
Figure 1. Roles for c-Met in GB response and resistance to Bevacizumab.
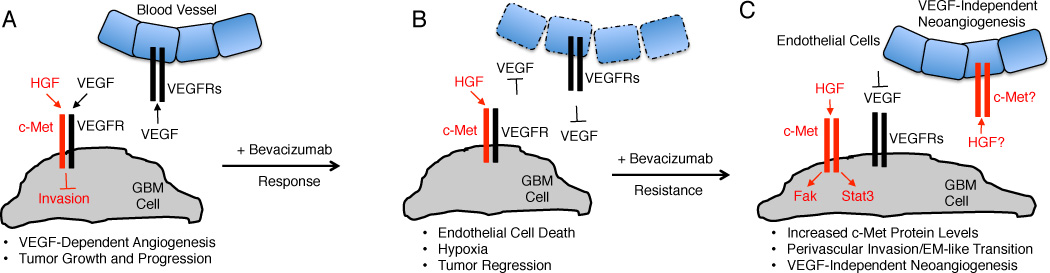
(A); Tumor cell proliferation and invasion are regulated via cross talk between the VEGF-A and HGF signaling pathways. During GB growth and progression c-Met and VEGFR-2 form heterodimeric complexes that suppress cell invasion via PTP1B-mediated Met tyrosine dephosphorylation. Meanwhile, VEGF-A produced by GB cells interacts with its receptors in endothelial cells and promotes angiogenesis and blood vessel permeability in the vascular niche. (B); Bevacizumab neutralizes VEGF-A signaling, leading to endothelial cell death, tumor regression, and transient improvements in overall and progression-free survival. (C); Responsiveness to Bevacizumab is followed by resistance, due to upregulation of c-Met gene expression in GB cells (acquired resistance) or selective survival of sub-populations of tumor cells that overexpress c-Met (intrinsic resistance). Met forms homodimers that bind HGF/SF and via an autocrine signaling loop promote Stat3 and Fak phosphorylation, promoting a mesenchymal-like phenotypes and perivascular tumor cell invasion. In Bevacizumab resistant tumors, paracrine HGF/SF signaling via Met in endothelial cells may also promote VEGF-independent neoangiogenesis.