

Abstract
One of the key advances in genome assembly that has led to a significant improvement in contig lengths has been improved algorithms for utilization of paired reads (mate-pairs). While in most assemblers, mate-pair information is used in a post-processing step, the recently proposed Paired de Bruijn Graph (PDBG) approach incorporates the mate-pair information directly in the assembly graph structure. However, the PDBG approach faces difficulties when the variation in the insert sizes is high. To address this problem, we first transform mate-pairs into edge-pair histograms that allow one to better estimate the distance between edges in the assembly graph that represent regions linked by multiple mate-pairs. Further, we combine the ideas of mate-pair transformation and PDBGs to construct new data structures for genome assembly: pathsets and pathset graphs.
Key words: algorithms, cancer genomics, combinatorics, computational molecular biology, evolution, genetic algorithms, genomics, genomic rearrangements, graphs and networks, next generation sequencing, sequence analysis
1. Introduction
Current High Throughput Sequencing (HTS) technologies have reduced the time and cost of genome sequencing and have enabled new experimental opportunities in a variety of applications. However, as sequencing technologies improve, the challenges in designing software to assemble genomes become harder. Current genome assemblers face the challenge of assembling billions of short reads. When the length of a repeat is longer than the read length, correctly matching up its upstream and downstream flanking regions is difficult. Fortunately, all current sequencing platforms are able to produce mate-pairs—pairs of reads whose separation in the genome (called the insert size) is approximately known. Because insert sizes may be much longer than the read length, mate-pairs may span over long repeats and match up their flanking regions.
Mate-pair information has played an important role in most genome projects, and many HTS assemblers incorporate mate-pair information to increase the contigs length in a post-processing step (Pevzner and Tang, 2001; Chaisson and Pevzner, 2008; Zerbino and Birney, 2008; Butler et al., 2008; Simpson et al., 2009; Chaisson et al., 2009; Li et al., 2010). Most of these assemblers rely on the same basic observation (Pevzner and Tang, 2001): If there is a unique path of suitable length in the assembly graph that connects the left and right reads of a mate-pair, the gap in the mate-pair can be filled in with the nucleotides spelled by this path (mate-pair transformation). However, when multiple paths exist between the left and right reads within a mate-pair, it remains ambiguous which path should be used to fill in the gap. Unfortunately, mate-pairs generated by existing HTS protocols are characterized by rather large variations in insert sizes, leading to multiple paths for a significant fraction of mate-pairs (since the range of suitable path lengths is wide), making it difficult to utilize such mate-pairs.
Recently, Medvedev et al. (2011) introduced the notion of the paired de Bruijn graph, which incorporates mate-pair information into the graph structure rather than using it for mate-pair transformations in a post-processing step. Similar methods were also developed independently (Chikhi and Lavenier, 2011; Donmez and Brudno, 2011). Unfortunately, the performance of these methods deteriorates as the variation in the insert size increases.
Below we show that while reducing variation in the insert size remains a difficult experimental problem, it can be addressed computationally by aggregating the mate-pair information for all mate-pairs linking a pair of edges in the condensed de Bruijn graph. This transforms mate-pairs into edge-pair histograms, consisting of pairs of edges together with distances aggregated from mate-pair information.
Using the collection of edge-pair histograms, we further combine the ideas of mate-pair transformations and paired de Bruijn graphs into new data structures for genome assembly, called pathsets and pathset graphs. We further compare the performance of our assembler (based on the pathsets approach) to other assemblers on various bacterial datasets.
2. Methods
2.1. Assumptions
There are many sources of error and variation in Next Generation Sequencing technologies. This paper focuses on issues arising from paired reads. For theoretical development of our methods, in this section we will assume all reads are perfect, with no local errors like point mutations, insertions, or deletions. We will also assume perfect coverage, with a k-mer starting at every position in the genome. Our focus is on the complexities of using paired reads in assembly: (1) insert size variation; (2) repeats for which a read pair maps as a pair to two or more places in the genome, resulting in multiple ways to fill in the region between those two reads; and (3) chimeric read pairs.
In Sec. 2.9 and Results, we demonstrate that our approach can be adapted to work well with various bacterial datasets where read errors and imperfect coverage are present.
2.2. De Bruijn graphs
We represent genomes as circular strings over {A,T,G,C}, the alphabet of nucleotides. An n-mer is a string of length n. Given a n-mer 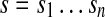 , we define prefix
, we define prefix
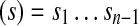 and suffix
and suffix
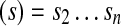 .
.
Let k be a fixed parameter. Given a set A of k-mers, the standard de Bruijn graph has a directed edge (prefix(s), suffix(s)) for each k-mer 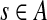 . The condensed de Bruijn graph CG(A,k) is obtained from the standard de Bruijn graph by replacing every maximal non-branching path1
P by a single edge e of length ℓ(e) equal to the number of edges in P (Fig. 1a,b). Below we work with condensed (rather than standard) de Bruijn graphs and refer to them simply as de Bruijn graphs.
. The condensed de Bruijn graph CG(A,k) is obtained from the standard de Bruijn graph by replacing every maximal non-branching path1
P by a single edge e of length ℓ(e) equal to the number of edges in P (Fig. 1a,b). Below we work with condensed (rather than standard) de Bruijn graphs and refer to them simply as de Bruijn graphs.
FIG. 1.

