverse tubule
- TEM
transmission electron microscopy
Footnotes
DISCLOSURES None
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Excitation-contraction Coupling and Cardiac Contractile Force. 2nd ed Kluwer; Dordrecht: 2001. [Google Scholar]
- 3.Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358. doi: 10.1161/01.RES.0000060027.40275.A6. [DOI] [PubMed] [Google Scholar]
- 4.Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–729. doi: 10.1152/physrev.1997.77.3.699. [DOI] [PubMed] [Google Scholar]
- 5.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 6.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 7.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. doi: 10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 8.Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, Bai Y, Hao XM, Han Q, Zhang Y, Wang SQ. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, Ma J, Wehrens XH, Ackerman MJ. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail. 2011;4:214–223. doi: 10.1161/CIRCHEARTFAILURE.110.958694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu HD, Xu M, Li RC, Guo L, Lai YS, Xu SM, Li SF, Lu QL, Li LL, Zhang HB, Zhang YY, Zhang CM, Wang SQ. Ultrastructural Remodeling of Ca2+ Signaling Apparatus in Failing Heart Cells. Cardiovasc Res. 2012 doi: 10.1093/cvr/cvs195. doi: 10.1093/cvr/cvs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson P, Kiriakidou M, Sharma A, Maniataki E, Mourelatos Z. The microRNA world: small is mighty. Trends Biochem Sci. 2003;28:534–540. doi: 10.1016/j.tibs.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 14.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–730. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 16.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J Physiol. 1998;512:677–691. doi: 10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q, Huang Z, Xue H, Jin C, Ju XL, Han JD, Chen YG. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood. 2008;111:588–595. doi: 10.1182/blood-2007-05-092718. [DOI] [PubMed] [Google Scholar]
- 18.Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O’Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O, Becker KG, Gorospe M, Hide W, Lieberman J. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Mol Cell. 2009;35:610–625. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, Lieberman J, Lagna G, Hata A. Molecular basis for antagonism between PDGF and the TGFbeta family of signalling pathways by control of miR-24 expression. EMBO J. 2010;29:559–573. doi: 10.1038/emboj.2009.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah AM, Mann DL. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet. 2011;378:704–712. doi: 10.1016/S0140-6736(11)60894-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.