

Summary
Cancer stem cells are self-renewing, tumorigenic cells at the apex of tumor hierarchies, and postulated to be quiescent in many tumor types. This issue of Cancer Discovery highlights a study that links the presentation of kinetochores within mitosis to an essential requirement for BUB1B/BubR1, broadening our understanding of the cell-cycle machinery in cancer stem cells.
Cancer can no longer be viewed through a myopic lens as cells simply undergoing rampant growth. It is now appreciated that a tumor develops in a calculated and self-serving manner, using the tumor microenvironment to regulate its growth and resistance to therapy as well as establishing a cellular hierarchy that provides for tumor maintenance. Cancer stem cells (CSC) reside at the top of neoplastic hierarchies and harbor the unique ability (as compared with all other neoplastic cells) to establish and drive tumor progression in orthotopic xenograft transplantation models. A central unresolved issue with CSCs has been their proliferative status. In normal hematopoiesis, a long-term, quiescent, self-renewing stem cell maintains the bone marrow over the life of the animal. Many leukemias display a parallel hierarchy with a quiescent leukemia stem cell that maintains the disease and exhibits therapeutic resistance. The situation in solid tissues is far less clear. Recent studies in reporter mice have shown that some tissues, notably the gut, contain at least 2 distinct pools of stem cells with at least one quiescent and another proliferative (1). Furthermore, these pools can replenish each other in response to injury, a logical mechanism to support organ stability if one compartment is selectively targeted by toxins, infection, and so forth. Evolving data in solid tumor stem cells suggest that multiple CSC pools exist within these tumors as well (2). Thus, CSCs may not have a single proliferative state or control mechanism. These unresolved issues and the dynamic heterogeneous nature of CSCs contribute to our inability to achieve cancer cures.
Glioblastoma [World Health Organization (WHO) grade 4] is one such tumor type for which a cellular hierarchy has been well established. Glioblastoma is the most common and deadly primary brain tumor, with a median survival of 12 to 15 months despite clinical intervention of surgical resection, radiotherapy, and chemotherapy with the DNA alkylating agent temozolomide (3). This survival rate has changed little in decades, underscoring the urgent need to develop novel treatment paradigms. It has been postulated that CSCs within glioblastoma, or glioblastoma stem cells (GSC), can directly contribute to tumor recurrence (4). Although recent studies have provided insight into the signaling pathways integral to GSC proliferation and survival, we know very little about the intrinsic cell-cycle machinery controlling this cell growth.
The mitotic phase of the cell cycle involves duplication of the genetic material of the cell and equal division from a mother cell into 2 daughter cells. As this inheritance may direct the cell fate of the daughter cells via the asymmetric cell division linked to stem cells, mitosis likely serves in maintenance of the cellular hierarchy. Transitioning through mitosis is required to establish the sustained proliferation hallmark of aggressive anaplastic malignancies, providing rationale for developing therapies to target mitosis. The first generation of antimitotics (e.g., paclitaxel, docetaxel, and vincristine) was developed to disrupt the microtubule dynamics that are required for the proper distribution of the genetic material. These drugs are still commonly used for cancer treatment, but their limitations have inspired the pursuit of more targeted therapies. Mitosis progresses as a series of finely orchestrated events that requires the coordinated action of a multitude of enzymatically active proteins, thereby providing numerous targets for small-molecule inhibitor development. The mitotic kinases (e.g., Aurora B and Plk1) are one such class of molecules that have moved forward as a target for anticancer drugs. Despite advances in treating other solid tumors, targeted antimitotics have not progressed beyond limited clinical trials for glioblastoma. This lack of progress is especially surprising given that the histopathologic assessment in diagnosing glioblastoma includes the identification of a high mitotic index. In addition, it has been reported that increased expression of mitotic genes is predictive for poor survival in patients with glioma (5).