From de Bruijn graph to pathset graph. (a) A standard de Bruijn graph and the corresponding mapping of mate-pairs. The number on top of each node is the node ID. The smaller blue numbers below/beside each node are the IDs of the corresponding paired right nodes. The bold red, blue, and green paths show how the genome traverses the graph. (b) The condensed de Bruijn graph with edges corresponding to non-branching paths in the standard de Bruijn graph. The dotted red lines indicate edge-pairs. (c) Pathset graph. Initially there are eight pathsets: C1 = {e1e3e5, e1e4e5}, C2 = {e1e3}, C3 = {e3e5}, C4 = {e2e3e5, e2e4e5}, C5 = {e2e3e6, e2e4e6}, C6 = {e4e5}, C7 = {e4e6}, and C8 = {e2e4}. Using the edge-pair information, we find phantom paths (indicated in boldface) and remove them. After removal of all prefix pathsets (C2 and C8), the pathset graph has six nodes and consists of three edges: C1 → C3 (red path), C4 → C6 (green path), and C5 → C7 (blue path). Each edge in the pathset graph corresponds to a contig; e.g., C1 → C3 spells out the red path (AAACAATCGGCCGCTTTAG).
Each k-mer 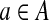 maps to an edge e = edge (a) in CG (A,k) at position offset(a) (1≤offset(a) ≤ ℓ(e)). For a path
maps to an edge e = edge (a) in CG (A,k) at position offset(a) (1≤offset(a) ≤ ℓ(e)). For a path 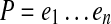 in a graph CG (A,k), we define
in a graph CG (A,k), we define 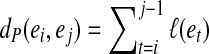 . We further define the length of P as dP(e1, en), i.e., the total length of its edges, excluding the last edge.
. We further define the length of P as dP(e1, en), i.e., the total length of its edges, excluding the last edge.
Given a parameter d, a pair of k-mers a, b form a (k,d)-mer (a|b) of string 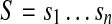 if there are instances of
if there are instances of 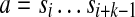 and
and 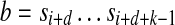 in S whose starting positions differ by d nucleotides. Given parameters d and Δ, a pair of k-mers (a|b) is called a (k, d, Δ)-mer of S if it is a (k, d0)-mer of S for some
in S whose starting positions differ by d nucleotides. Given parameters d and Δ, a pair of k-mers (a|b) is called a (k, d, Δ)-mer of S if it is a (k, d0)-mer of S for some 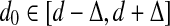 .
.
We call a and b the left and right k-mers of the pair (a|b), respectively, and we refer to the distance between them as the distance of (a|b). In particular, error-free pairs of reads of length l with exact insert size s form (l,s−l)-mers (Fig. 2). For a set B of k-mer pairs, we let left(B) (resp., right(B)) be the set of left (resp., right) k-mers appearing in B.
FIG. 2.
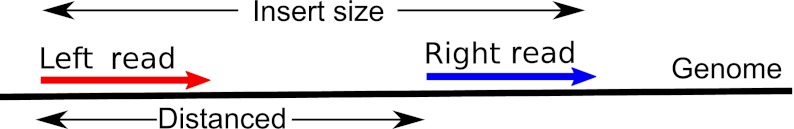
A mate-pair is a pair of reads with distance d between their starting positions. The insert size is the distance from the start of the left read to the end of the right read. Each platform has reads oriented a particular way, but for presentation purposes, we canonically reorient them as indicated.
We transform each pair of reads of length l into a sequence of l−k+1 consecutive k-mer pairs. For the set B of resulting k-mer pairs, we define the de Bruijn graph of reads as CG(left(B) ∪ right(B),k).
2.3. From k-mer pairs to edge-pair histograms.
We assume for simplicity that the genome defines a genomic walk W that passes through all edges in the de Bruijn graph of reads.2 Since some edges may appear multiple times in the genomic walk, we distinguish between the edge e and its instance 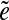 . Every two instances
. Every two instances 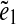 and
and 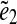 of edges e1 and e2 define a genomic distance
of edges e1 and e2 define a genomic distance
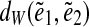 between edges e1 and e2. This in turn yields a triple
between edges e1 and e2. This in turn yields a triple 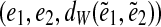 called a genomic edge-pair. Note that a pair of edges may have multiple genomic distances if one of these edges appears multiple times in the genomic walk.
called a genomic edge-pair. Note that a pair of edges may have multiple genomic distances if one of these edges appears multiple times in the genomic walk.
When the genomic walk W is unknown, genomic edge-pairs can be computed from the set of k-mer pairs B when the genomic distance between k-mers in each k-mer pair of B is known exactly. In practice, such distances are estimated rather than exact. Below, we define edge-pair histograms and use them for more accurate approximation of genomic edge-pairs.
For a set B of k-mer pairs, a pair of edges (e1, e2) in the de Bruijn graph CG(left(B) ∪ right(B), k) is called B-bounded if there exists at least one k-mer pair in B whose left (resp., right) k-mer maps to the edge e1 (resp., e2).
Below we assume that parameters d0 (estimated distance between reads within read-pairs) and Δ (maximum error in distance estimates) are fixed. Given a set of read-pairs whose distances fall in the range d0 ± Δ, one can generate a set B of all (k, d0, Δ)-mers (extracted from all read-pairs), and then compute the set of B-bounded edge-pairs. For a B-bounded edge-pair (e1, e2), we define the edge-pair histogram (e1, e2, h), where h is a histogram with h(x) equal to the number of (k, d0, Δ)-mers in B that support genomic distance x between e1 and e2:
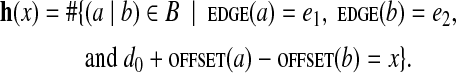 |
While HTS machines produce large numbers of read-pairs (e.g., over 107 for the E. coli datasets we analyze below), the de Bruijn graph of reads contains a small number of edges (several thousand for our E. coli datasets). Thus, edge-pair histograms are typically supported by many k-mer pairs, which allows one to accurately estimate the genomic distance(s) between e1 and e2.
