

Abstract
Histone deacetylase inhibitors (HDACi) can modulate innate antiviral responses and render tumors more susceptible to oncolytic viruses (OVs); however, their effects on adaptive immunity in this context are largely unknown. Our present study reveals an unexpected property of the HDACi MS-275 that enhances viral vector-induced lymphopenia leading to selective depletion of bystander lymphocytes and regulatory T cells while allowing expansion of antigen-specific secondary responses. Coadministration of vaccine plus drug during the boosting phase focuses the immune response on the tumor by suppressing the primary immune response against the vaccine vector and enhancing the secondary response against the tumor antigen. Furthermore, improvement of T cell functionality was evident suggesting that MS-275 can orchestrate a complex array of effects that synergize immunotherapy and viral oncolysis. Surprisingly, while MS-275 dramatically enhanced efficacy, it suppressed autoimmune pathology, profoundly improving the therapeutic index.
Introduction
Oncolytic viruses (OVs) are promising therapeutics applicable to a variety of malignancies. One of the mechanisms defining the tumor selectivity of OVs is the fact that cancer cells frequently acquire defects in cellular innate antiviral responses, such as the type I interferon (IFN) pathway.1,2 As a result, it has been shown that IFN sensitive viruses such as vesicular stomatitis virus (VSV) are highly effective in targeting and killing tumor cells while sparing normal tissues.3,4 However, the extent of IFN nonresponsiveness is variable in cancer cell lines and patient tumors, which represents an obstacle to effective OV therapy.5,6 Histone deacetylase inhibitors (HDACi) are small molecules that are currently being evaluated clinically for the treatment of cancer but are also known to prevent the transcriptional activation of antiviral genes after IFN stimulation or virus infection.7,8,9 We have recently demonstrated that several HDACi can markedly enhance the susceptibility of tumor cells to VSV killing, providing a pharmacological strategy to potentially increase the spectrum of malignancies amenable to OV therapy.10
However, HDACi are also under investigation as anti-inflammatory and immunosuppressive drugs. Evidence from different animal models indicates that HDACi therapy ameliorates inflammatory/autoimmune diseases, enhances allograft survival and induces immune tolerance in graft-versus-host disease.11,12,13,14 Thus, although HDACi may enhance viral oncolysis, it is unclear whether such a benefit would be at the expense of the optimal development of antitumor immunity that may be required to synergize and/or sustain virus-induced tumor regression.
We have recently demonstrated that OVs can be engineered to express tumor-associated antigens and used as oncolytic vaccines in tumor-bearing hosts.15 In particular, when combined with a priming vaccine that expresses the same tumor antigen, oncolytic vaccines can lead to both tumor debulking by the virus and a large boost of tumor-specific cytotoxic T lymphocytes (CTL) in primed animals. Furthermore, the replicating oncolytic vector is amplified in the tumor leading to a larger boost in tumor-bearing animals leading to significantly enhanced numbers of antigen-specific tumor-infiltrating lymphocytes. We reasoned that the combination of HDACi and oncolytic vaccines would allow simultaneous investigation of the impact of HDACi on viral oncolysis and adaptive immunity against the virus and the tumor.
In the current study, we confirmed that MS-275, an inhibitor of class I HDACs, led to modestly prolonged viral replication in the tumor but dramatically enhanced tumor-free survival. Surprisingly, this drug was able to impair primary immune responses directed at the oncolytic vaccine vector while allowing for potent secondary immune responses focused on the tumor antigen transgene. In fact, coadministration of MS-275 with an oncolytic VSV booster vaccine led to a severe but selective lymphopenia leaving the boosted antitumor lymphocytes intact while depleting both conventional lymphocytes and regulatory T cells (Treg). More strikingly, this combination therapy not only led to enhanced therapeutic efficacy but also suppressed vaccine-associated autoimmune pathology, yielding a profound improvement of the therapeutic index. Our data indicate that immunomodulation by HDACi occurs at multiple levels and their therapeutic benefit depends on the context and timing.
Results
MS-275 extends oncolytic VSV activity in tumors
We have previously demonstrated that MS-275 can facilitate VSV replication in different types of tumors by modifying IFN-responsiveness both in vitro and in vivo.10 To test for such an activity in our current tumor model, we treated mice bearing 5-day-old intracranial B16-F10 melanomas with intravenous (i.v.) injection of an oncolytic VSV expressing the Firefly luciferase (VSV-Luc). MS-275 or vehicle was given intraperitoneally (i.p.) at a dose of 100 µg/mouse on a daily basis for 5 days as optimized previously.10 This confirmed that MS-275 coadministration extended VSV-Luc activity in these tumors (Supplementary Figure S1).
