

Background: Typical FeFe and MnFe cofactors bind to numerous enzymes such as ribonucleotide reductases. Crystallographic data suggest x-ray photoreduction (XPR) effects.
Results: Rapid XPR-induced cofactor changes were monitored using time-resolved x-ray absorption spectroscopy.
Conclusion: The XPR-induced cofactor states differ significantly from the native configurations, but comply with crystallographic structures.
Significance: Structure determination for high-valent dimetal-oxygen cofactors requires free electron-laser protein crystallography combined with x-ray spectroscopy.
Keywords: Kinetics, Metalloenzymes, Protein Crystallization, Ribonucleotide Reductase, X-ray Absorption Spectroscopy, Chlamydia trachomatis, MnFe and FeFe Cofactors, X-ray Photoreduction
Abstract
Prototypic dinuclear metal cofactors with varying metallation constitute a class of O2-activating catalysts in numerous enzymes such as ribonucleotide reductase. Reliable structures are required to unravel the reaction mechanisms. However, protein crystallography data may be compromised by x-ray photoreduction (XRP). We studied XPR of Fe(III)Fe(III) and Mn(III)Fe(III) sites in the R2 subunit of Chlamydia trachomatis ribonucleotide reductase using x-ray absorption spectroscopy. Rapid and biphasic x-ray photoreduction kinetics at 20 and 80 K for both cofactor types suggested sequential formation of (III,II) and (II,II) species and similar redox potentials of iron and manganese sites. Comparing with typical x-ray doses in crystallography implies that (II,II) states are reached in <1 s in such studies. First-sphere metal coordination and metal-metal distances differed after chemical reduction at room temperature and after XPR at cryogenic temperatures, as corroborated by model structures from density functional theory calculations. The inter-metal distances in the XPR-induced (II,II) states, however, are similar to R2 crystal structures. Therefore, crystal data of initially oxidized R2-type proteins mostly contain photoreduced (II,II) cofactors, which deviate from the native structures functional in O2 activation, explaining observed variable metal ligation motifs. This situation may be remedied by novel femtosecond free electron-laser protein crystallography techniques.
Introduction
Dinuclear transition-metal cofactors play important roles in small molecule conversions by a broad range of biological enzymes. A prominent example are the prototypic so-called dimetal-oxygen cofactors, which are found, for example, in ribonucleotide reductases (RNRs)5 (1–3), methane monooxygenases (4, 5), and in various other oxidases (6–8). RNRs are essential for DNA synthesis in all organisms (9). In these enzymes, the metal cofactor is located in the R2 subunit, whereas the R1 subunit houses the active site of ribonucleotide reduction. In the classical R2 of Escherichia coli class-Ia RNR, two iron atoms form the cofactor, which are bound to the protein by the amino acid side chains of two histidine residues, three glutamates, and one aspartate, in a highly conserved binding motif (10, 11).
The metal cofactor in R2 proteins functions in dioxygen (O2) reduction (12, 13). Activation of the enzymes is achieved by O2 reduction at the initially divalent metal ions. Thereby the O-atoms become incorporated for example, as metal-bridging μO or μOH species (12–14) so that a high-valent Fe(IV)Fe(III) site (intermediate X) is formed (15–17). Subsequent oxidation of a nearby tyrosine residue to a radical (Y•) leaves the cofactor in an Fe(III)Fe(III) state. The catalytic cycle starts with the generation of a cysteine radical at the ribonucleotide binding site in R1 via long-range proton-coupled electron transfer to Y•, initiating ribonucleotide reduction, in the course of which Y• is restored by reverse ET for further turnover (18–23).
Recently, a new class-Ic RNR has been described in the human pathogenic bacterium Chlamydia trachomatis (Ct), in which a hetero-dinuclear MnFe cofactor is assembled instead of an FeFe site and furthermore, the radical-forming tyrosine is replaced by a redox-inert phenylalanine (Phe-127) (24–27). Extensive investigations on the Ct enzyme have revealed that a Mn(IV)Fe(III) site, as formed after O2 reduction at divalent manganese and iron ions, is the target of the proton-coupled electron transfer from R1 (25–30). Also in CtR2 under certain conditions an FeFe cofactor can be incorporated, but shows significantly lower activity (31, 32). Similar MnFe sites meanwhile have been found in a number of related enzymes (7, 8, 33–36) and in addition, MnMn cofactors were described in the tyrosine-radical forming class-Ib RNRs (37–39). An important common feature of all three types of cofactors is the participation of high-valent intermediates, for example, M(III)M(III) and M(IV)M(III), in the catalytic reactions (25, 28, 35, 36).
The Protein Database was used for structures. Most R2 structures are for FeFe- or MnMn-containing proteins. Only very recently, three structures have been reported for CtR2 including a MnFe cofactor (41, 42). In previous studies (see Refs. 29 and 30), we have surveyed the R2 structures. In none of the structures did the apparent mean metal oxidation state exceed ∼2.5 and the average value was close to 2, suggesting the predominance of M(III)M(II) and Mn(II)M(II) sites. The latter holds true also for structures that have been obtained for initially oxidized R2 proteins. In addition, considerable observed variability in metal coordination by oxygen species and amino acid groups is unexplained on a functional basis. Important structural parameters, such as the metal-metal distance, which is determined, e.g. by the type of metal-bridging oxide species (4, 30, 43, 44), are not correlated to the ligation environment or to the increasing crystallographic resolution over the last 20 years. Typical metal(II)-ligand bond lengths are often observed (29, 30).
These observations suggest that the metal sites in the crystal structures in fact do not correspond to high-valent states (29, 30). The likely reason for this is x-ray photoreduction (XPR) of initially high-valent cofactors during diffraction data collection (29, 30, 45). However, for well founded attributions, for example, on the O2-activation process, reliable structural information is also required for the high-valent cofactors.
X-ray irradiation induced modifications are a long-known problem in protein crystallography (46–49). XPR of metal cofactors often occurs at x-ray doses that are orders of magnitudes lower than used in x-ray crystallography causing radiation damage of the protein matrix (50–62). XPR therefore has even been used to study reduced states of metal centers (51–53, 56). However, because of a limited, but significant mobility of atoms in proteins at the cryogenic temperatures (around 100 K) usually employed in crystallography, XPR-induced and native structures of cofactors in reduced states may be expected to be different (58, 60).
By x-ray absorption spectroscopy (XAS), structural (metal-ligand bond lengths, metal-metal distances) and electronic (oxidation state) parameters also for high-valent metal sites can be determined (63–65) and furthermore, this technique allows studies of the XPR kinetics (58–60, 66). Using XAS, for CtR2 we have shown previously that under high-intensity x-ray irradiation the Mn(IV)Fe(III) cofactor becomes reduced by XPR within seconds to minutes even at 20 K (30). However, quantitative relationships between XPR-induced structural changes and metal oxidation states so far have not been obtained for the dimetal-oxygen cofactors. This is necessary to reconcile the site configurations in the crystal data with deviating structural features, for example, usually much shorter metal-metal distances, as determined by XAS (29, 30, 66–68). In principle, this would allow for the reversion of modifications due to XPR in the crystal structures in silico, using quantum chemical calculations (64, 69, 70), to obtain improved models of the high-valent sites.
In the present investigation, we used XAS to monitor XPR in CtR2 initially containing Fe(III)Fe(III) or Mn(III)Fe(III) cofactors at cryogenic temperatures. The XPR kinetics were determined by time-resolved XANES measurements and the structural changes, as derived from EXAFS analysis, were put in relationship to the XPR-induced metal oxidation states. Rapid XPR was observed for both cofactor types. This gave rise to site configurations, which differ from the ones in the native oxidized or reduced CtR2 proteins, but are in agreement with crystallographic data.
MATERIALS AND METHODS
Protein Sample Preparation
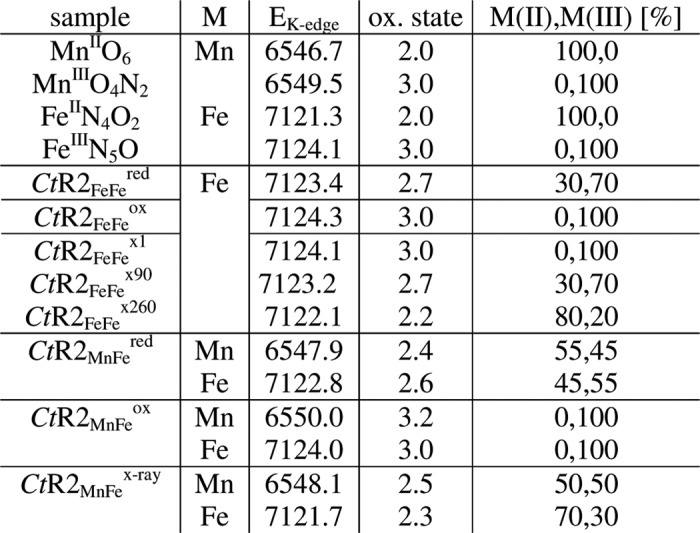
Recombinant Ct R2 protein was overexpressed in E. coli and R2 protein was purified and concentrated as described previously (see Refs. 29 and 30 and references therein). R2 protein containing an FeFe cofactor was obtained without addition of metal ions to the TB growth medium. R2 protein containing predominantly a MnFe cofactor was obtained by the addition of 30 μm MnCl2 to the LB medium (iron concentration 8 μm) after induction with isopropyl isopropyl-β-d-thiogalactopyranoside. Reduction of R2 was achieved by the addition of 100 μm TCEP (Tris(2-carboxyethyl)phosphine hydrochloride) to the cell lysis buffer. R2 protein (monomer) concentrations were derived photometrically using an extinction coefficient at 280 nm of 57,750 m−1 cm−1 for Ct R2 (27). The following protein samples were prepared. (a) Oxidized MnFe R2 protein, as aerobically purified from cells grown in manganese-enriched medium, further on is denoted CtR2MnFeox; (b) chemically reduced MnFe R2 is denoted CtR2MnFered; (c) FeFe R2 protein, as aerobically purified from cells grown in TB medium (14 μm iron) (71), is denoted CtR2FeFeox; and (d) chemically reduced FeFe R2 is denoted CtR2FeFered. XPR studies were carried out on CtR2MnFeox and CtR2FeFeox samples and resulted in R2 states denoted CtR2MnFe/FeFexi (xi specifies the mean x-ray irradiation period in min, i.e. CtR2FeFex90 for 90 min of x-ray exposure).
Metal Content Quantification
Metal contents of R2 samples were quantified by total-reflection x-ray fluorescence analysis (TXRF) (29, 30, 72) on a PicoFox spectrometer (Bruker) using a gallium metal standard (Sigma) and the respective R2 polypeptide concentrations.