Since insert sizes typically follow a Gaussian distribution (Chen et al., 2009), the histogram (e1, e2, h) either represents a sample from a single Gaussian distribution (if e1 and e2 appear once in the genomic walk) or a mixture of Gaussian distributions (if e1 and e2 appear multiple times in the genomic walk). Inferring each individual Guassian distribution from a sample of mixture distributions represents a challenging problem (Moitra and Valiant, 2010). Here we sketch a simple approach to address this problem (see Bankevich et al., 2012 for details of a more advanced analysis). We smooth the histogram, choose a threshold, and focus on the regions where the values of the histogram exceed the threshold (above the red line in Fig. 3b). The edge-pair histogram is thus transformed into one or more non-overlapping edge-pair intervals, each corresponding to one or more genomic edge-pairs with similar distances.
FIG. 3.

Estimating genomic distances between edges within edge-pairs using the edge-pair histogram. (a) The genome traverses the graph from e1 to e2 through P1, P2, and P3. (b) The smoothed edge-pair histogram (inferred from mate-pairs mapping to e1 and e2) supports various genomic distances between e1 and e2. Setting the threshold to 200 (red line) splits the histogram into two edge-pair intervals: (e1, e2,[130, 144]), supporting the traversal via paths P1 and P2, and (e1, e2,[156, 164]), supporting the traversal through path P3.
An edge-pair interval (e1, e2, [a, b]) is called correct if there exists a genomic edge-pair (e1, e1, D) such that a ≤ D ≤ b. A properly chosen threshold should maximize the number of correct edge-pair intervals, while still separating the genomic edge-pairs with different distances.
Figure 3a shows a pair of edges (e1, e2) that is traversed three times through the paths P1, P2, and P3. The first two traversals, e1P1e2 and e1P2e2, have similar lengths while the third, e1P3e2, has significantly larger length. Setting the threshold to 200 results in two edge-pair intervals: (e1, e2, [130,144]) and (e1, e2, [156,164]). The first interval supports two genomic edge-pairs, (e1, e2,134) and (e1, e2, 140), while the second interval supports a single genomic edge-pair, (e1, e2, 160).
Edge-pair intervals have two advantages over mate-pairs:
• With proper choice of thresholds, estimates of distances between edges in edge-pair intervals are more accurate than estimates of distances between individual mate-pairs. Better estimates may result in better performance of existing methods for resolving repeats, including mate-pair transformations (Pevzner et al., 2001), paired de Bruijn graphs (Medvedev et al., 2011), and mate-pair graphs (Donmez and Brudno, 2011).
• Since the edge-pair intervals compactly represent the mate-pair data, many redundant operations can be avoided; for instance, multiple function calls to perform mate-pair transformations for all mate-pairs corresponding to the same edge-pair interval (e.g., in EULER-SR; Chaisson and Pevzner, 2008) can be replaced by a single mate-pair transformation of the corresponding edge-pair interval.
Below, we use edge-pair intervals to define the notions of pathset and pathset graph.
2.4. From edge-pair intervals to pathsets
We define a pathset as any set of paths between a fixed pair of edges.3 Every edge-pair interval (e1, e2, [a, b]) corresponds to a pathset pathset(e1, e2, [a, b]) formed by all paths starting at e1, ending at e2, and having lengths in the interval [a, b]. For example, in Figure 1b, pathset(e1, e5, [9,11]) = {e1e3e5, e1e4e5}.
As a by-product of transforming edge-pair intervals to pathsets, the set of possible genomic distances between edges in each edge-pair interval can be further reduced. Namely, the interval [a, b] in an edge-pair interval (e1, e2, [a, b]) can be replaced by a list of lengths of paths in the corresponding pathset. This is referred to as an edge-pair distance set, or simply an edge-pair, and denoted (e1, e2, d). If all paths in a pathset have the same length, this length reveals the genomic distance between e1 and e2.
A path in the de Bruijn graph is called a genomic path if it corresponds to a substring of the genome (i.e., is a subpath of the genomic walk), and a phantom path otherwise. In general, a pathset may contain multiple genomic and phantom paths. Given a collection of pathsets obtained from edge-pairs, our goal is to remove the phantom paths in each pathset and to further split pathsets with t genomic paths into t pathsets consisting of singleton paths. While it is not always possible to accomplish this task (since it is not clear how to separate phantom and genomic paths), below we describe some steps towards this goal.
We note that all edge-pairs are disjoint, i.e., for every two distinct edge-pairs (e1, e2, d) and (e1, e2, d′) formed by the same two edges e1 and e2, the sets d and d′ are disjoint. A pair of edges e1 and e2 in the genomic walk is called d0-bounded if at least one of its genomic distances falls in the range [d0−ℓ(e2), d0 + ℓ(e1)]. A set of edge-pairs is representative if for each instance of a d0-bounded pair of edges e1 and e2 (at genomic distance D), there exists a supporting edge-pair (e1, e2, d) with 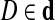 . For the sake of simplicity, we assume that a set of edge-pairs is representative and correct. Then the corresponding pathsets are also (a) disjoint (i.e., do not overlap as sets), (b) correct (i.e., each contains a genomic path), and (c) representative (i.e., every genomic path between d0-bounded instances of two edges belongs to some pathset). These conditions are important for constructing the pathset graph.4 Below we show how to remove phantom paths and split the pathsets into smaller pathsets while preserving conditions (a–c).