MS-275 dramatically improves the therapeutic outcome in combination with an oncolytic booster vaccine
We have recently demonstrated that by engineering VSV to express human dopachrome tautomerase (VSV-hDCT), we could turn it into a very potent booster vaccine while retaining its oncolytic properties in vivo.15 Combining a recombinant adenoviral vector (Ad) expressing hDCT (Ad-hDCT) and VSV-hDCT in a prime-boost manner dramatically enhanced therapeutic efficacy. The potency of this combination strategy prompted us to investigate whether coadministration of MS-275 with the oncolytic vaccine could further enhance this therapeutic strategy. Five days after intracranial inoculation of B16-F10 cells, mice were treated sequentially with Ad-hDCT and VSV-hDCT at a 14-day interval as described previously.15 MS-275 was administered 2 hours before the VSV-hDCT oncolytic vaccine and then given once daily for 5 consecutive days, which coincided with the persistence of VSV and the peak of the boosted CTL response.15,16 Vaccination with the Ad-hDCT vaccine alone prolonged animal survival to a median of 25 days, and this therapeutic effect was further enhanced by VSV-hDCT boosting (Figure 1). Despite the improvement of the survival rate, however, most animals treated with the prime-boost regimen ultimately succumbed to tumor progression. MS-275 alone had no therapeutic effect in this cancer model. Remarkably, concomitant treatment with MS-275 at the time of VSV-hDCT delivery dramatically enhanced the efficacy of the combination treatment and cured 64% (n = 11) of the mice. No further benefit was observed by giving MS-275 for more than 5 days (data not shown).
Figure 1.

MS-275 dramatically enhances the efficacy of oncolytic booster vaccination in a very stringent therapeutic intracranial melanoma model. C57BL/6 mice (n = 10–12 for each treatment, pooled data from two experiments) were injected intracranially with 1,000 B16-F10 cells. Five days later, they were vaccinated with 1 × 108 pfu of Ad-hDCT i.m. Following a 14-day interval, mice were boosted with 1 × 109 pfu of VSV-hDCT i.v., with or without five consecutive daily treatments with 100 µg of MS-275 or vehicle i.p. beginning 2 hours prior to injection of VSV. Controls either received Ad-BHG (empty vector) alone or Ad-hDCT ± MS-275 on day 5. An additional control group received five consecutive daily treatments with MS-275 alone beginning on day 5 treatment. Ad, adenoviral; hDCT, human dopachrome tautomerase; VSV, vesicular stomatitis virus.
Interestingly, when MS-275 was administered with Ad-hDCT at the priming step, we actually observed an attenuation of the CD8+ therapeutic effect (Figure 1) suggesting that correct timing (i.e., at boosting phase) of MS-275 treatment is critical.
MS-275 preserves secondary tumor-specific CTL and antibody responses but attenuates primary adaptive immunity against VSV
As MS-275 coadministration provided a rather mild extension of VSV-Luc activity in these tumors, we hypothesized that the drug might be serving to enhance the vaccine effects of our treatments which are key to efficacy in this model.15 We first examined the secondary immune response boosted by VSV by quantifying DCT-specific, IFN-γ-producing CD8+ T cells in the circulation at days 5 and 12 after VSV-hDCT booster vaccination. These time points were chosen based on our previous observation where the secondary T cell response induced by VSV-hDCT reached its peak at day 5 and declined after 12 days.15 Surprisingly, the magnitude of the DCT-specific CD8+ T cell response boosted by VSV vaccine was not further increased in the presence of MS-275 (Figure 2a). Similarly, DCT-specific IgG antibodies in plasma were significantly boosted regardless of MS-275 treatment (Figure 2b). Thus, MS-275 coadministration did not increase the secondary immune response to the tumor antigen transgene boosted by our oncolytic vaccine.
Figure 2.
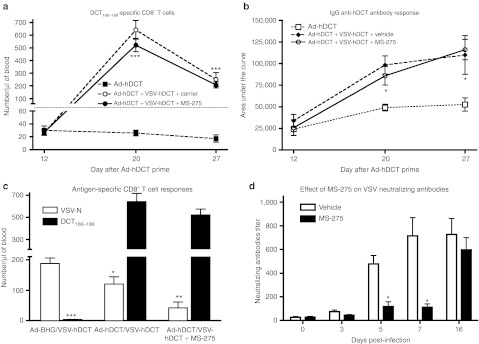
MS-275 inhibits primary immune responses while leaving secondary immune responses intact. B16-F10 tumor-bearing C57BL/6 mice (n = 10, pooled data from two experiments) were primed with 1 × 108 pfu of Ad-hDCT i.m. and boosted 14 days later with 1 × 109 pfu of VSV-hDCT i.v. with or without five consecutive daily treatments with 100 µg of MS-275 i.p. beginning 2 hours prior to injection of VSV. (a) On indicated days, tumor-specific CD8+ T cell responses were quantified in blood by flow cytometry after in vitro restimulation with the immunodominant epitope DCT180–188 and intracellular cytokine staining for IFN-γ. (b) Using an in-cell western assay, IgG anti-hDCT antibodies were quantified in plasma. (c) CD8+ T cell responses against the immunodominant viral epitope were compared to the immunodominant DCT epitope 5 days following boost. (d) Anti-VSV neutralizing antibodies were monitored. All graphs show means + SE, one- or two-way analysis of variance. *P < 0.05, **P < 0.01, ***P < 0.001. Ad, adenoviral; hDCT, human dopachrome tautomerase; IFN, interferon; VSV, vesicular stomatitis virus.