X-ray Absorption Spectroscopy
XAS was performed at the SuperXAS beamline of the Swiss Light Source (SLS) at Paul Scherrer Institute (Villigen, Switzerland) using a double-crystal Si[111] monochromator. Higher harmonics were suppressed by a silicon mirror in grazing incidence mode. The synchrotron was operated in top-up mode at 400 mA ring current, providing constant x-ray flux at the sample position, which was attenuated by carbon foil absorbers in the beam when necessary. A rapid shutter in front of the I0 ion chamber prevented x-ray irradiation of samples prior to the XAS scans. The spot size on the samples was ∼0.2 mm (vertical) × ∼1.0 mm (horizontal) as set by slits. Samples were positioned with micrometer precision in the beam by moving the cryostat on a computer-controlled stage. Kα fluorescence-detected XAS spectra at the iron and manganese K-edges were collected using an energy-resolving 13-element germanium detector (Canberra), which was shielded by 3-μm chromium foil (Mn) or 6-μm manganese foil (iron) against scattered x-rays. Samples were held in a liquid-helium cryostat (Oxford). Detector dead time-corrected XAS spectra were averaged after energy calibration using an iron metal foil (iron) or a KMnO4 powder sample (manganese) as energy standards (73, 74). XANES spectra were normalized and EXAFS oscillations were extracted as described previously (30). The duration of EXAFS scans up to a k value of 12.5 Å−1 was ∼22 min; the scan duration up to the K-edge was ∼1–2 min. EXAFS spectra were derived using E0 values of 7112 eV (iron) and 6540 eV (manganese); E0 was refined to 7120 ± 2 eV (iron) and 6547 ± 2 eV (manganese) in the least-squares simulations of unfiltered k3-weighted spectra (in-house program SimX (75), phase functions were calculated by FEFF8 (76, 77), amplitude reduction factors, S02, of 0.9 for iron and 0.85 for manganese). The error sum (RF) was defined as described elsewhere (75). Fourier transforms (FTs) of EXAFS spectra were calculated using k values of 2–12 Å−1 and cos2 windows extending over 10% at both k range ends. K-edge energies were determined at the 50% level of the normalized XANES. Time scans of the x-ray fluorescence were recorded at given monochromator energies at the K-edges to monitor XPR (60), using an acquisition period of 2 s/data point. Kinetic XPR data were simulated by sums of monoexponential functions.
Bond Valence Sum Calculations
BVS calculations (78) were done as previously described (29–30), using in-house software for the analysis of crystal structure files from the Protein Database, and included atoms within a radius of 2.7 Å around the metal ions. Given BVS values represent the average of calculations using R0 values for metal(II) and metal(III) species (30).
Density Functional Theory Calculations (DFT)
Spin-unrestricted geometry optimizations and calculations of electronic parameters of structural models of the metal sites were performed using the ORCA program package (79) as described previously (30, 80). Geometry optimizations involved the BP86 exchange-correlation functional (81) with a triple-ζ valence (TZVP) basis set (82). One set of polarization functions was used for all atoms. The resolution of identity approximation was used with the auxiliary TZV/J Coulomb fitting basis set (83) and a dielectric constant of ϵ = 4 in a COSMO solvation model (84). To derive the correct spin coupling of the two metal atoms, broken symmetry formalism using the flip-spin technique as implemented in ORCA (85, 86) was applied as described before (30). The initial geometry optimizations of (III,III) cofactors were based on crystal structure PDB code 1SYY of wild type Ct FeFe R2. For further restrictions in the DFT calculations see the “Results.”
RESULTS
Properties of Cofactors in CtR2 Crystals
Two crystal structures of the wild type CtR2MnFe protein have been published (24, 32, 41, 42). Earlier structures of CtR2 proteins produced in E. coli without addition of or reconstitution with manganese should relate to CtR2FeFe proteins (wt and the F127Y mutant) (24, 32, 41, 42). The structures were initially obtained using oxidized R2, and were expected to contain mainly Fe(III)Fe(III) or Mn(III)/(IV)Fe(III) cofactors. All structures show different coordination motifs of the metal ions, in particular with respect to metal-bridging oxides and orientation of the carboxylate group of Glu-227 (Fig. 1). BVS calculations reveal apparent oxidation states, which are close to the divalent level even for the 6-coordinated metal ions (Fig. 1, see legend); the average metal-ligand bond lengths thus are in the range for Mn(II) and Fe(II) species.
FIGURE 1.

Crystal structures of FeFe and MnFe cofactors in CtR2. The PDB entry numbers are shown (resolution and reference in parentheses): a, 1SYY (1.70 Å (24)); b, 2ANI (2.00 Å (25)); c, 4D8F (2.200 Å (42)); d, 4D8G (1.75 Å (42)). For c and d, one example of the 4 structures in the files is shown. The given metal-metal distances represent the mean values, the distance error may be on the order of ∼0.2 Å. Note varying metal bridging oxides and structural changes at Glu-227. 2ANI (b) corresponds to the F127Y mutant. The following mean BVS values for Fe1,Fe2 or Mn,Fe (numbers of ligands in parentheses) were obtained from the structures: a, 2.3,2.4 (6,6); b, 2.7,4.7 (6,6); c, 1.6,1.1 (4.5,5); d, 2.4,2.1 (6,6).
The metal-metal distances span about 3.07–3.47 Å (Fig. 1) and are seemingly unrelated to the metal coordination. The coordinate error may be expected to be on the order of at least 10% of the crystallographic resolution (87). This means that the metal-metal distances in the CtR2 structures at a resolution of ∼2 Å may be considered as equal within error limits. These distances are within the range of metal-metal distances observed in other R2 structures, which show an even broader distance distribution centered around ∼3.6 Å, and similar site heterogeneity has also been observed for related cofactors in other enzymes (see Refs. 29 and 30) and references therein). We consider these results as evidence that during diffraction data collection XPR altered the cofactor structures.
Metal and MnFe/FeFe Cofactor Contents
FeFe- and MnFe-containing CtR2 samples were prepared, which contained air-oxidized (ox) or chemically reduced (red) proteins and are denoted CtR2FeFeox, CtR2FeFered, CtR2MnFeox, and CtR2MnFered. The sample conditions (29, 30) were chosen to allow for comparison of the cofactor properties in the native oxidized and reduced states with those of XPR-induced species.
Metal contents were in the range of about 0.5–0.7 mm manganese and 0.9–3.2 mm iron in the XAS samples, which contained about 1–2 mm R2 polypeptide (Table 1). The binding sites in CtR2FeFe were occupied near quantitatively with close to two iron atoms per R2 polypeptide; manganese was at the detection limit. A slightly lower apparent occupancy was observed for CtR2MnFe, in which the manganese content was ∼55% of the iron content. The metal contents of oxidized and reduced samples were rather similar. These observations are in agreement with previous results (25, 29, 30, 71). An elevated Zn content was found only in the CtR2red samples. The maximal relative amounts of MnFe and FeFe sites were calculated from the metal and R2 concentrations (Table 1), yielding close to 100% FeFe sites in CtR2FeFe. In CtR2MnFe, sufficiently high amounts (70%) of MnFe sites were obtained (Table 1), which allowed for determination of the properties of the hetero-metal sites by XAS.
TABLE 1.
Metal and protein concentrations and relative amounts of cofactor species
Metal concentrations were determined by TXRF. Relative amounts of cofactor species represent maximal figures, which were calculated on the basis of the metal and R2 polypeptide concentrations and assuming that all Mn ions were incorporated in MnFe sites. Values in parentheses were calculated under the assumption that all zinc ions were included in FeZn sites (114). Other transition metal species were negligible in the R2 samples.
| Sample | R2 | Mn | Fe | Zn | Mn/R2 | Fe/R2 | Zn/R2 | MnFe | FeFe | FeZn |
|---|---|---|---|---|---|---|---|---|---|---|
| mm | mm | mm | mm | % | % | % | ||||
| CtR2FeFered | 0.8 | 0.04 | 1.37 | 0.44 | 0.05 | 1.71 | 0.55 | 6 (4) | 94 (49) | 0 (47) |
| CtR2FeFeox | 1.8 | 0.04 | 3.28 | 0.23 | 0.02 | 1.82 | 0.12 | 2 (1) | 98 (92) | 0 (7) |
| CtR2MnFered | 1.0 | 0.49 | 0.87 | 0.36 | 0.49 | 0.87 | 0.36 | 72 (57) | 28 (1) | 0 (42) |
| CtR2MnFeox | 1.3 | 0.72 | 1.32 | 0.21 | 0.55 | 1.02 | 0.16 | 71 (64) | 29 (17) | 0 (19) |
Structure and XPR of the FeFe Cofactor
XANES spectra at the Fe K-edge were recorded for the CtR2FeFe samples (Fig. 2A). They revealed K-edge energies, which indicated close to 100% Fe(III) in CtR2FeFeox, but about 70% Fe(III) and 30% Fe(II) in CtR2FeFered. These values were obtained by comparison to iron reference compounds, revealing a K-edge energy difference of ∼2.8 eV between the Fe(II) and Fe(III) levels (Table 2). Combining the XANES and TXRF data, ∼100% Fe(III)Fe(III) sites for CtR2FeFeox, but ∼57% Fe(III)Fe(III) and ∼43% Fe(III)Fe(II) for CtR2FeFered were calculated.
FIGURE 2.

XPR kinetics of the FeFe cofactor. A, iron XANES spectra of CtR2FeFeox (squares) and CtR2FeFered (open circles) of CtR2FeFe samples initially in the oxidized (ox) state after x-ray exposure at 20 K for ∼90 min (triangles) and ∼260 min (solid circles), and of iron oxidation state references (FeIIN4O2, dashed line; FeIIIN5O, solid line; see Table 3). Upper inset, K-edge spectra around the 50% level of CtR2FeFeox and CtR2FeFex260, the arrows mark the edge downshift (ΔE) and corresponding fluorescence intensity increase (ΔF) for an excitation energy of 7123 eV. Lower inset, expanded view of isolated pre-edge peaks in the XANES (asterisk). B, K-edge energies (triangles, left y axis) and x-ray fluorescence intensities at 7123 eV (circles, right y axis) at 20 K, together with the fit curves calculated using parameters in Table 3. Inset, time scan trace of the x-ray fluorescence intensity at 7123 eV at 80 K, K-edge data at 20 K, and fit curves (Table 3) on a logarithmic axis. Traces were normalized to unity amplitude after offset subtraction on the basis of the fit results. The given dose values (in Gy, gray = J kg−1) are approximate and correspond to the incident dose, the absorbed dose in 1 mm of water for an energy of 7125 eV is ∼77% of the incident dose. Dose values were calculated for an energy of 7125 eV = 1.14 × 10−14 joule, a flux (upper limit) of 1011 photons s−1, and an irradiated volume of 0.2 mm3 corresponding to 2.15 × 10−7 kg (aqueous buffer plus R2 protein contributing about 0.15 × 10−7 kg at 1.8 mm and a molecular mass of ∼43 kDa).
TABLE 2.