. For the sake of simplicity, we assume that a set of edge-pairs is representative and correct. Then the corresponding pathsets are also (a) disjoint (i.e., do not overlap as sets), (b) correct (i.e., each contains a genomic path), and (c) representative (i.e., every genomic path between d0-bounded instances of two edges belongs to some pathset). These conditions are important for constructing the pathset graph.4 Below we show how to remove phantom paths and split the pathsets into smaller pathsets while preserving conditions (a–c).
2.5. Removing phantom paths from pathsets
Since our pathsets are representative (condition (c)), for each instance of a d0-bounded pair of edges, there exists a supporting edge-pair. Therefore, if a path in a pathset does not have a supporting edge-pair, it represents a phantom path. Below we describe how to identify some (but not necessarily all) phantom paths.
A path 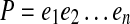 is supported by a set of edge-pairs EP if for every pair of d0-bounded edges e and e′ in P, there exists an edge-pair (e, e′, d)
is supported by a set of edge-pairs EP if for every pair of d0-bounded edges e and e′ in P, there exists an edge-pair (e, e′, d)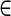 EP such that
EP such that 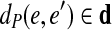 . The path P is strongly supported by a set of edge-pairs EP if it can be extended from both ends into a longer path
. The path P is strongly supported by a set of edge-pairs EP if it can be extended from both ends into a longer path 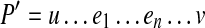 such that (i) P′ is supported by EP, (ii) the pair of edges u and e1 is d0-bounded, and (iii) the pair of edges en and v is d0-bounded. Paths in a pathset that are not strongly supported are classified as phantom paths and removed.5
such that (i) P′ is supported by EP, (ii) the pair of edges u and e1 is d0-bounded, and (iii) the pair of edges en and v is d0-bounded. Paths in a pathset that are not strongly supported are classified as phantom paths and removed.5
Indeed, if P is a genomic path, then it is a subpath of the genomic walk. In the genomic walk, there exists an edge u preceding P and an edge v succeeding P that satisfy the properties (ii) and (iii). The subpath starting at u and ending at v (denoted P′ above) clearly satisfies property (i).
2.6. Splitting pathsets
A pathset contains an edge e if there is a path in this pathset that contains e. For a pathset PS and sets of edges 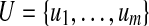 and
and 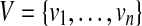 , we define
, we define 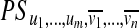 as the set of all paths in PS that contain all edges from U and no edges from V.
as the set of all paths in PS that contain all edges from U and no edges from V.
Two edges contained in a pathset are called independent if no path in this pathset contains them both. An edge e is essential for a pathset PS if there exists a genomic path in PS containing e. A set of essential edges in a pathset is called independent if every two edges in this set are independent. An independent set 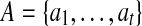 of essential edges contained in PS defines a split of PS into t disjoint pathsets:6
of essential edges contained in PS defines a split of PS into t disjoint pathsets:6
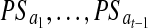 ,
, 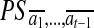 . We remark that each of these pathsets contains an essential edge from A and thus is correct. It is easy to check that the split operation preserves conditions (a–c).
. We remark that each of these pathsets contains an essential edge from A and thus is correct. It is easy to check that the split operation preserves conditions (a–c).
For example, for the pathset defined by edges e1 and e7 in Figure 4, all edges are essential. Edges e2 and e3 (as well as e5 and e6) form an independent set.
FIG. 4.
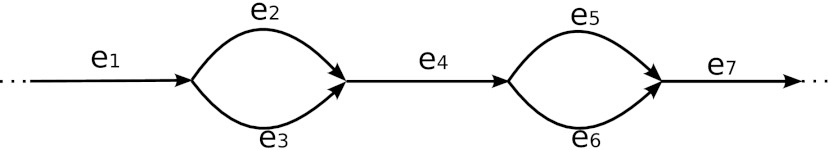
The pathset PS = pathset (e1, e7, d) corresponding to the edge-pair (e1, e7, d) contains four paths: PS={e1e2e4e5e7,e1e3e4e5e7,e1e2e4e6e7, e1e3e4e6e7}. Edges e2 and e3 (as well as edges e5 and e6) form an independent set. Therefore, PS can be split into two pathsets: PSe2= {e1e2e4e5e7,e1e2e4e6e7} and 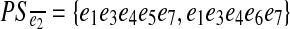 containing edges e2 and e3 respectively.
containing edges e2 and e3 respectively.
2.7. Identifying essential edges
To split a pathset, one has to identify essential edges. Consider an instance 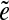 of an edge e in the genomic walk. A subpath of this walk
of an edge e in the genomic walk. A subpath of this walk 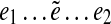 is called an
is called an 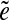 -subpath if (i) the pair of edges e1 and
-subpath if (i) the pair of edges e1 and 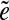 is d0-bounded, and (ii) the pair of edges
is d0-bounded, and (ii) the pair of edges 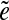 and e2 is d0-bounded. A maximal
and e2 is d0-bounded. A maximal 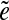 -subpath (i.e., a subpath containing all other
-subpath (i.e., a subpath containing all other 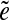 -subpaths) is called a span of
-subpaths) is called a span of 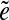 . For an edge e, we define Span(e) as a set of the spans of all instances
. For an edge e, we define Span(e) as a set of the spans of all instances 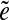 of the edge e.
of the edge e.
We refer to a pathset as PS(a, b) if all paths in this pathset start at edge a and end at edge b. Given paths 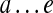 and
and 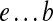 (i.e., starting and ending at the same edge e), we define their concatenation as the path
(i.e., starting and ending at the same edge e), we define their concatenation as the path 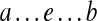 . Given a collection of pathsets and a fixed edge e, consider all pathsets PS(a, e) and PS(e, b) and all concatenations of paths from PS(a, e) with paths from PS(e, b) (for all possible choices of a and b). Define Spread(e) as the set of all supported paths in this set. Obviously, Span(e) ⊆ Spread(e).