However, in the course of our immune analysis, we discovered that primary responses against the oncolytic vaccine vector were largely disabled by coadministration of drug. To evaluate this, we first measured CD8+ T cell responses against an immunodominant epitope from the N-protein of VSV at day 7 after VSV-hDCT inoculation. Consistent with our previous report, preimmunization with Ad-hDCT allowed a dramatic boost of DCT-specific secondary T cell response while decreasing the primary response against VSV (Figure 2c). VSV-reactive CD8+ T cells were further reduced in the presence of MS-275 confirming that MS-275 differentially influences expansion of memory and naïve CD8+ T cells. Importantly, neutralizing antibodies against VSV were strongly inhibited by MS-275 coadministration where induction of antibodies against the virus was delayed until after drug administration was halted (Figure 2d).
Altogether, these results highlight a very curious property of this drug as it is able to impede the generation of primary immune responses while leaving secondary responses entirely intact. This differential immunosuppression appears to allow for an immune response following oncolytic vaccine administration largely focused on the tumor antigen transgene.
MS-275 enhances and sustains lymphopenia induced by VSV booster vaccination
It is well known that intravenous administration of VSV induces a transient lymphopenia.17,18 We have also noted that intravenous injection with VSV induced a rapid and severe lymphopenia with cell counts hitting a minimum at 24 hours (Figure 3a) as reported by others.17 The lymphocyte counts were recovered in 3–5 days and often led to a transient increase over the normal level. Surprisingly, although MS-275 alone had a moderate effect on lymphocyte numbers, it dramatically delayed the reconstitution when concomitantly administered with VSV (Figure 3a). Using an inactive analogue of MS-27519 (Supplementary Figure S2), we confirmed that the exacerbation of VSV-induced lymphopenia required HDAC inhibition. As VSV-induced lymphopenia has been shown to depend on type I IFN signaling in lymphocytes,17,20 we wondered whether polyIC, a classic inducer of type I IFN, would also generate this effect when combined with MS-275. Indeed, data in Supplementary Figure S3 indicate that polyIC + MS-275 yielded an identical, extended lymphopenia though the mechanisms underlying the effect of MS-275 remain to be determined.
Figure 3.

VSV induces a transient lymphopenia that is significantly extended by MS-275 coadministration. Ad-hDCT-primed mice (n = 12, pooled data from three experiments) were boosted with VSV-hDCT in the presence of MS-275, MS-275 analogue or carrier. As controls, mice received MS-275 or were untreated. (a) Numbers of cells per µl of blood are displayed over a 30-day period post-treatment. Horizontal dotted line represents average count for untreated mice. (b) CD4+, (c) CD8+ T cell, and (d) B cell counts are also indicated. Ad, adenoviral; hDCT, human dopachrome tautomerase; VSV, vesicular stomatitis virus.
A closer examination indicated that both CD4 and CD8 T cell subsets and the B cell population were profoundly affected by the addition of MS-275 (Figure 3b–d). T cells were largely recovered in 2 weeks, but B cell recovery was much slower suggesting that B cells were more sensitive to the combination therapy. This treatment significantly reduced CD4+CD8+ double positive T cells in the thymus (Supplementary Figure S4a). Further analysis in the bone marrow indicated that pre-B and immature B cells were almost completely eliminated by the combination treatment as early as day 3 (Supplementary Figure S4b). These results suggest that MS-275 and VSV may have a synergistic effect on the survival of precursors leading to the delayed reconstitution of the peripheral T cells and B cells and a significantly extended lymphopenia in response to IFN.
MS-275 improves CTL quality
We next sought to determine whether lymphopenia might actually provide a favorable environment (e.g., more cytokines) which would be predicted to promote the functionality of boosted CTL. Indeed, compared to Ad:VSV prime:boost alone, the addition of MS-275 significantly increased the frequency of CD8+ T cells that coexpressed tumor necrosis factor-α and IFN-γ (Figure 4a) and the increased intensity of their production (Figure 4b,c), indicating a heightened response to stimulatory peptide. To further determine the impact of MS-275 on CTL functionality, we tested the avidity of CD8+ T cells from mice boosted with or without MS-275. Figure 4d shows that approximately sixfold more CD8+ T cells could respond to the lowest concentration of the immunodominant peptide from DCT when mice had received MS-275 treatment, confirming that CTL developed in this environment displayed an enhanced functionality.
Figure 4.

Coadministration of MS-275 with oncolytic vaccine booster generates more functional tumor antigen-specific T cells. Blood-derived DCT180–188-specific CD8+ T cells were obtained 5 days after VSV boosting and their cytokine production and avidity were assessed. (a) The frequency capable of simultaneous production of IFN-γ and TNF-α and the amount of (b) IFN-γ and (c) TNF-α produced per cell (measured as mean fluorescence intensity). (d) The functional avidity of DCT180–188-specific CD8+ splenocytes was assessed by flow cytometric detection of intracellular IFN-γ after in vitro restimulation with different dilutions of cognate peptide (n = 5 for each treatment). DCT, dopachrome tautomerase; IFN, interferon; TNF-α, tumor necrosis factor-α VSV, vesicular stomatitis virus.
