

Abstract
Acyltransferase (AT) domains of modular polyketide synthases exercise tight control over the choice of α-carboxyacyl-CoA substrates, but the mechanistic basis for this specificity is unknown. We show that whereas the specificity for the electrophilic malonyl or methylmalonyl component is primarily expressed in the first half-reaction (formation of the acyl enzyme intermediate), the second half-reaction shows comparable specificity for the acyl carrier protein that carries the nucleophilic pantetheine arm. We also show that currently used approaches for engineering AT domain specificity work mainly by degrading specificity for the natural substrate rather than by enhancing specificity for alternative substrates.
The broad spectrum of medicinal properties of polyketide natural products is a result of their structural diversity. Many polyketides are produced in an assembly line fashion by multimodular polyketide synthases (PKSs) through repeated decarboxylative condensations of simple subunits derived from α-carboxyacyl-Coenzyme A (CoA) building blocks. The acyltransferase (AT) domain is responsible for selection of an appropriate building block in each chain elongation cycle, typically a malonyl- or methylmalonyl-CoA that is transferred to a dedicated acyl carrier protein (ACP) domain found within the same module. The ketosynthase (KS) domain then catalyzes chain elongation between the growing polyketide chain and the ACP-bound extender unit. Together, the AT, KS, and ACP domains comprise the minimal modular components necessary and sufficient for polyketide chain elongation.
Because the structural diversity of polyketides is strongly influenced by extender unit choices made by AT domains, these enzymes have been obvious targets for engineering of novel polyketides. Nearly all AT domains involved in polyketide chain elongation have high specificity for their cognate malonyl- or methylmalonyl-CoA substrate.1 The biochemical basis for this strict specificity is not well understood. Previous efforts to engineer PKSs that regiospecifically incorporate unnatural extender units have focused on the swapping of entire AT domains2–5, often at the expense of essential protein-protein interactions that are required for kinetically efficient polyketide chain elongation.6 The structural basis for these problems is evident when one examines the X-ray crystal structures of [KS][AT] fragments of PKS modules, which reveal extensive protein-protein interfaces between the AT domain and other portions of the module, including the highly structured linker region between the KS and AT domains.7–8
A more conservative and increasingly popular approach to the engineering of AT specificity involves targeted mutagenesis of the active site or adjacent residues. Although previous attempts at such site-directed mutagenesis have shown altered product profiles in vivo9–10, the strategy has not been generalized, and the causes for frequently decreased titres of altered polyketides are poorly understood. We therefore sought to clarify the mechanistic basis for the malonyl- or methylmalonyl-CoA and the holo-ACP specificity of modular AT domains.
Loading of the electrophilic (methyl)malonyl group and subsequent transfer to the ACP occurs by a ping-pong bi bi mechanism involving the formation of an acyl enzyme intermediate that is then subject to nucleophilic attack by the thiol residue of the pantetheinyl side chain of the ACP11 (Scheme 1, Figure S2). The specific steps in this catalytic mechanism at which specificity for the electrophilic extender unit and the nucleophilic ACP are controlled have not been identified. Two important questions also remain unanswered--How do the (methyl)malonyl-CoA and ACP substrates contribute individually to overall AT specificity? And what role does competing hydrolytic off-loading of the α-carboxyacyl-CoA substrate play in the determination of AT substrate specificity? These questions have been difficult to answer due to the challenges of analyzing the rapid kinetics of AT-domain-catalyzed reactions independent of the activities of the remainder of the modular PKS.
Scheme 1.
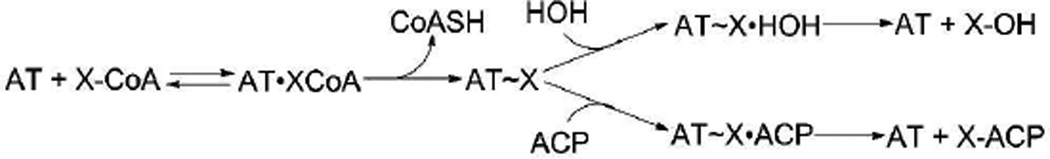
Ping-pong mechanism of the AT-catalyzed transacylation of a (methyl)malonyl-CoA (XCoA) substrate to a (methyl)malonyl-ACP (X-ACP). Transacylation requires holo-ACP, which contains the nucleophilic phosphopantetheine prosthetic group. Non-productive partitioning of the acyl enzyme intermediate by competing hydrolysis is shown in the top branch of the mechanism. (See Scheme S2 for detailed kinetic model.)