. Given a collection of pathsets and a fixed edge e, consider all pathsets PS(a, e) and PS(e, b) and all concatenations of paths from PS(a, e) with paths from PS(e, b) (for all possible choices of a and b). Define Spread(e) as the set of all supported paths in this set. Obviously, Span(e) ⊆ Spread(e).
An edge e is called (e1, e2)-constrained if all paths in Spread(e) have the form 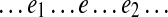 and edges e1 and e2 in all such paths are d0-bounded. It is easy to see that every (e1, e2)-constrained edge e is essential in some pathset from PS(e1, e2)s. Indeed, since the genomic walk contains each edge e in the graph, there exists a genomic path P in Span(e). Since Span(e) ⊆ Spread(e) and since e is (e1, e2)-constrained, P has the form
and edges e1 and e2 in all such paths are d0-bounded. It is easy to see that every (e1, e2)-constrained edge e is essential in some pathset from PS(e1, e2)s. Indeed, since the genomic walk contains each edge e in the graph, there exists a genomic path P in Span(e). Since Span(e) ⊆ Spread(e) and since e is (e1, e2)-constrained, P has the form 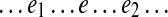 , where edges e1 and e2 are d0-bounded. Therefore, the subpath
, where edges e1 and e2 are d0-bounded. Therefore, the subpath 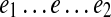 of P belongs to some pathset from PS(e1, e2), implying that e is an essential edge.
of P belongs to some pathset from PS(e1, e2), implying that e is an essential edge.
2.8. Pathset graph
Given a collection of pathsets satisfying conditions (a–c), the genome assembly problem becomes similar to traditional genome assembly with each pathset playing a role of a single (long) read. Below, we define the pathset graph with nodes corresponding to pathsets and non-branching paths corresponding to assembly contigs.
Path p is a prefix (resp., suffix) of path q if q can be obtained from p by concatenating some non-empty path to the end (resp., start) of p. Pathset PS is called a prefix of pathset PS′ if each path of PS is a prefix of a path in PS′. Path q follows path p if they have the following form: 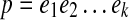 and
and 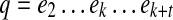 , where t ≥ 0. Pathset PS′ follows pathset PS if there exists paths
, where t ≥ 0. Pathset PS′ follows pathset PS if there exists paths 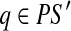 and
and 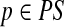 such that q follows p.
such that q follows p.
A collection of pathsets is called prefix-free if no pathset in this collection is a prefix of another pathset. Given a collection of pathsets, we remove phantom paths, perform splits, and remove prefix pathsets to obtain a prefix-free collection of pathsets. We then construct the pathset graph by representing each remaining pathset as a node and forming a directed edge PS → PS′ if PS′ follows PS (similarly to the classical overlap-layout-consensus approach to fragment assembly).
Figure 1c illustrates the pathset graph as a toy example. After removing phantom paths, we obtain six singleton pathsets. The pathset graph consists of six vertices and three edges (non-branching paths), corresponding to three contigs.
The pathset graph approach can be summarized as follows:
Input: Set of mate-pairs
1: Construct the de Bruijn graph from individual reads in mate-pairs.
2: Transform read-pairs into a set of edge-pair histograms.
3: Transform edge-pair histograms into edge-pair intervals.
4: Transform edge-pairs intervals into pathsets.
5: For each pathset:
6: Remove phantom paths.
7: Construct an independent edge-set in each pathset.
8: Split the pathset over the independent edge-set.
9: Remove prefix pathsets from the resulting collection of pathsets.
10: Construct the pathset graph on the resulting prefix-free collection of pathsets.
11: Output contigs as non-branching paths in the pathset graph.
2.9. Adaptations for imperfect coverage and read errors
In contrast to the paired de Bruijn graph approach by Medvedev et al. (2011), which requires us to have a pair of k-mers starting at each position of the genome, the pathset approach only requires us to have a pathset starting at each condensed edge. In the two bacterial datasets in our Results section, we observed very few cases where there is no such pathset. In genomes with complicated repeat structures, the number of such cases may increase. This raises two obstacles for the current approach: (1) some genomic paths may be removed; (2) some pathsets may not be extended. To address the first obstacle, we do not remove paths in singleton pathsets, and we retain the most supported path in each pathset if all of its paths are identified as phantom paths. For the second obstacle, if a pathset can not be extended, the algorithm finds the best possible extension pathset and extends the contig.
Additionally, some mate-pairs can have aberrant insert sizes or may contain errors that are not corrected by error correction programs. Therefore, some pathsets may not contain any genomic path. Our implementation has a procedure for removing pathsets that are not connected to any other pathsets and that have very few mapped mate-pairs.
3. Results
We implemented Pathset as a module in the new assembler SPAdes (Bankevich et al., 2012). The source code is released under the GNU General Public License and is available at http://bioinf.spbau.ru/spades/pathset. To evaluate the performance of Pathset, we compared it with various assemblers on two Illumina E. coli datasets. The first dataset (EMBL-EBI Sequence Read Archive ERA000206, which we refer to as EC215) consists of 28 million paired reads of length 100 bp and mean insert size ≈215 bp, while the second (EMBL-EBI Sequence Read Archive ERR022075, which we refer to as EC500) consists of 44 million paired reads of length 100 bp and mean insert size ≈500 bp. The reads in each dataset were error-corrected with Quake (Kelley et al., 2010).