Manganese and iron K-edge energies and relative amounts of M(II) and (III) ions
K-edge energies reflect the 50% level of XANES spectra (Figs. 2 and 5), the error was ∼0.15 eV. Oxidation state references are octahedral metal species, namely Mn(II) O6 in hexaquo-Mn(II) in a 3 mm solution of MnCl2, Mn(III)O4N2 in a powder sample of a synthetic Mn2 complex (115), Fe(II)N4O2 in the non-heme iron site of bacterial photosynthetic reaction center protein (98), and Fe(III)N5O in the heme of oxidized bovine hemoglobin in 15 mm solution. Indices xi specify the mean x-ray exposure in min. Metal (M) site amounts were calculated using linear relations between K-edge energies and oxidation states (Figs. 2B and 4; for CtR2MnFeox neglecting a minor Mn(IV) contribution). CtR2MnFex-ray denotes a state reached after ∼130 min of XPR.
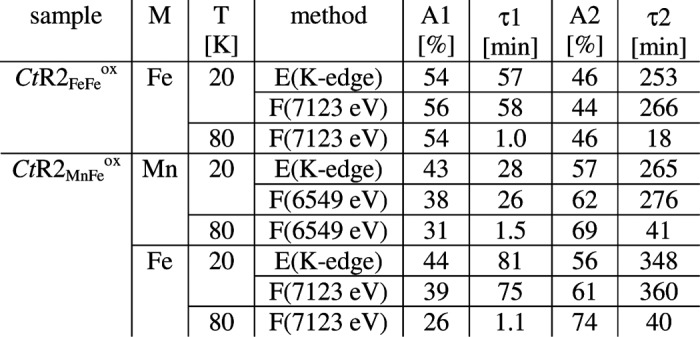
For monitoring of XPR of the initial Fe(III)Fe(III) cofactor, a series of consecutive. XAS scans were performed on single spots of CtR2FeFeox samples. Respective XANES spectra for about 2, 90, and 265 min of x-ray exposure at 20 K (Fig. 2A) revealed K-edge energies, which decreased from an initial mean oxidation state of 3 to a level of about 2.2, suggesting ∼80% of Fe(II) after prolonged XPR (Table 2). A plot of the K-edge energies of all obtained XANES spectra versus the x-ray exposure periods revealed biphasic XPR kinetics at 20 K (Fig. 2B). A similar behavior was observed when the normalized x-ray fluorescence level at a constant excitation energy of 7123 eV in the iron K-edge was used as an alternative measure of XPR (Fig. 2B). Both data sets were well simulated using sums of two exponential functions with similar time constants (τ) of about 60 and 260 min and amplitudes close to 50% each (Fig. 2B, Table 3). We attribute these kinetic phases (i) to the more rapid formation of Fe(III)Fe(II) states, terminated after about 1.5 h, and (ii) to the slower reduction to the Fe(II)Fe(II) level, finished after about 7 h at 20 K. Kinetic XPR data for CtR2FeFeox at an increased temperature of 80 K still revealed biphasic behavior, but acceleration of both reduction phases compared with the 20 K data (Fig. 2B, inset); so that τ1 and τ2 were decreased to ∼1 and ∼18 min, respectively (Table 3).
TABLE 3.
XPR kinetics for manganese and iron
Relative amplitudes (A) and time constants (τ) resulted from double-exponential simulations of K-edge energy (E) and X-ray fluorescence intensity (F) data for the two metals (Figs. 3B and 5), using appropriate offset values.
Changes of the coordination geometries of the iron sites were assayed by analysis of the pre-edge features in the XANES (88) (Fig. 2A, inset). For CtR2FeFeox, the small pre-edge amplitude suggested predominately 6-coordinated Fe(III) ions, whereas the increased pre-edge peak at ∼0.4 eV lower energy for CtR2FeFered suggested 5-coordinated iron at least in the 30% Fe(II) fraction. A pronounced increase and shift to lower energy of the pre-edge was observed for increasing x-ray exposure periods. Accordingly, this was explained by a change from 6-coordinated Fe(III) to 5-coordinated Fe(II) in the major sample fraction after prolonged XPR.
EXAFS on the native oxidized and reduced states, and on photoreduced CtR2FeFe samples was performed to study changes of interatomic distances at the metal cofactor (Fig. 3). The FTs of the EXAFS spectra (Fig. 3A, inset) of CtR2FeFeox and CtR2FeFered revealed relatively small differences, e.g. changes of the FT maxima reflecting Fe-Fe distances. Visual inspection of the FTs for increasing x-ray exposure periods revealed systematic changes, for example, an initial shift to shorter distances and later diversification of the main FT maximum due to Fe-Fe distances and shifts of the main FT maximum due to Fe-ligand interactions (Fig. 3A).
FIGURE 3.

EXAFS for XPR of the FeFe cofactor. A, FTs (thin lines) of EXAFS spectra (inset) and simulation curves (thick lines) based on parameters in Table 4 (Fe-O,N distances); and B (Fe-Fe distances) for the indicated CtR2FeFe states and mean x-ray exposure periods. Spectra were vertically displaced for comparison. B, coordination numbers (NFe-Fe) for EXAFS data in A of 3 Fe-Fe distances for oxidized (ox) and reduced (red) CtR2FeFe samples (depicted at “0” levels, meaning for negligible XPR) and for CtR2FeFeox after the indicated mean XPR periods. Fe-Fe distance margins (in parentheses) and vertical bars denote the full range of R and N variations for different fit approaches (simulations of individual spectra or joint simulations of all spectra including variations in the fit restraints for Fe-O,N shells (40)). Inset, mean Fe-O,N distances calculated from the data in Table 4. Smooth curves show fits of the N values by sums of two exponential functions using time constants in Table 3 and appropriate offsets and amplitude signs. Given dose values are approximate and were calculated as detailed in the legend of Fig. 2.
By simulation of the EXAFS spectra (curve fitting) the structural changes were quantified (Fig. 3A). The main difference in CtR2FeFered in the first iron coordination sphere was an increase of the Fe-O,N bonds lengths and a slightly increased coordination number (N) but largely increased Debye-Waller parameter (2σ2) for the shorter bonds, compared with CtR2FeFeox (Table 4). In particular the 2σ2 increase suggested significant amounts of 5-coordinated iron ions in CtR2FeFered. Fe-Fe distances (R) of about 2.95, 3.05, and 3.40 Å readily accounted for the Fe-Fe interactions. The 3.05-Å distance represented the main species in CtR2FeFeox (∼70%), whereas for CtR2FeFered it was the ∼3.40 Å distance (∼55%) (Fig. 3B). The ∼2.95-Å distance (∼10%) was at the detection limit. The Fe-Fe distance changes and coordination numbers in oxidized and reduced samples suggested that in the Fe(III)Fe(III) site the iron atoms are separated by ∼3.05 Å and ∼3.40 Å distance accounts for an Fe(III)Fe(II) site (29, 30).
TABLE 4.
EXAFS simulation parameters for the first coordination shells of manganese and iron
N, coordination number, R, interatomic distance; 2σ2, Debye-Waller factor, RF, fir error sum (75) calculated for reduced distances of FTs in the range of 1.0–3.5 Å. (*) Values that were kept constant in the fit procedures, (#) respective 2σ2 values were coupled to yield the same values for both coordination shells. For respective fit curves to the EXAFS spectra see Figs. 3A and 6.
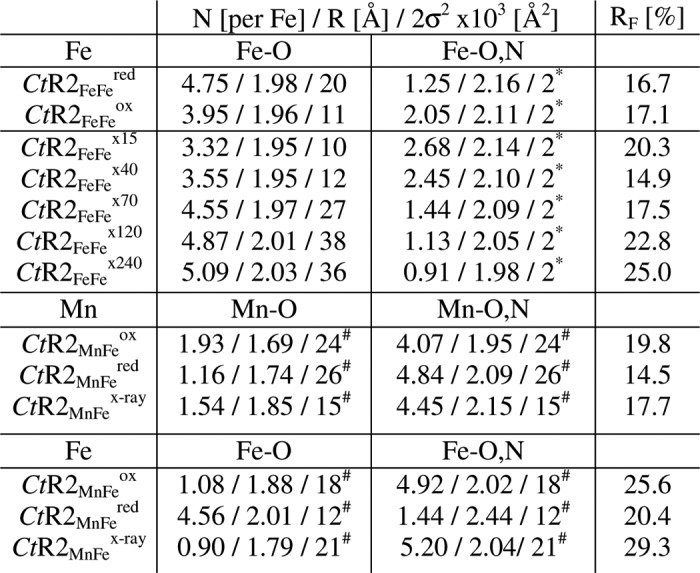
Simulations of a series of EXAFS spectra for the x-ray-exposed CtR2FeFe samples revealed the following (Fig. 3B, Table 4). The structural parameters for ∼15 min XPR were similar to those for CtR2FeFeox, revealing a predominant contribution of the ∼3.05 Å Fe-Fe distance. For increasing x-ray exposure periods up to ∼240 min, for the main Fe-O shell a simultaneous increase of the coordination number (N), the Debye-Waller parameter (2σ2), and the bond-length (R) were determined, whereas for the initially longer second Fe-O,N interaction decreased N and R values (for constant 2σ2) were obtained (Table 4). The mean Fe-ligand bond lengths, however, revealed an initial decrease up to ∼100 min XPR and an increase for longer periods (Fig. 3B, inset). Also for CtR2FeFered, increased N and 2σ2 values for the Fe-O shell compared with CtR2FeFeox were observed. These effects were explained by accumulation of the 5-coordinated Fe(II) during XPR.
For the Fe-Fe distances (Fig. 3B), an initial increase of the relative coordination number of the ∼2.95 Å distance to ∼50% after ∼70 min of x-ray exposure occurred at the expense of the ∼3.05 Å distance. It was followed by a decrease of the ∼2.95-Å contribution and a concomitant increase of a distance close to ∼3.45 Å to ∼55% after 240 min. The kinetic behavior of the three Fe-Fe distances was reasonably well described using the same time constants as determined for XPR from the XANES analysis (Fig. 3B). This suggested an Fe-Fe distance shortening from ∼3.05 Å to ∼2.95 Å for the initial Fe(III)Fe(III) → Fe(III)Fe(II) reduction step, which was followed by an elongation from ∼2.95 Å to ∼3.45 Å upon the Fe(III)Fe(II) → Fe(II)Fe(II) transition.
Structure and XPR of the MnFe Cofactor
The XPR kinetics of CtR2MnFeox samples were monitored using similar approaches as described above (Fig. 4). Biphasic reduction behavior was observed both at the manganese and iron K-edges. Kinetic simulations of the 20 K data showed that the first phase was by a factor of about 5–10 faster than the second phase for manganese and iron (Table 3). The time constants of the two phases for manganese reduction were by factors of about 1.5–2.5 smaller than the ones for iron reduction. At 80 K, pronounced acceleration of reduction resulted in quite similar kinetics for both metal species (Table 3). Overall, the time constants for reduction of manganese and iron for CtR2MnFe and CtR2FeFe samples, initially containing mostly Mn(III) and Fe(III), were similar within a factor of ∼2 at both 20 and 80 K.