MS-275 reduces Tregs, especially those that express a high level of Foxp3
The lymphopenia, especially the reduction of total CD4+ T cells, induced by MS-275 plus virus led us to assess it's direct impact on CD4+ Foxp3+ Tregs. The number of Tregs in the blood was significantly decreased during booster immunization, and it took 2 weeks for them to recover (Figure 5a). Notably, the intensity of Foxp3 expression by Tregs was significantly lower in mice coadministered MS-275 (Figure 5b) suggesting the drug may selectively remove Foxp3 high Tregs thought to have a stronger suppressive function.21,22 Downregulation of Treg in the context of an oncolytic booster vaccine increases the ratio of effectors to Tregs (Figure 5c) and may allow the secondary CD8+ T cell responses induced to function in a less stringently regulated environment. Consistent with observations in the periphery, reduction of Tregs and increase of antigen-specific effector T cells in the tumor were also evident in the group of combination therapy (Supplementary Figure S5).
Figure 5.
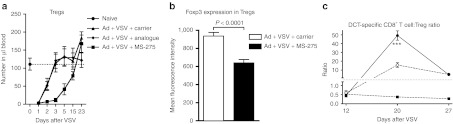
Coadministration of VSV and MS-275 depleted Treg numbers leaving residual Tregs with lower FoxP3 expression levels. Ad-hDCT-primed C57BL/6 mice were boosted with VSV-hDCT in the presence of MS-275, MS-275 analogue or carrier as detailed in Figure 2. (a) The kinetics of CD4+/FoxP3+ Tregs in blood following treatment. (b) The level of Treg expression of FoxP3 as measured by mean fluorescence intensity on day 7 after treatment. (c) The ratio of DCT-specific CD8+ T cells to CD4+/FoxP3+ Tregs in boosted animals plus/minus MS-275. ***P < 0.0001. Ad, adenoviral; hDCT, human dopachrome tautomerase; VSV, vesicular stomatitis virus.
MS-275 prevents vaccine-induced autoimmune vitiligo
We have noted that mice treated with the our oncolytic vaccine prime:boost regimen targeting DCT developed severe systemic vitiligo (depigmentation), an indication of autoimmune destruction of normal melanocytes expressing DCT. Remarkably, however, the induction of systemic vitiligo was almost completely abolished by concomitant treatment with MS-275, in contrast to it's effect on the enhancement of antitumor efficacy (Figure 6).
Figure 6.

Coadministration of MS-275 during boosting dramatically reduces autoimmunity. Tumor-bearing C57BL/6 mice were treated with Ad-hDCT + VSV-hDCT in the presence or absence of MS-275 and their vitiligo development was recorded (representatives of 30 mice for each treatment). The left panel displays five mice 2 months after Ad-hDCT + VSV-hDCT vaccination. The right panel displays five mice having been treated with Ad-hDCT + VSV-hDCT + MS-275 also 2 months after boosting. Note that MS-275 was administered during boosting and therefore failed to prevent vitiligo at surgical site on the head where Ad-hDCT priming alone was able to induce vitiligo localized to the site of surgical. Ad, adenoviral; hDCT, human dopachrome tautomerase; VSV, vesicular stomatitis virus.
Discussion
We have previously reported that HDACi can modulate IFN signaling pathways to enhance the susceptibility of tumor cells to OV killing, but their effect on the systemic immune responses in this context is unknown.10,23 Our current study demonstrates that coadministration of MS-275, a class I HDACi that is currently in clinical trials as anticancer agent, inhibits both the cellular and humoral immune that responses against the viral vector further supporting the combination strategy of HDACi and oncolytic therapy. However, MS-275 also downregulates the primary response against tumor antigens compromising the induction of antitumor immunity. This compromising effect can be avoided if coadministration of HDACi and oncolytic vaccine is carefully timed, namely at boosting phase, leading to synergy between oncolysis and T cell responses. Such a combination results in a selective lymphopenia that selectively reduces Tregs and naïve lymphocytes providing a favorable environment allowing a focused expansion of highly functional antitumor CTLs. As a consequence, enhanced oncolytic activity and antitumor immunity lead to a more than 60% durable cure rate in a very challenging cancer model. Most strikingly, while MS-275 dramatically enhanced efficacy, it suppressed autoimmune pathology, profoundly improving the therapeutic index.