We present here results obtained using a continuous, coupled enzymatic assay to examine steady-state AT kinetics. AT-catalyzed release of free Coenzyme A (CoASH) is coupled to the formation of NADH in the CoASH-dependent α-ketoglutarate dehydrogenase-catalyzed conversion of α-ketoglutarate to succinyl-CoA, thereby allowing continuous and sensitive fluorometric monitoring of AT activity (Scheme S1).12 Importantly, this assay enables accurate monitoring of reactions containing sub-micromolar enzyme concentrations and low micromolar concentrations of the (methyl)malonyl-CoA and ACP co-substrates under multiple turnover conditions. Assay signal was dependent on the presence of both the AT protein and its α-carboxyacyl-CoA substrate, and no activity was observed without all components of the coupled reaction (Figure S1). To our knowledge, this represents the first use of a continuous assay to examine the kinetics of the reaction catalyzed by an AT domain derived from an assembly line PKS. To validate the analytical method, we have performed an in-depth analysis of a representative AT domain from module 3 of the 6-deoxyerythronolide B synthase (DEBS). This domain has previously been structurally characterized (PDB ID: 2QO3),8 and can be expressed as a stand-alone protein. The AT3 protein utilized here includes the KS-AT linker but does not include the post-AT linker.13
Previous studies have highlighted the prevalence of AT-mediated substrate hydrolysis and suggested its potential role in determining the specificity of the AT for its natural malonyl or methylmalonyl-CoA substrate.14 We therefore sought to quantify the kinetic parameters for AT3-mediated hydrolysis of its native substrate, methylmalonyl-CoA (MMCoA), as well as several nonnative acyl-CoA substrates in the absence of the holo-ACP cosubstrate (Figure 1). Interestingly, the observed rates of hydrolysis, as measured by kcat/Km, were approximately 200-fold higher for the native MMCoA substrate compared to both malonyl-CoA (MCoA) and propionyl-CoA (PCoA), implying that substrate discrimination by AT3 is exercised in the formation of the acyl-enzyme, rather than by selective hydrolytic cleavage of incorrectly acylated protein.
Figure 1.

Hydrolysis of acyl-CoA substrates by DEBS AT3. Kinetic parameters were measured by varying acyl-CoA concentrations in the absence of a holo-ACP co-substrate. In one case the inactive apo-ACP surrogate was used to examine the influence of protein-protein interactions on hydrolysis rate (see text for details). N.D.: Not Determined.
The α-carboxyacyl-CoA specificity of DEBS AT3 during transacylation to ACP3, as measured by the relative values of kcat/Km, is also approximately 30-fold higher for MMCoA compared to MCoA (Figure 2). This is expected, as the value of kcat/Km is not influenced by the ACP-dependent rate of breakdown of the acyl-enzyme intermediate in the simple model of the ping-pong reaction. On the other hand, the kcat for transacylation of MMCoA to its cognate holo-ACP exceeds that of hydrolysis by at least 1–2 orders of magnitude. Since the presence of the holo-ACP will not affect the rate of formation of the methylmalonyl (or malonyl)-enzyme, the observed increase in the rate of formation of CoASH indicates that during transacylation the major determinant of kcat must be the rate of formation of the acyl enzyme. By contrast, the major influence on the much slower kcat for hydrolysis is the hydrolytic cleavage of this acyl enzyme intermediate. (See the SI for the detailed kinetic model.)
Figure 2.

Acyl-CoA-limited transacylation by DEBS AT3 with its cognate holo-ACP.
Incorporation of a polyketide extender unit is also highly dependent on the interaction of the AT domain with its cognate ACP. As seen in Figure 3, DEBS AT3 prefers its cognate ACP3 by a factor of 25 in kcat/Km over the ACP from module 6. This highlights the need for proper protein-protein interactions during the second half-reaction of AT-catalyzed transacylation, and is directly reminiscent of KS-ACP interactions that are important during both chain translocation15 and chain elongation.16
Figure 3.

ACP-limited transacylation by DEBS AT3 with its cognate holo-ACP and DEBS holo-ACP6.
To determine whether AT-ACP interactions also influence competing hydrolysis as well as transacylation, we examined the kinetics of AT3-mediated hydrolysis in the presence of the inactive apo form of ACP3 (Figure 1). The observed steady-state rate of hydrolysis of MMCoA was not significantly perturbed by the presence of this protein, implying that the highly evolved protein-protein interactions between an AT and its cognate ACP exclusively benefit the rate at which the extender unit is transferred to the phosphopantetheinyl arm of the ACP.
To gain further insight into the AT3-catalyzed reaction, we examined two mutants--Q652L and the Y751H/S753F double mutant (2QO3 numbering). All three of these residues are virtually invariant in MMCoA-specific AT domains.9 Mutation of these residues in DEBS AT4 has enabled incorporation of a MCoA extender unit and production of the predicted 6-desmethyl-6-dEB analog in vivo.9 The effects of these mutations on the rates of either acyl-enzyme formation or net transacylation are unknown. Unexpectedly, variants of DEBS AT3 harboring these mutations have drastically (>100 fold) attenuated values of kcat for both hydrolysis and transacylation with either substrate, MMCoA or MCoA (Table 1). The rates of hydrolysis and transacylation with both substrates are similar to the rates of the corresponding reactions with wild-type AT3 in the presence of MCoA. Thus, the loss of substrate specificity appears to be due to a drastic decrease in catalytic power as a result of the mutations, rather than any enhanced preference for the unnatural MCoA substrate.
Table 1.