FIGURE 4.

XPR kinetics of the MnFe cofactor. Data represent fluorescence intensity levels at energies in the manganese or iron K-edges, derived from XANES spectra (at 20 K, see Fig. 5) or time scan traces (80 K) and are shown on a logarithmic time axis. Lines (solid, Mn; dashed, Fe) represent double-exponential fits with time constants in Table 3. Inset, data at 80 K on a linear time axis. The incident dose values are approximate (the absorbed dose in 1 mm of water for 6550 eV is ∼85% of the incident dose); dose values were calculated using an energy of 6550 eV = 1.05 × 10−14 joule, an irradiated volume of 2.11 × 10−7 kg (buffer plus about 0.11 × 10−7 kg of R2 protein at 1.3 mm, and further parameters given in the legend of Fig. 2.
We note that a minor Mn(IV) contribution in CtR2MnFeox was suggested by the XANES spectrum (see below). However, the initial reduction phase of Mn(IV)Fe(III) sites has been found to be ∼150 faster than the next step (30). This suggested τ values of about 0.3 and 0.01 min for Mn(IV)Fe(III) reduction at 20 and 80 K under the present conditions, which was at the kinetic resolution limit. Small amplitudes due to Mn(IV)Fe(III) reduction accordingly were not discernable for CtR2MnFe.
XANES spectra at the manganese and iron K-edges for CtR2MnFeox, CtR2MnFered, and for a state obtained after prolonged XPR denoted CtR2MnFex-ray are shown in Fig. 5. The spectrum of CtR2MnFex-ray resulted from summation of spectra for about 30–210 min x-ray exposure (mean of ∼130 min) and thus represents the average over several manganese and iron oxidation states. Compared with manganese reference compounds, the manganese and iron edge energies of CtR2MnFeox were close to the trivalent level; a minor Mn(IV) contribution (∼20%) was implied by the somewhat higher manganese edge energy (Table 2). For CtR2MnFered, the edge energies suggested about ∼50% of each Mn/Fe(III) and Mn/Fe(II), neglecting the Mn(IV) contribution. CtR2MnFex-ray accordingly contained Mn/Fe(III) and Mn/Fe(II) in about 50/30% and 50/70% proportions.
FIGURE 5.

Reduction of the MnFe cofactor monitored in the XANES. Left, manganese K-edge spectra, and right, iron K-edge spectra of CtR2MnFe in the oxidized (ox) and reduced (red) states and after a mean x-ray exposure of CtR2MnFeox of ∼130 min (x-ray), together with spectra of respective oxidation state references (Table 2). Insets, corresponding isolated pre-edge features in magnification.
Metal site geometry changes again were deduced from the pre-edge features (Fig. 5, insets). Small pre-edge amplitudes for CtR2MnFeox suggested predominantly 6-coordinated (i.e. near-octahedral) Mn(III) and Fe(III) ions. The diminished manganese and iron pre-edge peaks at >1 eV lower energies for CtR2MnFered implied a further symmetrization of the coordination of the divalent metal ions. At variance with the latter behavior, CtR2MnFex-ray showed increased manganese and iron pre-edge features at lower energies compared with CtR2MnFeox. This suggested formation of 5-coordinated manganese and iron ions after prolonged XPR.
Visual inspection of the EXAFS spectra of the three CtR2MnFe states (Fig. 6) revealed that the main FT maximum of the manganese EXAFS due to first-sphere Mn-O,N distances was at higher distances in CtR2MnFered and CtR2MnFex-ray compared with CtR2MnFeox, suggesting an overall increase of the bond lengths for Mn(II). Less pronounced changes of the main FT maximum due to Fe-O,N bonds were observed. In the manganese EXAFS, two FT maxima at reduced distances of 2–3 Å were attributed to Mn-Fe interactions. For CtR2MnFeox the first maximum was larger than the second one, whereas for CtR2MnFered this was reversed. For CtR2MnFex-ray the main Mn-Fe FT peak was even at larger distances than for the other two samples. The iron EXAFS of CtR2MnFeox showed one main Fe-Mn,Fe distance, whereas for CtR2MnFered and CtR2MnFex-ray, several Fe-Mn,Fe distances were resolved (Fig. 6).
FIGURE 6.

EXAFS of the MnFe cofactor. FTs (thin lines) of manganese (A) and iron (B) EXAFS spectra (insets) of CtR2MnFeox, CtR2MnFeox, and CtR2MnFex-ray, together with simulation curves (thick lines) based on the parameters indicated in Table 4 (Mn/Fe-O,N distances) and Fig. 7 (metal-metal distances).
The simulation results for the manganese and iron EXAFS spectra are summarized in Table 4 (first coordination sphere) and Fig. 7 (metal-metal distances). In CtR2MnFeox, Mn-O,N distances of ∼1.70 Å likely were attributable to bonds between manganese and metal-bridging μO species; the ∼1.95 Å distances reflected μO(H) bridges and terminal Mn-O,N bonds (30). The Mn-O,N bond lengths for CtR2MnFered and CtR2MnFex-ray were increased, accompanied by moderate coordination number changes. The iron EXAFS indicated that the Fe-O,N bonds overall were ∼0.2 Å longer than the Mn-O,N bonds (Table 4). For CtR2MnFeox the two Fe-O,N bond lengths differed by ∼0.14 Å. In CtR2MnFered the shorter Fe-O,N bonds were elongated compared with CtR2MnFeox to even ∼2.4 Å, possibly suggesting elongation of Fe-NHis bonds. The first-sphere iron coordination of CtR2MnFex-ray was more similar to CtR2MnFeox, suggesting relatively minor changes of the Fe-O,N bonds.
FIGURE 7.

Metal-metal coordination numbers and distances for MnFe sites. Data correspond to simulations of EXAFS data for three CtR2MnFe states in Fig. 6. Vertical and horizontal bars denote the full variation range of N and R values as obtained for different fit approaches (individual or joint EXAFS simulations and variations in the fit restraints for the Mn/Fe-O,N shells; see the text).
At least three metal-metal distances were discernable both in the manganese and iron EXAFS spectra (Fig. 7). The main Mn-Fe distance in CtR2MnFeox was ∼2.90 Å (∼55%) and minor contributions of a ∼3.30-Å distance (∼35%) were observed. CtR2MnFered showed opposite magnitudes (∼25%, ∼50%) of contributions from these two distances. Similar metal-metal distances in CtR2MnFe have previously been attributed to Mn(IV)/(III)Fe(III) (∼2.90 Å) and Mn(II)Fe(III) (∼3.3 Å) sites (29, 30, 66). Here, the ∼2.90 Å distance mostly reflected the Mn(III)Fe(III) cofactor, because Mn(IV) contributions were small. We note that in the iron EXAFS the ∼3.3 Å distance was less well defined than in the manganese EXAFS so that several equivalent fits with distances around 3.1–3.4 Å were possible. This reflects contributions from the minor amounts of FeFe species (compare Table 1 and Fig. 3B).
In CtR2MnFex-ray, the main metal-metal distance was ∼3.65 Å (∼55%) and contributions from the other two distances apparently were small (Fig. 7). A ∼3.65 Å distance was close to the detection limit in CtR2MnFeox and CtR2MnFered. Accordingly, we assign the ∼3.65 Å distance to the main XPR-induced state of the MnFe site in CtR2MnFex-ray. Comparison of the TXRF, XANES, and EXAFS results revealed that the ∼3.65 Å distance most likely reflects a Mn(II)Fe(II) state of the cofactor, as preferably produced by XPR at cryogenic temperatures. That the ∼2.9 Å distance in CtR2MnFex-ray remained at a level, which was comparable with CtR2MnFered, may be explained by a similar initial metal-metal distance shortening as observed for CtR2FeFe.
Model Structures for XPR from DFT
Density functional theory calculations were employed to generate model structures for the native and XPR-modified cofactors. The restricted mobility of amino acids during XPR at cryogenic temperatures was simulated by fixation of carbon atoms during geometry optimization after addition of 1 electron to the initial Fe(III)Fe(III) and Mn(III)Fe(III) sites or 2 electrons to the singly reduced structures. Structures were calculated for additions of electrons only or for balancing of respective surplus negative charges by protons.
Comparison of more than 20 DFT-optimized structures revealed the following (supplemental Table S1, Fig. 8). When only one C-atom of the side chains (C1 in Fig. 8) was fixed during geometry optimizations of single and double reduced states and no protons were added, the resulting structures showed excessive changes, for example, breaking the Glu-120 carboxylate and μO(H) bridges, detachment of histidine ligands, and pronounced Glu ligand reorientations, leading, e.g. to 4-coordinated manganese and iron ions. We consider such large scale geometry changes as unlikely at cryogenic temperatures. Fixation of two C-atoms resulted in less severe structural changes, but the tendency for carboxylate bridge breaking and histidine detachment remained, still causing mostly low coordination numbers as well as metal-metal distance elongation already for single-electron reduction in some cases. The structural changes after additions of electrons and protons depended on the actual protonation site. As a trend, less pronounced changes were observed so that higher metal coordination numbers prevailed (supplemental Table S1).
FIGURE 8.

DFT structures for oxidized and single or double reduced FeFe and MnFe sites. The (III,III) states (top) were calculated on the basis of the PDB 1SYY structure of wild type CtR2FeFe (replacing Fe2 by Mn, right). Middle and bottom structures were calculated after additions of 1/2 electron(s) and 1/2 proton(s) (dotted circles); Fe2/Mn-OH2 and Fe1/Fe-OH distances in the (II,II) states are >3 Å. (III,III) and (III,II) structures show the localizations of the lowest energy unoccupied molecular orbitals (LUMO) with β spin orientation. The α-spin LUMOs were largely located at Fe2 for the FeFe sites and at manganese for the MnFe sites (not shown). The (II,II) structures in stick representation (most protons omitted for clarity) exemplify the positions of hetero-atoms in all structures. Atoms denoted C1 and C2 for Glu-120 were fixed at their crystallographic positions for all amino acid ligands in the DFT calculations. Approximate metal-metal distances are indicated, for further parameters, see supplemental Table S1.
Best agreement with the structural parameters from XAS was achieved by proton addition first to terminal OH groups at Fe2 or manganese in the single reduced sites and second to μO(H) bridges in the double reduced sites, both for the FeFe and MnFe structures (Fig. 8). In this case, the first reduction for both structures resulted in a shortening of the metal-metal distance by ∼0.1 Å in the mixed-valent states, similar to the experiment. This was followed by elongations to >3 Å of Fe-μO(H) and Fe/Mn-OH2 bonds, resulting in 5-coordinated Fe(II) and Mn(II) ions after the second reduction step and in a pronounced elongation of the metal-metal distance to about 3.4–3.5 Å, as also inferred from the experimental data (Fig. 8). For a brief account on the electronic configurations of the DFT structures and their possible relationship to the redox potentials see supplemental Table S1.