It is well known that intravenous delivery of VSV induces a transient lymphopenia that has been attributed to the coincident induction of type I IFNs.17,20 Some studies suggest that memory CD8+ T cells are more sensitive to IFN-dependent early attrition than naive CD8+ T cells, but others have argued that memory T cells have lowered expression of IFN receptor and STAT1, leading to reduced sensitivity to IFN-mediated depletion.24,25,26,27 Another group reported that both naïve and memory CD8+ T cells could be sensitized by IFN at the early stage of viral infection but antigen-specific T cells were rescued by subsequent antigenic stimulation while bystander T cells died after initial nonspecific activation.28 Our demonstration that pre-existing tumor antigen-specific T cells could be dramatically boosted by oncolytic vaccine during lymphopenia appears to support the latter two possibilities. Interestingly, coadministration of MS-275 prolonged the lymphopenia without affecting the expansion of secondary T cell responses, further extending a favorable environment for the development of antitumor immunity while reducing/delaying antiviral responses. The drug alone did not significantly affect circulating lymphocytes suggesting that its effect may lie in the delay of reconstitution. This speculation is supported by the observations that the virus:drug combo selectively eliminated lymphocyte precursors in the bone marrow and thymus, consistent with an important role for HDAC1/2 in lymphocyte development.29
It has been demonstrated in other cancer immunotherapeutic settings that elimination of unwanted immune cells can provide supportive cytokines for the functional development of tumor antigen-specific CD8+ T cells particularly during adoptive cell therapy.30,31 The fact that a higher frequency of CD8+ T cells that can produce more IFN-γ and tumor necrosis factor-α was found in animals cotreated with MS-275 supports this notion.32 It is notable that these T cells with enhanced cytokine profiles performed better in the functional avidity assay. Having CTL that respond more strongly to cognate antigen should lead to a more potent antitumor immune response. Our finding may also have important implications for adoptive cellular therapy because this IFN:drug combo is able to generate a transient lymphopenia while preserving the desired immune responses.33
Treg plays an important role in maintaining immunological tolerance to self/tumor antigens, and depletion of Treg is a key mechanism underlying the effectiveness of cancer immunotherapy.34 In contrast to SAHA and VPA, which expand Tregs, MS-275 can downregulate Treg function.35 We demonstrate that in the context of VSV infection, MS-275 reduces the number of Treg, especially those that express high levels of Foxp3,21,22 revealing a novel aspect of MS-275 as a strong immunomodulator. It is likely that the removal of Treg-mediated immune suppression contributes to the enhanced antitumor efficacy following the combination therapy. Among all cell populations, B cell depletion appeared to be most profound. This correlated with a reduction of neutralizing antibodies against the oncolytic vector, likely contributing to the enhanced viral replication that was observed in the tumor by in vivo imaging on days 4 and 5 after infection when neutralizing antibodies have begun to appear in the absence of drug. Interestingly, tumor-specific antibodies could still be boosted in the presence of the HDACi, suggesting that memory B cells must be resistant to elimination. Several recent studies suggest that naïve B cells or certain subsets of B cells negatively regulate antitumor immunity and depletion of B cells increases the efficacy of cancer vaccination.36 We speculate that removal of B cells together with Treg reduction may further antagonize inhibitory networks.37
Another novel and very important finding in this study is the prevention of vaccine-induced vitiligo by MS-275. Autoimmune pathology has been observed in both preclinical and clinical studies and has been considered an unavoidable outcome following cancer immunotherapy against a self/tumor antigen.38,39,40 In fact, the association of autoimmune pathology with enhanced clinical responses has even led to it being identified as a positive prognostic factor.41,42 Using vitiligo as a read-out, we have previously demonstrated that inflammatory signals in the skin are essential to the recruitment of these DCT-specific T cells and to render normal melanocytes more susceptible to destruction (upregulation of major histocompatibility complex/self antigens or stress molecules).43,44 Strikingly, disseminated vitiligo was dramatically diminished when MS-275 was codelivered with virus. To the best of our knowledge, this represents the first time that antimelanoma efficacy was dramatically enhanced with a simultaneous reduction in vitiligo. Type I IFN has been implicated as a key factor in triggering autoimmune tissue damage45 and the ability of MS-275 to modify IFN signaling may contribute to reduced autoimmunity. MS-275 also has known anti-inflammatory properties46,47,48, and we speculate that MS-275 administration may suppress these inflammatory signals and thus reduce the recruitment of effector cells into the skin. This notion is supported by other observations where MS-275 has a strong anti-inflammatory effect in ameliorating arthritis through inhibition of proinflammatory cytokines and immune cell recruitment.49 Our finding may offer a pharmacological strategy to enhance antitumoral immunity while preventing unwanted autoimmune sequelae.
In summary, we sought to combine an oncolytic vaccine therapy with an HDACi previously demonstrated to enhance viral oncolysis by modifying IFN signaling and found that this combination also mediated significant modification of both antiviral and antitumor acquired immunity. By reducing antiviral responses while focusing the immune response on the tumor, we were able to extend viral oncolysis, enhance antitumor destruction and even reduce autoimmune sequelae. The selective immunosuppressive actions of this drug imply that it should not be combined with therapies aimed at generating primary immune responses including not only vaccination but potentially also viral oncolysis as the drug may impair in situ vaccine effects even while enhancing viral replication. Our results provide the evidence that MS-275 can function as a selectively immunosuppressive drug that can enhance the potency of an oncolytic vaccine booster. Importantly, this differential immunosuppression can be induced in combination with nonviral IFN inducers such as Poly I:C. This study highlights the importance of monitoring immunity while combining chemotherapy and biologics as unexpected interactions may occur.
Materials and Methods
Mice. Female, age-matched (8–10 weeks old at initiation of experiments) C57BL/6 were purchased from Charles River Laboratories (Wilmington, MA) and housed in a controlled environment in the Central Animal Facility at McMaster University with food and water provided ad libitum. All animal experimentation was approved by McMaster University's Animal Research Ethics Board and complied with the Canadian Council on Animal Care guidelines.
Viral vectors. Ad-hDCT is a replication-deficient, E1/E3-deleted Ad vector containing the full-length hDCT transgene.44 Replication-competent VSV-hDCT and VSV-Luc carry transgenes encoding hDCT and firefly luciferase, respectively, and have been described.15,50 The Ad-BHG and VSV-MT were control vectors, lacking a transgene.