In this issue of Cancer Discovery, Ding and colleagues (6) use RNA interference to screen for kinases central to the viability of glioblastoma cells grown under conditions to maintain a poorly differentiated state and tumorigenicity, that is, brain tumor–initiating cells (BTIC), but dispensable for non-neoplastic fetal neural stem cells (NSC; Fig. 1). More than 700 human kinases were targeted, with about 7% of kinases having representative short hairpin RNAs (shRNA) depleted in BTICs, but not in NSCs, indicating a specific requirement for cell growth in BTICs. These hits were then further paired down using an elegant bioinformatics approach to yield the mitotic checkpoint protein BubR1 (encoded by the BUB1B, Budding Uninhibited by Benzimidazoles 1 Homolog Beta, gene) as the top-scoring kinase. The authors validated the functional impact of BUB1B/BubR1 loss in BTICs with interrogation of cells isolated by the validated GSC marker SSEA1/CD15. Future studies will define the importance of these kinases in the full cellular hierarchy with a direct, upfront comparison with the differentiated tumor cells following prospective isolation based on surface marker expression for the GSCs and minimized culture. It is possible that screening the full cellular hierarchy may identify additional targets. As BUB1B/BubR1 is a kinase, targeted therapeutics may be readily developed and provide both a direct therapeutic benefit and a sensitization to cytotoxic therapies. This project used the control of a fetal tissue–specific stem cell, as these cells are most freely available, but changes in proliferation and mitotic control occur during development, suggesting that adult stem cells may have a distinct regulation of mitosis. Thus, preclinical trials will elucidate the impact that targeting BUB1B/BubR1 has on adult NSCs in vivo.
Figure 1.
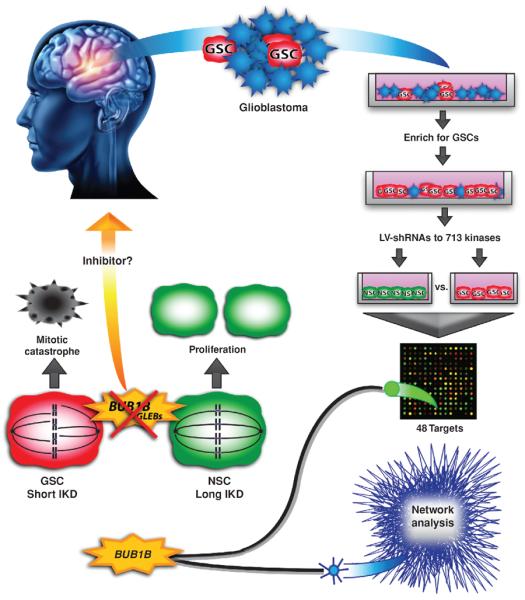
Schematic of experimental flow and results as described by Ding and colleagues (6). Primary human glioblastoma cells were taken from resected tumors and grown in conditions that enrich for GSCs. These cells, along with NSCs, were infected with a shRNA library that targeted the human kinome. Both cell types were then expanded for 21 days before isolating RNA and submitting it for microarray. Microarray analysis revealed 48 candidate targets that were depleted in GSCs, but not fetal NSCs. Network analysis was done on The Cancer Genome Atlas (TCGA) expression data to inform the results of the shRNA screen. By combining both methods, the authors were able to identify BUB1B/BubR1 as their top candidate. BUB1B/BubR1 is a protein that monitors proper spindle microtubule attachment to the kinetochore, and whose GLEBs domain is specifically required when the IKD is short, as is the case in GSCs. Knockdown of BUB1B/BubR1 in cells with a short IKD leads to mitotic catastrophe and cell death. However, in NSCs the IKD is longer and BUB1B/BubR1 is not required in this process, allowing for normal proliferation even in the presence of BUB1B/BubR1 knockdown. This feature makes BUB1B/BubR1, or potentially other proteins involved in this process, promising candidates for targeted therapies.
The authors validate a unique requirement in glioblastoma BTICs and RasV12-transformed astrocytes for BUB1B/BubR1 that is driven by the distance between the kinetochores of sister chromatids. Kinetochores are large protein complexes assembled at each centromere on each chromosome. Mitosis will not progress through anaphase, the actual separation of the genetic material to the daughter cells, until the kinetochores of every sister chromatid pair are captured by microtubules originating from opposite poles of the bipolar spindle. BUB1B/BubR1 plays a central role in this process. The authors show that a decrease in the interkinetochore distance (IKD) by only approximately 0.10 μm can alter the sensitivity of a cell to loss of BUB1B/BubR1. Moreover, they show that the domain within BUB1B/BubR1 required for kinetochore localization, the GLEBs (Gle2-binding sequence) domain, is essential only in cells that have short IKDs. The authors propose an intriguing model that reconciles differential reports regarding the requirement of the GLEBs domain and provokes additional questions. Specifically, further evaluation of the mechanism will be revealing, especially in regard to the role of Bub3, the binding partner of the GLEBs domain within BUB1B/BubR1, in cells with short IKDs.