3.1. From mate-pairs to edge-pair intervals
For each dataset, we constructed the de Bruijn graph for k = 55 and transformed the set of input mate-pairs into a set EP of edge-pair intervals. To evaluate this transformation, we further extracted from the E. coli genome a set of k-mer pairs at fixed distance d0. The mapping of these k-mer pairs to edges of the graph defines a set of genomic edge-pairs, EP0. Table 1 demonstrates that for insert size d0 = 215, most genomic edge-pairs in EP0 (97%) are also present7 in EP and very few (0.8%) of the edge-pair intervals in EP are incorrect. We also observed that for d0 = 500, the proportion of incorrect edge-pairs in EC500 only slightly increases as compared to EC215.
Table 1.
Comparison of Edge-Pairs from E. coli Genome vs. from Paired Reads
These correspond to E. coli datasets EC215 and EC500.
Edge-pairs determined from the reference genome; these are correct by definition.
Edge-pairs determined by mapping mate-pairs to the assembly graph.
The False Positive Rate is the fraction of edge-pair intervals in EP that are incorrect.
The False Negative Rate is the fraction of genomic edge-pairs in EP0 that are missing in EP.
3.2. From edge-pair intervals to pathsets
For each edge-pair interval 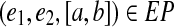 , we constructed its pathset and further applied phantom path removal and pathset splitting. For EC215 (resp., EC500), we generated 6178 (resp., 18571) pathsets before removing prefix pathsets and 1430 (resp., 2037) pathsets after removing prefix pathsets. Approximately 90% (resp., 74%) of all pathsets that remained represented singletons.
, we constructed its pathset and further applied phantom path removal and pathset splitting. For EC215 (resp., EC500), we generated 6178 (resp., 18571) pathsets before removing prefix pathsets and 1430 (resp., 2037) pathsets after removing prefix pathsets. Approximately 90% (resp., 74%) of all pathsets that remained represented singletons.
Figure 5 shows the number of pathsets of each multiplicity before (red) and after (blue) phantom path removal and splitting. Before removing phantom paths and splitting, the largest pathset contained only 4 (resp., 27) paths for the EC215 (resp., EC500) dataset. As Figure 5 illustrates, most pathsets with multiple paths turn into singletons after phantom path removal and splitting.
FIG. 5.

The number of pathsets of each size in datasets (a) EC215 and (b) EC500. The red columns count initially constructed pathsets. The blue columns count pathsets after phantom path removal and splitting.
3.3. Comparing Pathset with other genome assemblers
We compared Pathset to Velvet (Zerbino and Birney, 2008), SOAPdenovo (Li et al., 2010), SPAdes (Bankevich et al., 2012), and IDBA (Peng et al., 2010) assemblers using Plantagora (Young et al., 2011), an assembly evaluation tool (see Table 2) For dataset EC215, Pathset improved on other assemblers in N50, N75, number of misassemblies,8 and the number of captured complete genes. For dataset EC500, Pathset outperformed the other assemblers in N75 and the number of captured genes. While Velvet had a larger N50 for the EC500 dataset, Velvet's assembly was compromised by the largest number of errors (5). SOAPdenovo produced the most accurate assembly (no assembly errors) but the smallest N50 (57167 as compared to 97971 for Pathset and 105637 for Velvet).
Table 2.
Comparison of Different Assemblers
| Assemblera | No. of contigs | N50 | N75 | Covered (%)b | MAc | MMd | CGe |
|---|---|---|---|---|---|---|---|
| EC215 dataset | |||||||
| Velvet | 198 | 82776 | 42878 | 99.93 | 4 | 1.2 | 4223 |
| SOAPdenovo | 192 | 62512 | 35069 | 97.72 | 1 | 26.1 | 4141 |
| IDBA | 246 | 48825 | 25483 | 99.60 | 3 | 1.3 | 4170 |
| SPAdes | 385 | 86548 | 42441 | 99.53 | 2 | 3.7 | 4223 |
| SPAdes single read | 458 | 54858 | 30309 | 99.56 | 0 | 0.7 | 4239 |
| Pathset | 360 | 91829 | 56830 | 99.56 | 1 | 2.1 | 4249 |
| EC500 dataset | |||||||
| Velvet | 169 | 105637 | 57172 | 99.28 | 5 | 2.5 | 4095 |
| SOAPdenovo | 982 | 57167 | 31582 | 99.88 | 0 | 0.2 | 4196 |
| IDBA | 227 | 57827 | 34421 | 99.26 | 1 | 4.2 | 4158 |
| SPAdes | 215 | 95454 | 46490 | 99.80 | 4 | 1.7 | 4223 |
| SPAdes single read | 493 | 54666 | 35158 | 99.88 | 0 | 0.9 | 4215 |
| Pathset | 320 | 97971 | 58548 | 99.46 | 2 | 2.0 | 4252 |
For each column, the best assembler by each criteria is indicated in bold.
Percent of genome covered is the ratio of total number of aligned bases in the assembly to the genome size.
MA: Misassemblies are locations on an assembled contig where the left flanking sequence aligns over 1 kb away from the right flanking sequence on the reference.
MM: Mismatch (substitution) error rate per 100 kbp is measured in the correctly assembled contigs.
CG: Complete genes is the number of genes contained completely within assembly contigs (using E. coli gene annotations from www.ecogene.org).
4. Discussion
In this paper, we presented the pathset data structure for assembling genomes using mate-pair data. Instead of using mate-pair transformations on a set of mate-pairs directly, which is computationally expensive and susceptible to failure when the insert size variation is high, we first transform mate-pairs into edge-pairs. We aggregate the distance estimates from all mate-pairs mapping to each pair of edges to make distance estimates more accurate.
As compared with the traditional mate-pair transformation approach, where it is required to have a unique path between paired reads in the de Bruijn graph, our approach stores all suitable paths in a pathset data structure and later uses the paired information to remove phantom paths and further split pathsets. We also introduce the pathset graph, which allows one to construct contigs from the pathsets. Multiple libraries with different insert sizes can be utilized in the pathset data structure. The paired information in different libraries can be used to remove invalid paths in each pathset.