Cells and culture conditions. B16-F10 cells were grown at 37 °C in a humidified atmosphere with 5% CO2 in F11-minimum essential medium containing 10% fetal bovine serum, 2 mmol/l L-glutamine, 5 ml sodium pyruvate, 5 ml minimum essential medium nonessential amino acids, 5 ml vitamin solution, 55 µmol/l 2-mercaptoethanol and antibiotics (all cell culture reagents from Invitrogen, Grand Island, NY).
Prime-boost protocol. Mice were primed by intramuscular injection of 1 × 108 pfu of Ad. For boosting, 1 × 109 pfu of VSV was injected intravenously 14 days later.
Cancer model. To establish intracranial tumors, mice received intracranial injections of 1 × 103 B16-F10 cells in 1 µl of phosphate-buffered saline (PBS). Under anesthetic, mice were placed in a stereotactic instrument (Xymotech Biosystems Inc, Mont-Royal, Quebec, Canada) and an incision made in the scalp to expose the skull. A small burr hole was drilled through the skull at the injection site. Cells were injected with a 26-gauge needle mounted on a 10 µl Hamilton syringe (Hamilton Company, Reno, NV) at the following site in the right hemisphere of the brain (relative to bregma): 0.62 mm anterior, 2.25 mm lateral, and 4.0 mm deep. Cells were injected over a period of 1 minute and the needle was left in place for 2 minutes prior to withdrawal to minimize reflux along the injection tract. The scalp incision was closed with stainless steel clips that were removed 7–10 days later.
Peptides. The immunodominant peptide from DCT that binds to H-2Kb (DCT180–188, SVYDFFVWL) was synthesized by PepScan Systems (Lelystad, The Netherlands). The H-2Kb-restricted epitope from the N protein of VSV (RGYVYQGL) was purchased from Biomer Technologies (Hayward, CA).
Antibodies. Monoclonal antibodies recognizing the following targets were used for flow cytometry assays: CD16/CD32 (Fc Block), CD3 (clone 145-2C11), CD4 (RM4-5), CD8 (53–6.7), IFN-γ (XMG1.2), tumor necrosis factor-α (MP6-XT22), CD19 (1D3), B220 (RA3-6B2) (BD Biosciences, Mississauga, Ontario, Canada), and Foxp3 (FJK-16s) (eBioscience, San Diego, CA).
Detection of antigen-specific T cell responses. Peripheral lymphocytes were restimulated with peptides (1 µg/ml) at 37 °C for 5 hours with brefeldin A (Golgi Plug, 1 µg/ml; BD Biosciences) added after 1 hour. Cells were treated with Fc block, stained for surface expression of CD3 and CD8 and then fixed, permeabilized (Cytofix/Cytoperm, BD Biosciences), and stained for intracellular IFN-γ and tumor necrosis factor-α. Data were acquired using a FACSCanto with FACSDiva software (BD Biosciences) and analyzed with FlowJo software (Tree Star, Ashland, OR).
T cell functional avidity assay. The functional avidity of T cells was determined with the same method used to assess antigen-specific responses (above) with modifications. Specifically, lymphocytes were stimulated in vitro with tenfold serial peptide dilutions ranging from 1 µg/ml to 10 pg/ml. The frequency of CD8+ T cells that produced IFN-γ at each peptide concentration was determined. These results were then plotted as the proportion of cell responding relative to the response induced by the highest concentration (i.e., 1 µg/ml) of peptide.
Detection of VSV-neutralizing antibodies. Serum or plasma samples were acquired from blood. Vero cells were seeded into a flat-bottom 96-well culture plate (BD Biosciences) at a density of 12,500 cells/well. Twenty-four hours later, serum or plasma samples were diluted 1/50 in serum-free medium. This, plus subsequent 1:2 serial dilutions were made in a separate 96-well plate (50 µl per well). To each well, 2 × 105 pfu of VSV in 50 µl of serum-free medium was added and allowed to incubate for 1 hour at 37 °C. Each aliquot of serum/plasma + VSV was then transferred to a well of confluent Vero cells and incubated for 48 hours at 37 °C. Cell viability was assessed by alamar blue staining (Invitrogen) and detection using a fluoroskan reader (Thermo, Nepean, Ontario, Canada). The neutralizing antibody titer was defined as the serum/plasma dilution at which cell viability remained 50% of the cells-only positive control.
Quantification of DCT-specific antibodies. DCT-specific antibodies were detected using an in-cell western blotting assay. U-2OS cells were seeded into a 96-well flat-bottom culture plate and cultured at 37 °C. Once confluent, they were infected with a vaccinia vector expressing hDCT at a multiplicity of infection of 5 for 6 hours. Infected cells were fixed in the plates with 100 µl of 4% paraformaldehyde for 15 minutes at 37 °C, followed by the addition of 100 µl of ice-cold acetone:methanol (1:1) for 5 minutes at room temperature. Cells were rehydrated with PBS. Plates were blocked with 1% BSA (wt/vol) in PBS overnight at 4 °C or for 1 hour at room temperature. Blocking solution was removed and the plates washed three times with 0.1% Tween-20 (v/v) in PBS. Serum/plasma samples were diluted (six serial dilutions per sample, done in duplicate) in a separate 96-well plate and then transferred onto the fixed cells for 1 hour at room temperature on a shaker or overnight at 4 °C. Plates were washed three times and Alexa Fluor 680-conjugated goat antimouse secondary antibody diluted 1:2,000 in PBS was added to each well for 1 hour at room temperature on a shaker. Plates were washed three times and fluorescence detected using an Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE). Average background fluorescence (cells + secondary antibody only) was subtracted from values obtained from samples. The adjusted optical density values were then plotted for all dilutions for each sample. The area under the curve was determined for each sample and used to plot the magnitude of the antibody response.