DISCUSSION
Structures of FeFe and MnFe Cofactors
Our results suggest the following native structures of FeFe and MnFe cofactors in CtR2. For the trivalent sites (L = Glu/Asp and His ligands), L3(HO)Fe(III)(μO)(μOH)Fe(III)L4 (Fe-Fe ∼3.05 Å) and L3(HO)Mn(III)(μO)(μOH)Fe(III)L4 (Mn-Fe ∼2.90 Å) core configurations are proposed (Fig. 9). This is in agreement with previous assignments (29, 30, 44). Chemical reduction of the (III,III) states by one electron at room temperature likely induces breaking of a μO(H) bridge, presumably accompanied by a protonation, which explains the ∼0.4 Å metal-metal distance increase (4, 43, 63, 75, 89) and metal-ligand bond elongations in the (III,II) cofactors. Fe(III) reduction leads to 5-coordinated Fe(II), at least in the FeFe site, whereas in particular for the MnFe site, Mn/Fe(II) formation may lead to the (partial) loss of one ion and to a remaining 6-coordinated site. For the (II,II) sites, the absence of μO(H) metal bridges is likely, resulting in metal-metal distances exceeding 3.7 Å (Fig. 9), similar to the distances observed in crystal structures of reduced R2 proteins (29, 30).
FIGURE 9.

Proposed structural and redox changes at FeFe and MnFe cofactors for chemical reduction at ambient temperatures and XPR at cryogenic temperatures. Assignments represent a combination of our XAS and DFT results (M = metal, L = ligands). Approximate metal-metal distances (average of FeFe and MnFe sites): R(III,III), ∼3.0 Å; R′(III,II), ∼3.4 Å (ambient temperatures) and ∼2.9 Å (XPR); R″(II,II), ≥3.7 Å (ambient temperatures) and ∼3.5 Å (XPR). Dashed lines denote a sixth ligand (e.g. a water molecule), which possibly is bound upon reduction at ambient temperatures (for example, when one metal ion is lost in the (II,II) states). Positions and numbers of protonation sites and O(H1,2) groups are tentative, but in plausible agreement with the features of the (III,III) states and the structural changes upon reduction.
The structural differences between the FeFe and MnFe sites in CtR2 presumably are relatively subtle. On the structural basis, the reasons for the preferential use of differently metallated sites in the class-Ia RNRs (FeFe), in the class-Ib RNRs (MnMn), and in the tyrosine-radical lacking R2 protein of class-Ic Ct RNR (MnFe) therefore remain elusive. Tuning of the redox potential, for example, by different protonation states of bridging oxides (29, 30, 44, 90, 91), thus may play an important role in the functional diversification of the dimetal-oxygen cofactors.
Structural Changes during XPR
The main structural changes at the FeFe and MnFe cofactors of CtR2 that are induced by XPR at cryogenic temperatures apparently differ from the changes occurring during chemical reduction at room temperature (Fig. 9). At low temperatures, single electron reduction of the (III,III) cofactors causes even a ∼0.1 Å shortening of the metal-metal distance and only relatively minor bond length changes in the first-sphere metal coordination, but leaves the μO(H) bridging intact. A possible explanation for this behavior is the associated protonation, for example, of a terminal OH group. Reduction of the (III,II) states induces more severe changes, such as formation of 5-coordinated Fe/Mn(II) ions, which likely is explained by the breaking of at least one μO(H) bridge and/or of a terminal metal-OH2 bond. This leads to a ≥0.5 Å elongation of the metal-metal distances in the (II,II) states (Fig. 9). Apart from minor differences in the overall site geometries, this sequence of events seems to hold for FeFe and MnFe cofactors.
The structural changes during XPR at 20 K have to involve movements of metal ions and (first-sphere) ligand atoms on the order of 1 Å or less, to explain the observed bond length changes. Changes of similar magnitude have been inferred from XPR studies of other metalloproteins and synthetic complexes (53, 54, 58, 60, 61, 92–94). XPR even has been suggested as a tool for studying reduced states of metal cofactors (52, 53, 56, 57). In the case of CtR2, our data suggest that both the mixed valence and fully reduced states of the FeFe and MnFe cofactors differ from their counterparts formed at ambient conditions. Presumably, this reflects the limited mobility of amino acid groups and water species in the frozen state. The XPR-induced states of dimetal-oxygen cofactors thus may have limited physiological relevance.
Our DFT results suggest that protonation events may play a role in the compensation of XPR-induced metal-centered charges even at 20 K. Indeed, proton movements in proteins and model systems can occur at low temperatures at least within a single hydrogen bond, if not over longer distances within extended hydrogen-bonded networks (95–97). This may be taken as evidence that terminal and/or metal-bridging O(H) ligands in CtR2 are integrated in hydrogen-bonding interactions, for example, with water molecules. Water molecules in hydrogen-bonding distance, for instance, to metal-binding carboxylates, indeed were found in high-resolution R2 structures (39, 99, 100). The proton distribution in XPR-induced structures thus may significantly differ from the respective native oxidation states.
XPR Kinetics and Redox Potentials
Metal site reduction during XPR mostly is anticipated to result from electron transfer from radical sites, which have captured photoelectrons produced after core level excitations mainly of C-, O-, and N-atoms of the protein matrix (46). In addition, the XPR rates for the R2 cofactors may be expected to be proportional to their redox midpoint potentials (Em) (30, 60, 101).
The XPR rates for the Fe(III)Fe(III) and Mn(III)Fe(III) cofactors differ by less than a factor of 2 (30), pointing to Em values for the FeFe and MnFe cofactors, which are similar within about 150 mV (74, 102–103). The absolute Em values for the Mn(IV)Fe(III) states presumably are on the order of 1 V (30) and the slower XPR of the (III,III) states thus suggests a significant Em drop (104). A similar Em implies relatively minor structural differences between the FeFe and MnFe sites at least in the (III,III) states, as observed.
Biphasic XPR kinetics were found for both the FeFe and MnFe sites. For the Fe(III)Fe(III) cofactor, this is straightforwardly explained by sequential Fe(III)Fe(II) and Fe(II)Fe(II) formation. For the MnFe cofactor, similar XPR rates for Mn(III) and Fe(III), in particular at 80 K, imply reduction of either ion at about equal probability. At first glance, the Mn(III) and Fe(III) ions thus seem to possess a similar Em. However, our DFT results suggested reduction first of the iron ion (supplemental Table S1). The even faster manganese reduction at 20 K therefore may be explained by the ∼1.5-times larger absorption of the protein at the lower manganese K-edge energies, i.e. creation of more reducing sites within a given XPR period. This could lead to the apparent inversion of manganese and iron reduction rates.
Implications for X-ray Crystallography of Dimetal-Oxygen Cofactors
Our present and previous data (29, 30, 66) indicate that XPR of high-valent FeFe and MnFe sites in CtR2 and also in the standard type R2 protein from mouse can occur within seconds to minutes already under XAS conditions. The respective doses (at 80 K) are at least 100 times smaller than those resulting in radiation damage of amino acid groups (46, 105).
Using an x-ray flux of ∼1011 photons s−1 at the iron and manganese K-edges as the upper limit and a spot size on the sample of ∼0.2 mm2, time constants of about 1–1.5 min (mean of ∼70 s) for the (III,III) states and 20–40 min (mean of ∼2000 s) for the (III,II) states were observed at 80 K, which is close to the 100 K often used in crystallography. A linear dose-rate relationship for the R2 cofactor reduction is suggested by the ∼10-fold decrease of the time constant from ∼3500 to ∼350 s (at 20 K) of (III,III) reduction for a flux increase from ∼1011 photons s−1 used here to ∼1012 photons s−1 used previously (30). A typically more focused beam of 50 μm diameter for crystallography, i.e. a spot size of 0.002 mm2, and a similar flux of ∼1011 photons s−1 therefore are expected to cause about 100 times faster XPR, i.e. time constants of 0.7 s for the (III,III) states and 20 s for the (III,II) states. A flux of ∼1013 photons s−1 (or more) of undulator crystallography beamlines thus decreases both time constants to far below 1 s. That these estimates are realistic is exemplified by the observed reduction of the Mn(IV)Fe(III) site in CtR2 at 20 K within ∼2 s at ∼1012 photons s−1 (30).
These results imply that for typical x-ray crystallography conditions, XPR of dimetal-oxygen cofactors is unavoidable. Moreover, this will not improve much for the use of liquid-helium temperatures, radical quenching agents (106), acquisition of only a few frames per crystal spot, moderately diminished doses, and faster detector readout. Accordingly, the initial high-valent sites in the crystals were mostly reduced to the (II,II) level. The respective metal-metal distance spread thus may in part reflect the remnants of (III,II) states. Further evidence for this view comes from our result that the Fe/Mn-Fe distances of 3.4–3.7 Å of the XPR-induced (II,II) states match the distances in most R2 structures, irrespective of the initial valence state (29, 30). Notably, the XAS-derived metal-metal distances for the (III,III) states of ∼3 Å are considerably shorter than most of the crystallographic distances even for an upper-limit coordinate error of ∼0.4 Å. The broad distribution of metal coordination motifs in the crystals may thus be due to the superimposition of several XPR-induced conformations.
Crystallographic information for initial (II,II) sites, which will not become further reduced, should be more reliable. However, in these states the metal complex is labilized, leading to single-metal sites and/or to heterogeneity in amino acid and oxygen species ligation geometries. The question arises why the significant XPR-induced structural changes at the metal site still allow for high-resolution crystallographic coordinates. The likely reason for this is that XPR is terminated already during the first few seconds of diffraction data collection and thereafter, the low-valence configuration of metal ions and ligands is stable.
In conclusion, crystal structures of higher valence states exceeding (II,II) or perhaps (III,II) levels of highly oxidizing dimetal-oxygen cofactors cannot be reliably obtained by conventional crystallography. A similar situation has been encountered for other types of high-potential metal cofactors (70, 107). The recent inauguration of fourth generation synchrotron radiation sources based on free electron-laser techniques seems to offer a way to overcome the XPR problem (108–110). Using femtosecond x-ray pulses, XPR may be outrun by termination of the scattering process prior to initiation of the photoelectron cascade. The first structures for the free electron-laser approach using protein nanocrystals at room temperature have been reported even for radiation-sensitive systems, albeit at moderate resolution (111, 112). However, there is no principal limitation toward high resolution structures (113). Femtosecond crystallography on the superfamily of enzymes containing dimetal-oxygen cofactors appears to be an excellent scientific case for this emerging technique to obtain structures of high-valent intermediates.
Acknowledgments
We thank Dr. M. Nachtegaal at SuperXAS of the Swiss Light Source (SLS at Paul Scherrer Institut, Villigen, Switzerland) for excellent support. We are indebted to Dr. R. Schlesinger (FU-Berlin, Physics Department) for kind support in molecular biology.

This article contains supplemental Table S1.