One should note that the pathset algorithms were designed and tested for Illumina paired-end reads with short insert sizes (typically 200 bp to 500 bp). Without further developments, the current pathset algorithms will not perform well on Illumina mate-pair (jumping) libraries with insert size in the thousands (typically 2 kb to 5 kb). These long libraries, while able to span longer repeats, possess multiple properties that make it difficult to use the current pathset algorithms: a) high variation in the insert size; b) low coverage; c) high rate of chimeric reads and read-pairs. The high variation of insert size together with its long range result in pathsets containing a very large number of paths. The low coverage violates the representative property of pathsets. The high rate of chimeric reads and read pairs introduces false paths in the graph. Adapting the pathset algorithm to jumping libraries faces many algorithmic challenges and requires further investigation.
5. Appendix: Compact representation of pathsets
5.1. Gapped pathsets
The number of paths of length d between two given edges may be exponential in d, so working with pathsets given explicitly may lead to computational difficulties in the case of large insert size, especially in highly repetitive regions. Below, we describe an implicit representation of pathsets to compactly represent such large sets. This has not yet been implemented, but we expect it will prove valuable for dealing with jumping libraries.
Also we assume below that a directed weighted graph G(V, E, ℓ) (where the weighting function ℓ()measures the length of edges) is fixed.
A gapped path in G is a tuple 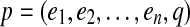 where
where 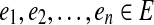 and q an (n-1)-dimensional integer vector. The edges
and q an (n-1)-dimensional integer vector. The edges 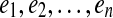 are called solid edges of p. A gapped path
are called solid edges of p. A gapped path 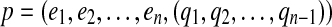 encodes a pathset consisting of all paths P that pass through the solid edges
encodes a pathset consisting of all paths P that pass through the solid edges 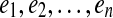 in order such that
in order such that 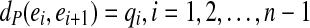 . We denote the corresponding pathset by pathset(p).
. We denote the corresponding pathset by pathset(p).
A gapped pathset is a set of gapped paths. For a gapped pathset g, we let 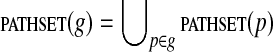 .
.
Initially we transform given edge-pairs into gapped pathsets with two solid edges. Namely, an edge-pair (e1, e2, q) is transformed into a gapped pathset 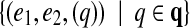 .
.
5.2. Counting paths
For 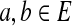 and a nonnegative integer d, define NumPaths(a, b, d) as the number of paths of length d starting at a and ending at b.
and a nonnegative integer d, define NumPaths(a, b, d) as the number of paths of length d starting at a and ending at b.
Since we will use this function extensively, for efficiency purposes, we precompute and store its values in a table of size |V| × |V|×dmax, where dmax is the maximum value of d that we use.
Our dynamic programming algorithm is based on the following formula:
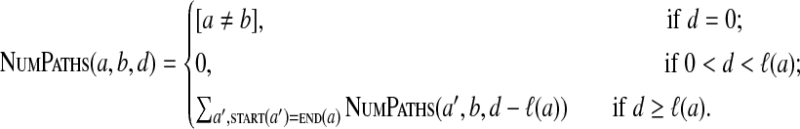 |
This formula allows one to efficiently fill up the table in O(|V|2·dmax) time.
We can easily extend NumPaths() to gapped paths:
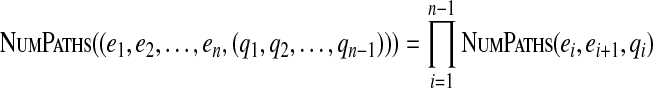 |
and further to gapped pathsets:
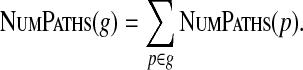 |
In particular, this can be used for detecting empty gapped pathsets (when the number of paths is zero) and gapped pathsets with a unique path (when the number of paths is one).
5.3. Identifying bridges
An edge e is called a bridge for a gapped pathset g if every path in pathset(g) passes through e.
We remark that e is a bridge for a gapped path (e1, e2, q) if and only if NumPaths(e1, e2, q) equals
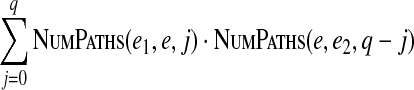 |
which can be easily tested. We further can detect that e is a bridge for a gapped path 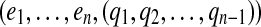 by testing whether e is a bridge for at least one of the gapped subpaths
by testing whether e is a bridge for at least one of the gapped subpaths 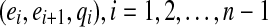 .
.
It is clear that e is a bridge for a gapped pathset g if e is a bridge for every gapped path in g.
5.4. Prefix testing
To test whether a gapped path (e1, e2, q) represents a prefix of a gapped path 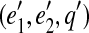 , we check that
, we check that 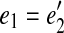 and
and 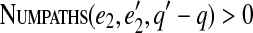 . This can be further extended to gapped paths with more than two solid edges and gapped pathsets (to be described elsewhere).
. This can be further extended to gapped paths with more than two solid edges and gapped pathsets (to be described elsewhere).
5.5. Finding essential edges
To find essential edges for gapped pathsets in a given set S, we first remark that if an edge e is (e1, e2)-constrained, then e1 represents a bridge in every gapped pathset ending with e and e2 represents a bridge in every gapped pathset starting with e. So to determine whether an edge e is essential in some gapped pathset from S, we start with searching for such bridges e1 and e2. Then for each possible pair of (e1, e2), we check whether it is d0-bounded. In this case, e represents an essential edge for every gapped pathset from S whose elements (i.e., gapped paths) contain e1, e2 in order as solid edges.