In vivo imaging of VSV infection. Mice bearing 5-day-old B16-F10 intracranial melanomas received i.v. injections of 4 × 108 pfu of VSV-Luc with or without cotreatment with MS-275. Each of the 4 days following treatment with VSV, mice were injected with 3 mg of d-luciferin (Molecular Imaging Products Company, Ann Arbor, MI) intraperitoneally. Under anesthetic, VSV infection was visualized with a 200 Series Imaging System (Xenogen Corporation, Hopkinton, MA). Data acquisition and analysis were performed using Living Image v2.5 software (Xenogen, Almeda, CA). Images were captured under identical exposure, aperture and pixel binning settings.
Statistical analyses. GraphPad Prism for Windows (GraphPad Software, San Diego, CA) was used for graphing. For statistical analyses, GraphPad Prism and Minitab Statistical Software (Minitab Inc., State College, PA) were used. If required, data were normalized by log transformation. Student's two-tailed t-test, one- or two-way analysis of variance or general linear modeling was used to query immune response data. Differences between means were considered significant at P ≤ 0.05. Means plus standard error bars are shown. Survival data were analyzed using the Kaplan–Meier method and the logrank test.
SUPPLEMENTARY MATERIAL Figure S1. Coadministration of MS-275 extends VSV-luc activity in B16-F10 tumors. Figure S2. Structure of MS-275 and inactive analogue. Figure S3. PolyI:C induces a lymphopenia that is extended by MS-275 coadministration. Figure S4. Coadministration of VSV and MS-275 depleted immature lymphocyte precursors in bone marrow and thymus. Figure S5. Combination therapy reduces Tregs but increases effector T cells in the tumor.
Acknowledgments
This work was supported by grants to Y.W. from the Canadian Institutes of Health Research [MOP-67066] and to B.D.L. and Y.W. from the Ontario Institute for Cancer Research and to B.D.L. from the Canadian Cancer Society Research Institute. The authors thank Natasha Khazdan for producing the rVSV vectors. The authors declared no conflict of interest.
Supplementary Material
REFERENCES
- Barber GN. VSV-tumor selective replication and protein translation. Oncogene. 2005;24:7710–7719. doi: 10.1038/sj.onc.1209042. [DOI] [PubMed] [Google Scholar]
- Parato KA, Senger D, Forsyth PA., and, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N.et al. (2000Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus Nat Med 6821–825. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S.et al. (2003VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents Cancer Cell 4263–275. [DOI] [PubMed] [Google Scholar]
- Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E.et al. (2010Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production Proc Natl Acad Sci USA 1071576–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo JS, Le Boeuf F, Lai F, Cox J, Vaha-Koskela M, Abdelbary H.et al. (2010A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers Mol Ther 181123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marié I.et al. (2004Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity Proc Natl Acad Sci USA 1019578–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minucci S., and, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Nusinzon I., and, Horvath CM. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol. 2006;26:3106–3113. doi: 10.1128/MCB.26.8.3106-3113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyên TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS.et al. (2008Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis Proc Natl Acad Sci USA 10514981–14986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard F., and, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases. Drug Discov Today. 2005;10:197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- Choi S., and, Reddy P. HDAC inhibition and graft versus host disease. Mol Med. 2011;17:404–416. doi: 10.2119/molmed.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL., and, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakespear MR, Halili MA, Irvine KM, Fairlie DP., and, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32:335–343. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E.et al. (2010Potentiating cancer immunotherapy using an oncolytic virus Mol Ther 181430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D.et al. (2006Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas J Natl Cancer Inst 981546–1557. [DOI] [PubMed] [Google Scholar]
- Kamphuis E, Junt T, Waibler Z, Forster R., and, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- McNally JM, Zarozinski CC, Lin MY, Brehm MA, Chen HD., and, Welsh RM. Attrition of bystander CD8 T cells during virus-induced T-cell and interferon responses. J Virol. 2001;75:5965–5976. doi: 10.1128/JVI.75.13.5965-5976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Ando T, Tsuchiya K, Fukazawa N, Saito A, Mariko Y.et al. (1999Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives J Med Chem 423001–3003. [DOI] [PubMed] [Google Scholar]
- Schattner A, Meshorer A., and, Wallach D. Involvement of interferon in virus-induced lymphopenia. Cell Immunol. 1983;79:11–25. doi: 10.1016/0008-8749(83)90046-1. [DOI] [PubMed] [Google Scholar]
- Chauhan SK, Saban DR, Lee HK., and, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol. 2009;182:148–153. doi: 10.4049/jimmunol.182.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Hennezel E, Yurchenko E, Sgouroudis E, Hay V., and, Piccirillo CA. Single-cell analysis of the human T regulatory population uncovers functional heterogeneity and instability within FOXP3+ cells. J Immunol. 2011;186:6788–6797. doi: 10.4049/jimmunol.1100269. [DOI] [PubMed] [Google Scholar]