Rates of transacylation and hydrolysis by DEBS AT3 and mutant AT domains. Rates were measured in the presence of 150 uM α-carboxyacyl-CoA substrate. Transacylation reactions were run in the presence of 100 uM DEBS holo-ACP3.
| AT Domain | Substrate | v/[E]o Transacylation (min−1) |
v/[E]o Hydrolysis (min−1) |
|---|---|---|---|
| DEBSAT3 | MMCoA | 30 | 2 |
| DEBSAT3-Q652L | MMCoA | 0.2 | 0.1 |
| DEBSAT3-Y751H,S753F | MMCoA | <0.1 | <0.1 |
| DEBSAT3 | MCoA | 0.9 | 0.8 |
| DEBSAT3-Q652L | MCoA | <0.1 | <0.1 |
| DEBSAT3-Y751H,S753F | MCoA | <0.1 | <0.2 |
Assuming that DEBS AT3 is representative of other AT domains from assembly line PKSs, there are several important implications. First, in the presence of physiologically relevant (low micromolar) concentrations of the cognate MMCoA substrate, AT-catalyzed hydrolysis is unlikely to be a significant process in a native PKS module. Second, while the observed MMCoA specificity of an AT domain is expressed in the first half-reaction (the formation of the acyl enzyme), the second halfreaction also exhibits specificity for the nature of the ACP domain that carries the nucleophilic pantetheine side chain. Third, AT domains in PKS have evolved to keenly discriminate between closely related ACP domains, as evidenced by significant discrimination between two ACP domains from the same PKS assembly line. Finally, it is evident that current active site engineering approaches for altering α-carboxyacyl-CoA specificity have serious shortcomings in that at best they suppress the catalytic specificity for the natural substrate, thereby imposing a substantial kinetic penalty for utilization of the unnatural MCoA substrate. Future AT domain engineering should take into consideration the importance of AT-ACP interactions as well as the effects of mutagenesis on the relative rates of hydrolysis and transacylation. The conceptual and methodological advances presented in this report provide a fundamentally new starting point for the protein engineer who seeks to understand and manipulate the structural determinants of AT substrate specificity.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the laboratories of Dr. James Swartz and Dr. Jennifer Cochran for use of their fluorescent plate readers.
Funding Sources
This research was supported by grants from the National Institutes of Health (GM 087934 to C.K. and GM 022172 to D.E.C.).
ABBREVIATIONS
- AT
acyltransferase
- CoA
Coenzyme A
- CoASH
free Coenzyme A
- PKSs
Polyketide Synthases
- ACP
Acyl Carrier Protein
- KS
Ketosynthase
- MMCoA
Methylmalonyl CoA
- MCoA
Malonyl CoA
- PCoA
Propionyl CoA
- NADH
Nicotinamide Adenine Dinucleotide, reduced
- DEBS
6-Deoxyerythronolide B Synthase
Footnotes
ASSOCIATED CONTENT
Supporting Information
Materials, Methods, Derivation of Kinetic Constants, and Supplemental Figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- 1.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Annu. Rev. Biochem. 1999;68:219–253. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 2.Oliynyk M, Brown MJ, Cortes J, Staunton J, Leadlay PF. Chem Biol. 1996;3:833–839. doi: 10.1016/s1074-5521(96)90069-1. [DOI] [PubMed] [Google Scholar]
- 3.Lau J, Fu H, Cane DE, Khosla C. Biochemistry. 1999;38:1643–1651. doi: 10.1021/bi9820311. [DOI] [PubMed] [Google Scholar]
- 4.Stassi DL, Kakavas SJ, Reynolds KA, Gunawardana G, Swanson S, Zeidner D, Jackson M, Liu H, Buko A, Katz L. Proc Natl Acad Sci U S A. 1998;95:7305–7309. doi: 10.1073/pnas.95.13.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruan X, Pereda A, Stassi DL, Zeidner D, Summers RG, Jackson M, Shivakumar A, Kakavas S, Staver M, Donadio S, Katz L. J. Bacteriol. 1997;179:6416–6425. doi: 10.1128/jb.179.20.6416-6425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. J Am Chem Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- 7.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. Proc. Natl. Acad. Sci. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang Y, Chen AY, Kim CY, Cane DE, Khosla C. Chem. Biol. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reeves CD, Murli S, Ashley GW, Piagentini M, Hutchinson CR, McDaniel R. Biochemistry. 2001;40:15464–15470. doi: 10.1021/bi015864r. [DOI] [PubMed] [Google Scholar]
- 10.Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. ACS Chem. Biol. 2012 doi: 10.1021/cb300505w. Article ASAP. [DOI] [PubMed] [Google Scholar]
- 11.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu. Rev. Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 12.Molnos J, Gardiner R, Dale GE, Lange R. Anal. Biochem. 2003;319:171–176. doi: 10.1016/s0003-2697(03)00327-0. [DOI] [PubMed] [Google Scholar]
- 13.Chen AY, Cane DE, Khosla C. Chem. Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonnett SA, Rath CM, Shareef AR, Joels JR, Chemler JA, Hakansson K, Reynolds K, Sherman DH. Chem. Biol. 2011;18:1075–1081. doi: 10.1016/j.chembiol.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu N, Cane DE, Khosla C. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]
- 16.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. J. Am. Chem. Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.