- RNR
- ribonucleotide reductase
- BVS
- bond valence sum
- Ct
- Chlamydia trachomatis
- DFT
- density functional theory
- EXAFS
- extended x-ray absorption fine structure
- FT
- Fourier transform
- R1/2
- subunits R1 and R2 of RNR
- TXRF
- total reflection x-ray fluorescence analysis
- XANES
- x-ray absorption near-edge structure
- XAS
- x-ray absorption spectroscopy
- XPR
- x-ray photoreduction
- TCEP
- Tris(2-carboxyethyl)phosphine hydrochloride
- ox
- oxidized
- red
- reduced.
REFERENCES
- 1. Cotruvo J. A., Stubbe J. (2011) Class I ribonucleotide reductases. Metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 80, 733–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordlund P., Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 3. Sjöberg B. M., Gräslund A. (1983) Ribonucleotide reductase. Adv. Inorg. Biochem. 5, 87–110 [PubMed] [Google Scholar]
- 4. Tinberg C. E., Lippard S. J. (2011) Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res. 44, 280–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Westerheide L., Pascaly M., Krebs B. (2000) Methane monooxygenase and its related biomimetic models. Curr. Opin. Chem. Biol. 4, 235–241 [DOI] [PubMed] [Google Scholar]
- 6. Lange S. J., Que L., Jr. (1998) Oxygen activating nonheme iron enzymes. Curr. Opin. Chem. Biol. 2, 159–172 [DOI] [PubMed] [Google Scholar]
- 7. Högbom M. (2010) The manganese/iron-carboxylate proteins. What is what, where are they, and what can the sequences tell us? J. Biol. Inorg. Chem. 15, 339–349 [DOI] [PubMed] [Google Scholar]
- 8. Krebs C., Matthews M. L., Jiang W., Bollinger J. M., Jr. (2007) AurF from Streptomyces thioluteus and a possible new family of manganese/iron oxygenases. Biochemistry 46, 10413–10418 [DOI] [PubMed] [Google Scholar]
- 9. Herrick J., Sclavi B. (2007) Ribonucleotide reductase and the regulation of DNA replication. An old story and an ancient heritage. Mol. Microbiol. 63, 22–34 [DOI] [PubMed] [Google Scholar]
- 10. Brignole E. J., Ando N., Zimanyi C. M., Drennan C. L. (2012) The prototypic class Ia ribonucleotide reductase from Escherichia coli. Still surprising after all these years. Biochem. Soc. Trans. 40, 523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kolberg M., Strand K. R., Graff P., Andersson K. K. (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta 1699, 1–34 [DOI] [PubMed] [Google Scholar]
- 12. Bollinger J. M., Jr., Krebs C. (2006) Stalking intermediates in oxygen activation by iron enzymes. Motivation and method. J. Inorg. Biochem. 100, 586–605 [DOI] [PubMed] [Google Scholar]
- 13. Que L., Jr. (1991) Oxygen activation at the diiron center of ribonucleotide reductase. Science 253, 273–274 [DOI] [PubMed] [Google Scholar]
- 14. Que L., Jr. (2004) The oxo/peroxo debate. A nonheme iron perspective. J. Biol. Inorg. Chem. 9, 684–690 [DOI] [PubMed] [Google Scholar]
- 15. Han W. G., Liu T., Lovell T., Noodleman L. (2006) Seven clues to the origin and structure of class-I ribonucleotide reductase intermediate X. J. Inorg. Biochem. 100, 771–779 [DOI] [PubMed] [Google Scholar]
- 16. Mitić N., Clay M. D., Saleh L., Bollinger J. M., Jr., Solomon E. I. (2007) Spectroscopic and electronic structure studies of intermediate X in ribonucleotide reductase R2 and two variants. A description of the FeIV-oxo bond in the FeIII-O-FeIV dimer. J. Am. Chem. Soc. 129, 9049–9065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burdi D. W., Willems J. P., Riggs-Gelasco P., Antholine W. E., Stubbe J., Hoffman B. M. (1998) The core structure of X generated in the assembly of the di-iron cluster of ribonucleotide reductase. 17O2 and H217O ENDOR. J. Am. Chem. Soc. 120, 12910–12919 [Google Scholar]
- 18. Reece S. Y., Hodgkiss J. M., Stubbe J., Nocera D. G. (2006) Proton-coupled electron transfer. The mechanistic underpinning for radical transport and catalysis in biology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1351–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adrait A., Ohrström M., Barra A. L., Thelander L., Gräslund A. (2002) EPR studies on a stable sulfinyl radical observed in the iron-oxygen-reconstituted Y177F/I263C protein R2 double mutant of ribonucleotide reductase from mouse. Biochemistry 41, 6510–6516 [DOI] [PubMed] [Google Scholar]
- 20. Licht S., Gerfen G. J., Stubbe J. (1996) Thiyl radicals in ribonucleotide reductases. Science 271, 477–481 [DOI] [PubMed] [Google Scholar]
- 21. Mao S. S., Holler T. P., Yu G. X., Bollinger J. M., Jr., Booker S., Johnston M. I., Stubbe J. (1992) A model for the role of multiple cysteine residues involved in ribonucleotide reduction. Amazing and still confusing. Biochemistry 31, 9733–9743 [DOI] [PubMed] [Google Scholar]
- 22. Prinz W. A., Aslund F., Holmgren A., Beckwith J. (1997) The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272, 15661–15667 [DOI] [PubMed] [Google Scholar]
- 23. Stubbe J., Riggs-Gelasco P. (1998) Harnessing free radicals. Formation and function of the tyrosyl radical in ribonucleotide reductase. Trends Biochem. Sci. 23, 438–443 [DOI] [PubMed] [Google Scholar]
- 24. Högbom M., Stenmark P., Voevodskaya N., McClarty G., Gräslund A., Nordlund P. (2004) The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass. Science 305, 245–248 [DOI] [PubMed] [Google Scholar]
- 25. Voevodskaya N., Lendzian F., Ehrenberg A., Gräslund A. (2007) High catalytic activity achieved with a mixed manganese-iron site in protein R2 of Chlamydia ribonucleotide reductase. FEBS Lett. 581, 3351–3355 [DOI] [PubMed] [Google Scholar]
- 26. Boal A. K., Cotruvo J. A., Jr., Stubbe J., Rosenzweig A. C. (2010) Structural basis for activation of class Ib ribonucleotide reductase. Science 329, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang W., Yun D., Saleh L., Barr E. W., Xing G., Hoffart L. M., Maslak M. A., Krebs C., Bollinger J. M., Jr. (2007) A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science 316, 1188–1191 [DOI] [PubMed] [Google Scholar]
- 28. Bollinger J. M., Jr., Jiang W., Green M. T., Krebs C. (2008) The manganese(IV)/iron(III) cofactor of Chlamydia trachomatis ribonucleotide reductase. Structure, assembly, radical initiation, and evolution. Curr. Opin. Struct. Biol. 18, 650–657 [DOI] [PubMed] [Google Scholar]
- 29. Voevodskaya N., Lendzian F., Sanganas O., Grundmeier A., Gräslund A., Haumann M. (2009) Redox intermediates of the Mn-Fe site in subunit R2 of Chlamydia trachomatis ribonucleotide reductase. An x-ray absorption and EPR study. J. Biol. Chem. 284, 4555–4566 [DOI] [PubMed] [Google Scholar]
- 30. Leidel N., Popović-Bijelić A., Havelius K. G., Chernev P., Voevodskaya N., Gräslund A., Haumann M. (2012) High-valent [MnFe] and [FeFe] cofactors in ribonucleotide reductases. Biochim. Biophys. Acta 1817, 430–444 [DOI] [PubMed] [Google Scholar]
- 31. Voevodskaya N., Lendzian F., Gräslund A. (2005) A stable FeIII-FeIV replacement of tyrosyl radical in a class I ribonucleotide reductase. Biochem. Biophys. Res. Commun. 330, 1213–1216 [DOI] [PubMed] [Google Scholar]
- 32. Voevodskaya N., Galander M., Högbom M., Stenmark P., McClarty G., Gräslund A., Lendzian F. (2007) Structure of the high-valent FeIIIFeIV state in ribonucleotide reductase (RNR) of Chlamydia trachomatis. Combined EPR, 57Fe-, 1H-ENDOR and x-ray studies. Biochim. Biophys. Acta 1774, 1254–1263 [DOI] [PubMed] [Google Scholar]
- 33. Schenk G., Boutchard C. L., Carrington L. E., Noble C. J., Moubaraki B., Murray K. S., de Jersey J., Hanson G. R., Hamilton S. (2001) A purple acid phosphatase from sweet potato contains an antiferromagnetically coupled binuclear Fe-Mn center. J. Biol. Chem. 276, 19084–19088 [DOI] [PubMed] [Google Scholar]
- 34. Andersson C. S., Högbom M. (2009) A Mycobacterium tuberculosis ligand-binding Mn/Fe protein reveals a new cofactor in a remodeled R2-protein scaffold. Proc. Natl. Acad. Sci. U.S.A. 106, 5633–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Högbom M. (2011) Metal use in ribonucleotide reductase R2, di-iron, di-manganese and heterodinuclear. An intricate bioinorganic workaround to use different metals for the same reaction. Metallomics 3, 110–120 [DOI] [PubMed] [Google Scholar]
- 36. Cotruvo J. A., Jr., Stubbe J. (2012) Metallation and mismetallation of iron and manganese proteins in vitro and in vivo. The class I ribonucleotide reductases as a case study. Metallomics 4, 1020–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boal A. K., Cotruvo J. A., Jr., Stubbe J., Rosenzweig A. C. (2012) The dimanganese(II) site of Bacillus subtilis class Ib ribonucleotide reductase. Biochemistry 51, 3861–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cotruvo J. A., Jr., Stubbe J. (2010) An active dimanganese(III)-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Biochemistry 49, 1297–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cox N., Ogata H., Stolle P., Reijerse E., Auling G., Lubitz W. (2010) A tyrosyl-dimanganese coupled spin system is the native metalloradical cofactor of the R2F subunit of the ribonucleotide reductase of Corynebacterium ammoniagenes. J. Am. Chem. Soc. 132, 11197–11213 [DOI] [PubMed] [Google Scholar]
- 40. Lambertz C., Leidel N., Havelius K. G., Noth J., Chernev P., Winkler M., Happe T., Haumann M. (2011) O2 reactions at the six-iron active site (H-cluster) in [FeFe]-hydrogenase. J. Biol. Chem. 286, 40614–40623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andersson C. S., Öhrström M., Popović-Bijelić A., Gräslund A., Stenmark P., Högbom M. (2012) The manganese ion of the heterodinuclear Mn/Fe cofactor in Chlamydia trachomatis ribonucleotide reductase R2c is located at metal position 1. J. Am. Chem. Soc. 134, 123–125 [DOI] [PubMed] [Google Scholar]