- MacTavish H, Diallo JS, Huang B, Stanford M, Le Boeuf F, De Silva N.et al. (2010Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors PLoS ONE 5e14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondi E, Rogge L, Lutfalla G, Uzé G., and, Pellegrini S. Down-modulation of responses to type I IFN upon T cell activation. J Immunol. 2003;170:749–756. doi: 10.4049/jimmunol.170.2.749. [DOI] [PubMed] [Google Scholar]
- Gil MP, Salomon R, Louten J., and, Biron CA. Modulation of STAT1 protein levels: a mechanism shaping CD8 T-cell responses in vivo. Blood. 2006;107:987–993. doi: 10.1182/blood-2005-07-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HD, Urban SL., and, Welsh RM. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J Virol. 2011;85:5929–5939. doi: 10.1128/JVI.02516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock CD, Kim SK., and, Welsh RM. Attrition of virus-specific memory CD8+ T cells during reconstitution of lymphopenic environments. J Immunol. 2003;171:655–663. doi: 10.4049/jimmunol.171.2.655. [DOI] [PubMed] [Google Scholar]
- Jiang J, Lau LL., and, Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol. 2003;171:4352–4358. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C.et al. (2010Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression Genes Dev 24455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Zhang H, Meadors J, Poon R, Guimond M., and, Mackall CL. Harnessing the physiology of lymphopenia to support adoptive immunotherapy in lymphoreplete hosts. Blood. 2009;114:3831–3840. doi: 10.1182/blood-2009-03-212134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionda F, Fry TJ, Milliron MJ, McKirdy MA, Tagaya Y., and, Mackall CL. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. J Clin Invest. 2005;115:1177–1187. doi: 10.1172/JCI23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Darrah PA., and, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- Weber J, Atkins M, Hwu P, Radvanyi L, Sznol M., and, Yee C, Immunotherapy Task Force of the NCI Investigational Drug Steering Committee White paper on adoptive cell therapy for cancer with tumor-infiltrating lymphocytes: a report of the CTEP subcommittee on adoptive cell therapy. Clin Cancer Res. 2011;17:1664–1673. doi: 10.1158/1078-0432.CCR-10-2272. [DOI] [PubMed] [Google Scholar]
- Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR.et al. (2005CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells J Immunol 1742591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P.et al. (2012Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models PLoS ONE 7e30815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaziz JD, Yanaba K., and, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev. 2008;224:201–214. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S.et al. (2010FcRgamma activation regulates inflammation-associated squamous carcinogenesis Cancer Cell 17121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi RR. Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol. 2008;8:970–976. doi: 10.1038/nri2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa E. The risk of autoimmunity associated with tumor immunotherapy. Nat Immunol. 2001;2:789–792. doi: 10.1038/ni0901-789. [DOI] [PubMed] [Google Scholar]
- Koon H., and, Atkins M. Autoimmunity and immunotherapy for cancer. N Engl J Med. 2006;354:758–760. doi: 10.1056/NEJMe058307. [DOI] [PubMed] [Google Scholar]
- Nanda NK., and, Sercarz EE. Induction of anti-self-immunity to cure cancer. Cell. 1995;82:13–17. doi: 10.1016/0092-8674(95)90047-0. [DOI] [PubMed] [Google Scholar]
- Overwijk WW., and, Restifo NP. Autoimmunity and the immunotherapy of cancer: targeting the “self ” to destroy the “other”. Crit Rev Immunol. 2000;20:433–450. [PMC free article] [PubMed] [Google Scholar]
- Bridle BW, Li J, Jiang S, Chang R, Lichty BD, Bramson JL.et al. (2010Immunotherapy can reject intracranial tumor cells without damaging the brain despite sharing the target antigen J Immunol 1844269–4275. [DOI] [PubMed] [Google Scholar]
- Lane C, Leitch J, Tan X, Hadjati J, Bramson JL., and, Wan Y. Vaccination-induced autoimmune vitiligo is a consequence of secondary trauma to the skin. Cancer Res. 2004;64:1509–1514. doi: 10.1158/0008-5472.can-03-3227. [DOI] [PubMed] [Google Scholar]
- Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S.et al. (2005Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease Nat Med 11138–145. [DOI] [PubMed] [Google Scholar]
- Adcock IM. Histone deacetylase inhibitors as novel anti-inflammatory agents. Curr Opin Investig Drugs. 2006;7:966–973. [PubMed] [Google Scholar]
- Nencioni A, Beck J, Werth D, Grünebach F, Patrone F, Ballestrero A.et al. (2007Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity Clin Cancer Res 133933–3941. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Zhang Z., and, Schluesener HJ. MS-275, an histone deacetylase inhibitor, reduces the inflammatory reaction in rat experimental autoimmune neuritis. Neuroscience. 2010;169:370–377. doi: 10.1016/j.neuroscience.2010.04.074. [DOI] [PubMed] [Google Scholar]
- Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L.et al. (2007Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents Br J Pharmacol 150862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD.et al. (2007Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity Mol Ther 15123–130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.