Of great importance, however, is the feasibility of moving BUB1B/BubR1 into the clinic as an antimitotic therapeutic target. In this study, the authors show a complete lack of tumor engraftment when BUB1B/BubR1 is depleted from BTICs before orthotopic injection. This promising result supports the clinical efficacy of targeting BUB1B/BubR1, although studying tumor response to BUB1B/BubR1 inhibition after the tumor has been established will be more clinically relevant. They also show applicability to a potentially broad patient cohort, as the impact of targeting BUB1B/BubR1 on cell growth was not dependent on recently described glioblastoma subtypes (7). The authors report that some level of intertumor variation exists, as certain cells, described as those with longer IKDs, are not sensitive to BUB1B/BubR1 inhibition. Additional complexity may be derived from intratumoral variation. Glioblastoma is a heterogeneous tumor composed of a CSC hierarchy. Further heterogeneity is likely to exist within the CSC and the neoplastic non-CSC compartments with regard to IKD, therefore influencing the use of targeting BUB1B/BubR1. Striking disparity in the response to antimitotics for individual cells within a cell line has been previously reported (8). The ultimate fate of each individual tumor cell upon prolonged mitotic arrest through BUB1B/BubR1 inhibition, and the potential contribution to cellular plasticity within the hierarchy, is therefore still an open area for investigation. The intratumoral variation could also have an impact on pathologic scoring, which in itself might be a technical challenge and a limitation to the use of IKD as a biomarker for sensitivity to BUB1B/BubR1 inhibition.
Another clinical consideration is how to target BUB1B/BubR1 outside of targeted gene therapy or the de novo development of small-molecule inhibitors to BUB1B/BubR1. Targeting the mitotic kinase Aurora B or the mitotic kinesin centromere protein E (CENP-E) might be attractive alternatives, as they are central to kinetochore dynamics, have functional ties to BUB1B/BubR1, and have small-molecule inhibitors at various stages of clinical development (9). Although the authors identified equal toxicity for Aurora B inhibition in BTICs and NSCs, the use of targeting Aurora B in a preclinical malignant glioma model has recently been reported (10). Targeting CENP-E, on the other hand, is completely unexplored in glioblastoma but does significantly correlate with glioma grade (5).
This report by Ding and colleagues (6) advances the glioblastoma field, and likely that of other cancer types, through the identification of a novel molecular mechanism for kinetochore dynamics in glioblastoma cells. More importantly, it provides the basis for moving forward in the basic science realm to further elucidate cell-cycle signaling unique to CSCs as well as pushing forward targeted antimitotic therapies for glioblastoma treatment.
Acknowledgments
The authors thank Dr. Matthew Summers for insightful discussion and comments and Dr. David Schonberg for critical manuscript review.
Grant Support Work in the Rich laboratory is supported by NIH grants CA154130 and CA1129958 and the James S. McDonnell Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interests were disclosed.
REFERENCES
- 1.Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–5. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piccirillo SG, Combi R, Cajola L, Patrizi A, Redaelli S, Bentivegna A, et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene. 2009;28:1807–11. doi: 10.1038/onc.2009.27. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 5.Bie L, Zhao G, Cheng P, Rondeau G, Porwollik S, Ju Y, et al. The accuracy of survival time prediction for patients with glioma is improved by measuring mitotic spindle checkpoint gene expression. PLoS ONE. 2011;6:e25631. doi: 10.1371/journal.pone.0025631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Y, Hubert CG, Herman J, Corrin P, Toledo CM, Skutt-Kakaria K, et al. Cancer-specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discov. 2013;3:198–211. doi: 10.1158/2159-8290.CD-12-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant sub-types of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Kaestner P, Bastians H. Mitotic drug targets. J Cell Biochem. 2010;111:258–65. doi: 10.1002/jcb.22721. [DOI] [PubMed] [Google Scholar]
- 10.Diaz RJ, Golbourn B, Shekarforoush M, Smith CA, Rutka JT. Aurora kinase B/C inhibition impairs malignant glioma growth in vivo. J Neurooncol. 2012;108:349–60. doi: 10.1007/s11060-012-0835-2. [DOI] [PubMed] [Google Scholar]