- 42. Dassama L. M., Boal A. K., Krebs C., Rosenzweig A. C., Bollinger J. M., Jr. (2012) Evidence that the beta subunit of Chlamydia trachomatis ribonucleotide reductase is active with the manganese ion of its manganese(IV)/iron(III) cofactor in site 1. J. Am. Chem. Soc. 134, 2520–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Que L., Jr., Tolman W. B. (2002) Bis(mu-oxo)dimetal “diamond” cores in copper and iron complexes relevant to biocatalysis. Angew. Chem. Int. Ed. Engl. 41, 1114–1137 [DOI] [PubMed] [Google Scholar]
- 44. Roos K., Siegbahn P. E. (2011) Oxygen cleavage with manganese and iron in ribonucleotide reductase from Chlamydia trachomatis. J. Biol. Inorg. Chem. 16, 553–565 [DOI] [PubMed] [Google Scholar]
- 45. Griese J. J., Högbom M. (2012) X-ray reduction correlates with soaking accessibility as judged from four non-crystallographically related diiron sites. Metallomics 4, 894–898 [DOI] [PubMed] [Google Scholar]
- 46. Garman E. F. (2010) Radiation damage in macromolecular crystallography. What is it and why should we care? Acta Crystallogr. D Biol. Crystallogr. 66, 339–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holton J. M. (2009) A beginner's guide to radiation damage. J. Synchrotron Radiat. 16, 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sommerhalter M., Lieberman R. L., Rosenzweig A. C. (2005) X-ray crystallography and biological metal centers. Is seeing believing? Inorg. Chem. 44, 770–778 [DOI] [PubMed] [Google Scholar]
- 49. Högbom M., Huque Y., Sjöberg B. M., Nordlund P. (2002) Crystal structure of the di-iron/radical protein of ribonucleotide reductase from Corynebacterium ammoniagenes. Biochemistry 41, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 50. Lindqvist Y., Huang W., Schneider G., Shanklin J. (1996) Crystal structure of δ(9) stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. EMBO J. 15, 4081–4092 [PMC free article] [PubMed] [Google Scholar]
- 51. Logan D. T., Su X. D., Aberg A., Regnström K., Hajdu J., Eklund H., Nordlund P. (1996) Crystal structure of reduced protein R2 of ribonucleotide reductase. The structural basis for oxygen activation at a dinuclear iron site. Structure 4, 1053–1064 [DOI] [PubMed] [Google Scholar]
- 52. Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G. (2000) The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622 [DOI] [PubMed] [Google Scholar]
- 53. Berglund G. I., Carlsson G. H., Smith A. T., Szöke H., Henriksen A., Hajdu J. (2002) The catalytic pathway of horseradish peroxidase at high resolution. Nature 417, 463–468 [DOI] [PubMed] [Google Scholar]
- 54. Adam V., Royant A., Nivière V., Molina-Heredia F. P., Bourgeois D. (2004) Structure of superoxide reductase bound to ferrocyanide and active site expansion upon x-ray-induced photoreduction. Structure 12, 1729–1740 [DOI] [PubMed] [Google Scholar]
- 55. Wuerges J., Lee J. W., Yim Y. I., Yim H. S., Kang S. O., Djinovic Carugo K. (2004) Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc. Natl. Acad. Sci. U.S.A. 101, 8569–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kühnel K., Jarchau T., Wolf E., Schlichting I., Walter U., Wittinghofer A., Strelkov S. V. (2004) The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc. Natl. Acad. Sci. U.S.A. 101, 17027–17032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hough M. A., Antonyuk S. V., Strange R. W., Eady R. R., Hasnain S. S. (2008) Crystallography with online optical and x-ray absorption spectroscopies demonstrates an ordered mechanism in copper nitrite reductase. J. Mol. Biol. 378, 353–361 [DOI] [PubMed] [Google Scholar]
- 58. Yano J., Kern J., Irrgang K. D., Latimer M. J., Bergmann U., Glatzel P., Pushkar Y., Biesiadka J., Loll B., Sauer K., Messinger J., Zouni A., Yachandra V. K. (2005) X-ray damage to the Mn4Ca complex in single crystals of photosystem II. A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. U.S.A. 102, 12047–12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Haumann M., Liebisch P., Müller C., Barra M., Grabolle M., Dau H. (2005) Photosynthetic O2 formation tracked by time-resolved x-ray experiments. Science 310, 1019–1021 [DOI] [PubMed] [Google Scholar]
- 60. Grabolle M., Haumann M., Müller C., Liebisch P., Dau H. (2006) Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by x-ray irradiation at 10–300 K. J. Biol. Chem. 281, 4580–4588 [DOI] [PubMed] [Google Scholar]
- 61. Yi J., Orville A. M., Skinner J. M., Skinner M. J., Richter-Addo G. B. (2010) Synchrotron X-ray-induced photoreduction of ferric myoglobin nitrite crystals gives the ferrous derivative with retention of the O-bonded nitrite ligand. Biochemistry 49, 5969–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Purwar N., McGarry J. M., Kostera J., Pacheco A. A., Schmidt M. (2011) Interaction of nitric oxide with catalase. Structural and kinetic analysis. Biochemistry 50, 4491–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dau H., Haumann M. (2008) The manganese complex of photosystem II in its reaction cycle. Basic framework and possible realization at the atomic level. Coord. Chem. Rev. 252, 273–295 [Google Scholar]
- 64. Cotelesage J. J., Pushie M. J., Grochulski P., Pickering I. J., George G. N. (2012) Metalloprotein active site structure determination. Synergy between X-ray absorption spectroscopy and X-ray crystallography. J. Inorg. Biochem. 115, 127–137 [DOI] [PubMed] [Google Scholar]
- 65. Shi W., Chance M. R. (2011) Metalloproteomics. Forward and reverse approaches in metalloprotein structural and functional characterization. Curr. Opin. Chem. Biol. 15, 144–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Younker J. M., Krest C. M., Jiang W., Krebs C., Bollinger J. M., Jr., Green M. T. (2008) Structural analysis of the Mn(IV)/Fe(III) cofactor of Chlamydia trachomatis ribonucleotide reductase by extended x-ray absorption fine structure spectroscopy and density functional theory calculations. J. Am. Chem. Soc. 130, 15022–15027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Riggs-Gelasco P. J., Shu L. J., Chen S. X., Burdi D., Huynh B. H., Que L., Stubbe J. (1998) EXAFS characterization of the intermediate X generated during the assembly of the Escherichia coli ribonucleotide reductase R2 diferric tyrosyl radical cofactor. J. Am. Chem. Soc. 120, 849–860 [Google Scholar]
- 68. Baldwin J., Krebs C., Saleh L., Stelling M., Huynh B. H., Bollinger J. M., Jr., Riggs-Gelasco P. (2003) Structural characterization of the peroxodiiron(III) intermediate generated during oxygen activation by the W48A/D84E variant of ribonucleotide reductase protein R2 from Escherichia coli. Biochemistry 42, 13269–13279 [DOI] [PubMed] [Google Scholar]
- 69. Petrie S., Gatt P., Stranger R., Pace R. J. (2012) Modelling the metal atom positions of the photosystem II water oxidising complex. A density functional theory appraisal of the 1.9-Å resolution crystal structure. Phys. Chem. Chem. Phys. 14, 11333–11343 [DOI] [PubMed] [Google Scholar]
- 70. Grundmeier A., Dau H. (2012) Structural models of the manganese complex of photosystem II and mechanistic implications. Biochim. Biophys. Acta 1817, 88–105 [DOI] [PubMed] [Google Scholar]
- 71. Popović-Bijelić A., Voevodskaya N., Domkin V., Thelander L., Gräslund A. (2009) Metal binding and activity of ribonucleotide reductase protein R2 mutants. Conditions for formation of the mixed manganese-iron cofactor. Biochemistry 48, 6532–6539 [DOI] [PubMed] [Google Scholar]
- 72. Klockenkämper R. (1996) Total Reflection X-ray Fluorescence Analysis, Wiley-VCH, London, UK [Google Scholar]
- 73. Stripp S., Sanganas O., Happe T., Haumann M. (2009) The structure of the active site H-cluster of [FeFe] hydrogenase from the green alga Chlamydomonas reinhardtii studied by x-ray absorption spectroscopy. Biochemistry 48, 5042–5049 [DOI] [PubMed] [Google Scholar]
- 74. Haumann M., Müller C., Liebisch P., Iuzzolino L., Dittmer J., Grabolle M., Neisius T., Meyer-Klaucke W., Dau H. (2005) Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3,4 →S0) characterized by x-ray absorption spectroscopy at 20 K and room temperature. Biochemistry 44, 1894–1908 [DOI] [PubMed] [Google Scholar]
- 75. Dau H., Liebisch P., Haumann M. (2003) X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers. Potential and questions examined with special focus on the tetra-nuclear manganese complex of oxygenic photosynthesis. Anal. Bioanal. Chem. 376, 562–583 [DOI] [PubMed] [Google Scholar]
- 76. Zabinsky S. I., Rehr J. J., Ankudinov A. L., Albers R. C., Eller M. J. (1995) Multiple-scattering calculations of x-ray absorption spectra. Phys. Rev. B 52, 2995–3009 [DOI] [PubMed] [Google Scholar]
- 77. Rehr J. J. (2006) Theory and calculations of X-ray spectra. XAS, XES, XRS, and NRIXS. Radiat. Phys. Chem. 75, 1547–1558 [Google Scholar]
- 78. Liu W. T., Thorp H. H. (1993) Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2. Refined distances and other enzymes. Inorg. Chem. 32, 4102–4105 [Google Scholar]
- 79. Neese F. (2008) ORCA. An ab initio, DFT, and semiempirical electronic structure package version 2.6.35. Theoretical Chemistry Group, Max-Planck Institute for Chemical Energy Conversion, Mühlheim, Germany [Google Scholar]
- 80. Chernev P., Zaharieva I., Dau H., Haumann M. (2011) Carboxylate shifts steer interquinone electron transfer in photosynthesis. J. Biol. Chem. 286, 5368–5374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Becke A. D. (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 [DOI] [PubMed] [Google Scholar]
- 82. Schäfer A., Huber C., Ahlrichs R. (1994) Fully optimized contracted Gaussian basis sets of triple ζ valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 [Google Scholar]
- 83. Weigend F. (2006) Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065 [DOI] [PubMed] [Google Scholar]
- 84. Sinnecker S., Rajendran A., Klamt A., Diedenhofen M., Neese F. (2006) Calculation of solvent shifts on electronic g-tensors with the conductor-like screening model (COSMO) and its self-consistent generalization to real solvents (Direct COSMO-RS). J. Phys. Chem. A 110, 2235–2245 [DOI] [PubMed] [Google Scholar]