5.6. Splitting pathsets
Assume that we have a set of independent essential edges 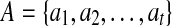 in a gapped path
in a gapped path 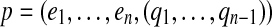 . The subset of pathset(p) that contains paths passing through a1 is encoded by a gapped pathset g constructed as follows: For every
. The subset of pathset(p) that contains paths passing through a1 is encoded by a gapped pathset g constructed as follows: For every 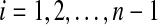 , we find all such
, we find all such 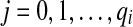 that
that 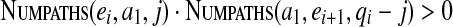 , and add a gapped path
, and add a gapped path
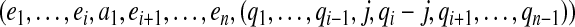 |
g. Gapped pathset representing a subset of pathset(p) consisting of pathsets containing ai
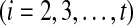 is constructed similarly.
is constructed similarly.
Construction of a gapped pathset representing pathsets not containing any edges from A is to be described elsewhere.
Footnotes
A path is non-branching if all its vertices (except possibly the first and last) have indegree and outdegree both equal to 1. The condensed de Bruijn graph may contain loops and multiple edges between the same pair of vertices.
While this assumption is unrealistic (e.g., gaps in coverage often make the de Bruijn graphs of reads disconnected), the analysis of fragment assembly in this idealized setting can be extended to real sequencing data; see the Results section.
The term “pathset” is also used in Donmez and Brudno, 2011, to describe the construction of a different graph that represents mate-pairs.
In the Results section, we demonstrate that conditions (a–c) are satisfied for the vast majority of pathsets constructed for our bacterial assembly datasets.
In Section 2.9, we describe how to adapt this approach for real datasets where the representativeness condition can be violated.
We remark that the resulting pathsets depend on ordering of elements in A (in particular, on the choice of “last” element at); different orderings may result in different splits.
A genomic edge-pair (ei, ej, dg) is present (resp., missing) in EP if there exists (resp., does not exist) an edge-pair interval 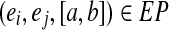 , such that a ≤ dg ≤ b.
, such that a ≤ dg ≤ b.
A misassembly is formed by concatenation of two sequences A and B that both align to the reference genome but with a gap between them larger than 1000 bases.
Acknowledgments
As members of the SPAdes assembler developers group (Bankevich et al., 2012) at the Algorithmic Biology Laboratory (Russian Academy of Science) and UCSD, the authors would like to thank the rest of the group for productive collaboration throughout the SPAdes and Pathset projects. We are especially indebted to Anton Bankevich, Mikhail Dvorkin, Alexey Gurevich, Alexey Pyshkin, Sergey Nikolenko, Sergey Nurk, Nikolay Vyahhi, and Viraj Deshpande for their support of the Pathset project, helpful comments, and thought-provoking discussions. The authors are grateful to Paul Medvedev for his insightful comments. This work was supported by grants from the National Institutes of Health, (NIH grant 3P41RR024851-02S1) and the Government of the Russian Federation (grant 11.G34.31.0018).
Disclosure Statement
No competing financial interests exist.
References
- Bankevich A. Nurk S. Antipov D., et al. SPAdes: a new genome assembler and its applications to single cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J. MacCallum I. Kleber M., et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M.J. Pevzner P.A. Short read fragment assembly of bacterial genomes. Genome Res. 2008;18:324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M.J. Brinza D. Pevzner P.A. De novo fragment assembly with short mate-paired reads: Does the read length matter? Genome Research. 2009;19:336–346. doi: 10.1101/gr.079053.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. Wallis J.W. McLellan M.D., et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhi R. Lavenier D. Localized genome assembly from reads to scaffolds: practical traversal of the paired string graph, 39–48. In: Przytycka T.M., editor; Sagot M.-F., editor. Algorithms in Bioinformatics. Springer-Verlag; Berlin Heidelberg: 2011. [Google Scholar]
- Donmez N. Brudno M. Hapsembler: An assembler for highly polymorphic genomes, 38–52. In: Donmez N., editor; Brudno M., editor. Research in Computational Molecular Biology. Springer Verlag; Berlin Heidelberg: 2011. [Google Scholar]
- Kelley D.R. Schatz M.C. Salzberg S.L. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. Zhu H. Ruan J., et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P. Pham S. Chaisson M., et al. Paired de Bruijn graphs: A novel approach for incorporating mate pair information into genome assemblers, 238–251. In: Bafina V., editor; Sahinalp S. C., editor. Research in Computational Molecular Biology. Springer, Verlag; Berlin Heidelberg: 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitra A. Valiant G. Settling the polynomial learnability of mixtures of gaussians, 93–102. Foundations of Computer Science (FOCS), 2010 51st Annual IEEE Symposium on IEEE; New York. 2010. [Google Scholar]
- Peng Y. Leung H.C.M. Yiu S.-M., et al. IDBA—A practical iterative de Bruijn graph de novo assembler, 426–440. In: Berger B., editor. Research in Computational Molecular Biology, 14th Annual International Conference, RECOMB 2010, Lisbon, Portugal, April 25–28, 2010. Proceedings.; Springer, Berlin Heidelberg. 2010. [Google Scholar]
- Pevzner P.A. Tang H. Fragment assembly with double-barreled data. Bioinformatics. 2001;17(suppl 1):S225–S233. doi: 10.1093/bioinformatics/17.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Pevzner P.A. Tang H. Waterman M.S. An Eulerian path approach to DNA fragment assembly. PNAS. 2001;98:9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T. Wong K. Jackman S.D., et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S. Barthelson R. McFarlin A., et al. Plantagora toolset. 2011. www.plantagora.org. [Sep 1;11 ]. www.plantagora.org
- Zerbino D.R. Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]