- 85. Seal P., Chakrabarti S. (2008) Magnetic interactions in alkyl substituted cyclohexane diradical systems. A broken symmetry approach. J. Phys. Chem. A 112, 3409–3413 [DOI] [PubMed] [Google Scholar]
- 86. Sinnecker S., Neese F., Noodleman L., Lubitz W. (2004) Calculating the electron paramagnetic resonance parameters of exchange coupled transition metal complexes using broken symmetry density functional theory. Application to a Mn-III/Mn-IV model compound. J. Am. Chem. Soc. 126, 2613–2622 [DOI] [PubMed] [Google Scholar]
- 87. Zwart P. H., Lamzin V. S. (2003) Distance distributions and electron density characteristics of protein models. Acta Crystallogr. D Biol. Crystallogr. 59, 2104–2113 [DOI] [PubMed] [Google Scholar]
- 88. Westre T. E., Kennepohl P., DeWitt J. G., Hedman B., Hodgson K. O., Solomon E. I. (1997) A multiplet analysis of Fe K-edge 1s → 3d pre-edge features of iron complexes. J. Am. Chem. Soc. 119, 6297–6314 [Google Scholar]
- 89. Visser H., Anxolabéhère-Mallart E., Bergmann U., Glatzel P., Robblee J. H., Cramer S. P., Girerd J. J., Sauer K., Klein M. P., Yachandra V. K. (2001) Mn K-edge XANES and K, XES studies of two Mn-oxo binuclear complexes. Investigation of three different oxidation states relevant to the oxygen-evolving complex of photosystem II. J. Am. Chem. Soc. 123, 7031–7039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roos K., Siegbahn P. E. (2009) Density functional theory study of the manganese-containing ribonucleotide reductase from Chlamydia trachomatis. Why manganese is needed in the active complex. Biochemistry 48, 1878–1887 [DOI] [PubMed] [Google Scholar]
- 91. Han W. G., Giammona D. A., Bashford D., Noodleman L. (2010) Density functional theory analysis of structure, energetics, and spectroscopy for the Mn-Fe active site of Chlamydia trachomatis ribonucleotide reductase in four oxidation states. Inorg. Chem. 49, 7266–7281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Beitlich T., Kühnel K., Schulze-Briese C., Shoeman R. L., Schlichting I. (2007) Cryoradiolytic reduction of crystalline heme proteins. Analysis by UV-Vis spectroscopy and x-ray crystallography. J. Synchrotron Radiat. 14, 11–23 [DOI] [PubMed] [Google Scholar]
- 93. Dubois L., Jacquamet L., Pecaut J., Latour J. M. (2006) X-ray photoreduction of a di(mu-oxo)Mn(III)Mn(IV) complex occurs at temperatures as low as 20 K. Chem. Commun. (Camb.) 43, 4521–4523 [DOI] [PubMed] [Google Scholar]
- 94. de Serrano V. S., Davis M. F., Gaff J. F., Zhang Q., Chen Z., D'Antonio E. L., Bowden E. F., Rose R., Franzen S. (2010) X-ray structure of the metcyano form of dehaloperoxidase from Amphitrite ornata. Evidence for photoreductive dissociation of the iron-cyanide bond. Acta Crystallogr. D Biol. Crystallogr. 66, 770–782 [DOI] [PubMed] [Google Scholar]
- 95. Kim J. H., Kim Y. K., Kang H. (2009) Proton transfer and H/D isotopic exchange of water molecules mediated by hydroxide ions on ice film surfaces. J. Chem. Phys. 131, 044705 [DOI] [PubMed] [Google Scholar]
- 96. Chen S. H., Loong C. K. (2006) Neutron scattering investigations of proton dynamics of water and hydroxyl species in confined geometries. Nuclear Engineer. Technol. 38, 201–224 [Google Scholar]
- 97. Rasaiah J. C., Garde S., Hummer G. (2008) Water in nonpolar confinement. From nanotubes to proteins and beyond. Annu. Rev. Phys. Chem. 59, 713–740 [DOI] [PubMed] [Google Scholar]
- 98. Hermes S., Bremm O., Garczarek F., Derrien V., Liebisch P., Loja P., Sebban P., Gerwert K., Haumann M. (2006) A time-resolved iron-specific X-ray absorption experiment yields no evidence for an Fe2+ → Fe3+ transition during QA → QB electron transfer in the photosynthetic reaction center. Biochemistry 45, 353–359 [DOI] [PubMed] [Google Scholar]
- 99. Högbom M., Galander M., Andersson M., Kolberg M., Hofbauer W., Lassmann G., Nordlund P., Lendzian F. (2003) Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-Å x-ray data. Proc. Natl. Acad. Sci. U.S.A. 100, 3209–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Voegtli W. C., Sommerhalter M., Saleh L., Baldwin J., Bollinger J. M., Jr., Rosenzweig A. C. (2003) Variable coordination geometries at the diiron(II) active site of ribonucleotide reductase R2. J. Am. Chem. Soc. 125, 15822–15830 [DOI] [PubMed] [Google Scholar]
- 101. Leidel N., Chernev P., Havelius K. G., Schwartz L., Ott S., Haumann M. (2012) Electronic structure of an [FeFe] hydrogenase model complex in solution revealed by x-ray absorption spectroscopy using narrow-band emission detection. J. Am. Chem. Soc. 134, 14142–14157 [DOI] [PubMed] [Google Scholar]
- 102. Dau H., Zaharieva I. (2009) Principles, efficiency, and blueprint character of solar-energy conversion in photosynthetic water oxidation. Acc. Chem. Res. 42, 1861–1870 [DOI] [PubMed] [Google Scholar]
- 103. Vass I., Styring S. (1991) pH-Dependent charge equilibria between tyrosine-D and the S states in photosystem II. Estimation of relative midpoint redox potentials. Biochemistry 30, 830–839 [DOI] [PubMed] [Google Scholar]
- 104. Magnuson A., Liebisch P., Högblom J., Anderlund M. F., Lomoth R., Meyer-Klaucke W., Haumann M., Dau H. (2006) Bridging-type changes facilitate successive oxidation steps at about 1 V in two binuclear manganese complexes. Implications for photosynthetic water oxidation. J. Inorg. Biochem. 100, 1234–1243 [DOI] [PubMed] [Google Scholar]
- 105. Gonzalez A., Nave C. (1994) Radiation damage in protein crystals at low temperature. Acta Crystallogr. D Biol. Crystallogr. 50, 874–877 [DOI] [PubMed] [Google Scholar]
- 106. Macedo S., Pechlaner M., Schmid W., Weik M., Sato K., Dennison C., Djinović-Carugo K. (2009) Can soaked-in scavengers protect metalloprotein active sites from reduction during data collection? J. Synchrotron Radiat. 16, 191–204 [DOI] [PubMed] [Google Scholar]
- 107. Umena Y., Kawakami K., Shen J. R., Kamiya N. (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 [DOI] [PubMed] [Google Scholar]
- 108. Schlichting I., Miao J. (2012) Emerging opportunities in structural biology with x-ray free-electron lasers. Curr. Opin. Struct. Biol. 22, 613–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Neutze R., Moffat K. (2012) Time-resolved structural studies at synchrotrons and x-ray free electron lasers. Opportunities and challenges. Curr. Opin. Struct. Biol. 22, 651–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fromme P., Spence J. C. (2011) Femtosecond nanocrystallography using x-ray lasers for membrane protein structure determination. Curr. Opin. Struct. Biol. 21, 509–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chapman H. N., Fromme P., Barty A., White T. A., Kirian R. A., Aquila A., Hunter M. S., Schulz J., DePonte D. P., Weierstall U., Doak R. B., Maia F. R., Martin A. V., Schlichting I., Lomb L., Coppola N., Shoeman R. L., Epp S. W., Hartmann R., Rolles D., Rudenko A., Foucar L., Kimmel N., Weidenspointner G., Holl P., Liang M., Barthelmess M., Caleman C., Boutet S., Bogan M. J., Krzywinski J., Bostedt C., Bajt S., Gumprecht L., Rudek B., Erk B., Schmidt C., Hömke A., Reich C., Pietschner D., Strüder L., Hauser G., Gorke H., Ullrich J., Herrmann S., Schaller G., Schopper F., Soltau H., Kühnel K. U., Messerschmidt M., Bozek J. D., Hau-Riege S. P., Frank M., Hampton C. Y., Sierra R. G., Starodub D., Williams G. J., Hajdu J., Timneanu N., Seibert M. M., Andreasson J., Rocker A., Jonsson O., Svenda M., Stern S., Nass K., Andritschke R., Schroter C. D., Krasniqi F., Bott M., Schmidt K. E., Wang X., Grotjohann I., Holton J. M., Barends T. R., Neutze R., Marchesini S., Fromme R., Schorb S., Rupp D., Adolph M., Gorkhover T., Andersson I., Hirsemann H., Potdevin G., Graafsma H., Nilsson B., Spence J. C. (2011) Femtosecond x-ray protein nanocrystallography. Nature 470, 73–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kern J., Alonso-Mori R., Hellmich J., Tran R., Hattne J., Laksmono H., Glöckner C., Echols N., Sierra R. G., Sellberg J., Lassalle-Kaiser B., Gildea R. J., Glatzel P., Grosse-Kunstleve R. W., Latimer M. J., McQueen T. A., DiFiore D., Fry A. R., Messerschmidt M., Miahnahri A., Schafer D. W., Seibert M. M., Sokaras D., Weng T. C., Zwart P. H., White W. E., Adams P. D., Bogan M. J., Boutet S., Williams G. J., Messinger J., Sauter N. K., Zouni A., Bergmann U., Yano J., Yachandra V. K. (2012) Room temperature femtosecond x-ray diffraction of photosystem II microcrystals. Proc. Natl. Acad. Sci. U.S.A. 109, 9721–9726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Boutet S., Lomb L., Williams G. J., Barends T. R., Aquila A., Doak R. B., Weierstall U., DePonte D. P., Steinbrener J., Shoeman R. L., Messerschmidt M., Barty A., White T. A., Kassemeyer S., Kirian R. A., Seibert M. M., Montanez P. A., Kenney C., Herbst R., Hart P., Pines J., Haller G., Gruner S. M., Philipp H. T., Tate M. W., Hromalik M., Koerner L. J., van Bakel N., Morse J., Ghonsalves W., Arnlund D., Bogan M. J., Caleman C., Fromme R., Hampton C. Y., Hunter M. S., Johansson L. C., Katona G., Kupitz C., Liang M., Martin A. V., Nass K., Redecke L., Stellato F., Timneanu N., Wang D., Zatsepin N. A., Schafer D., Defever J., Neutze R., Fromme P., Spence J. C., Chapman H. N., Schlichting I. (2012) High-resolution protein structure determination by serial femtosecond crystallography. Science 337, 362–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Danford J. J., Dobrowolski P., Berreau L. M. (2009) Thioester hydrolysis reactivity of an Fe(III)Zn(II) complex. Inorg. Chem. 48, 11352–11361 [DOI] [PubMed] [Google Scholar]
- 115. Lomoth R., Huang P., Zheng J., Sun L., Hammarström L., Akermark B., Styring S. (2002) Synthesis and characterization of a dinuclear manganese(III,III) complex with three phenolate ligands. Eur. J. Inorg. Chem. 2002, 2965–2974 [Google Scholar]