

Abstract
Chronic granulomatous disease, an inherited disorder of the NADPH oxidase in which phagocytes are defective in the generation of superoxide anion and downstream reactive oxidant species, is characterized by severe bacterial and fungal infections and excessive inflammation. Although NADPH oxidase isoforms exist in several lineages, reactive oxidant generation is greatest in neutrophils, where NADPH oxidase has been deemed vital for pathogen killing. In contrast, the function and importance of NADPH oxidase in macrophages are less clear. Therefore, we evaluated susceptibility to pulmonary aspergillosis in globally NADPH oxidase-deficient mice versus transgenic mice with monocyte/macrophage-targeted NADPH oxidase activity. We found that the lethal inoculum was more than 100-fold greater in transgenic versus globally NADPH oxidase-deficient mice. Consistent with these in vivo results, NADPH oxidase in mouse alveolar macrophages limited germination of phagocytosed Aspergillus fumigatus spores. Finally, globally NADPH oxidase-deficient mice developed exuberant neutrophilic lung inflammation and pro-inflammatory cytokine responses to zymosan, a fungal cell wall-derived product composed principally of particulate beta-glucans, whereas inflammation in transgenic and wildtype mice was mild and transient. Together, our studies identify a central role for monocyte/macrophage NADPH oxidase in controlling fungal infection and in limiting acute lung inflammation.
Keywords: Chronic Granulomatous Disease, NADPH oxidase, macrophage, Aspergillus
Introduction
The lung is an interface where host cells are regularly exposed to microbes and microbial products. Aspergillus-associated diseases encompass both invasive and allergic diseases in which pathogenesis is regulated by host factors (1). Alveolar macrophages are the first-line phagocytic cells that encounter inhaled fungi. Macrophages and other immune cells sense Aspergillus motifs by pathogen recognition receptors that include specific toll like receptors and dectin-1 (2), and initiate downstream inflammatory responses. The phagocyte NADPH oxidase (NOX2) generates reactive oxidant intermediates (ROIs) in response to specific microbial products, and is critical for host defense. We sought to evaluate the specific role of NADPH oxidase in macrophages in antifungal host defense and in regulating downstream inflammatory responses.
Dectin-1 is a receptor and immunomodulator of beta-glucans, which are cell wall constituents of fungi and plants. Beta-glucans in conidia (spores) of Aspergillus fumigatus are masked by cell wall surface proteins that blunt immune activation (3). Following transition to the germling stage, beta-glucans become exposed, and induce inflammatory responses in macrophages that are coordinated by toll like receptors and dectin-1 (4–6). Dectin-1 in macrophages is activated by particulate (but not soluble) beta-glucans, which, in nature, would occur following direct contact with microbes (7). In contrast, neutrophil NADPH oxidase is activated by Aspergillus hyphae largely independent of dectin-1 (8). Ligation of dectin-1 can stimulate NADPH oxidase activity and pro-inflammatory cytokines and chemokines (7, 9–11). The ability of innate immune cells to recognize fungal products displayed at different stages of fungal growth is likely important in calibrating the immune response to control the growth of inhaled fungi while averting excessive inflammation.
The important role of NADPH oxidase in host defense is demonstrated by chronic granulomatous disease (CGD), an inherited disorder of the NADPH oxidase characterized by severe bacterial and filamentous fungal infections. The phagocyte NADPH oxidase is the principal source of ROI generation in activated neutrophils and macrophages. Among CGD patients, residual neutrophil NADPH oxidase activity correlates with less severe illness and improved survival (12). Infections by Aspergillus species and other filamentous fungi are major causes of mortality in CGD (13, 14). CGD patients are prone to developing inflammatory complications, such as inflammatory bowel disease and obstructive granulomatous inflammation of the genitourinary tract (15). Engineered NADPH oxidase-deficient mice have a hyper-inflammatory phenotype to sterile products, including heat-killed Aspergillus hyphae (16, 17) and fungal cell wall-derived products (18, 19), emphasizing a key role of NADPH oxidase in limiting inflammation in addition to its antimicrobial activity.
NADPH oxidase activation requires translocation of cytosolic phox (phagocyte oxidase) proteins (p47phox, p67phox, p40phox) and rac to the membrane-bound flavocytochrome consisting of the gp91phox and p22phox heterodimer. NADPH oxidase activation results in conversion of oxygen to superoxide anion and generation of downstream reactive oxidant metabolites with antimicrobial activity, such as hydrogen peroxide, hydroxyl anion, and hypohalous acid. In neutrophils, NADPH oxidase activation is linked to activation of primary granule antimicrobial proteases and generation of neutrophil extracellular traps (20, 21). NADPH oxidase-generated ROIs and activation of neutrophil proteases have distinct roles in host defense against bacterial and fungal pathogens (22).
Although NADPH oxidase is critical for neutrophil-mediated host defense, the importance of NADPH oxidase in macrophages is unclear. The strongest evidence for the role of macrophage NADPH oxidase in host defense is from the finding that mutations in gp91phox that selectively affect macrophages lead to increased susceptibility to mycobacterial diseases (23). Prior studies have shown that alveolar macrophages ingest and kill Aspergillus spores, whereas neutrophils principally target the hyphal stage (24). However, there have been conflicting results as to the role of NADPH oxidase in macrophages in controlling the growth of A. fumigatus spores (25, 26).
Our major goal was to delineate the specific role of NADPH oxidase in macrophages in mediating host defense against A. fumigatus and in regulating the inflammatory response to fungal components. Transgenic mice with CD68 promoter-driven gene expression have been widely used to study monocyte/macrophage lineage-restricted production of targeted proteins (27). To address the specific role of macrophage NADPH oxidase in mediating antifungal host defense and inflammation, we used mice with monocyte/macrophage-targeted NADPH oxidase activity that have a naturally acquired disabling mutation of Ncf1 (which encodes the p47phox protein) and harbor a transgene containing wildtype Ncf1 under the control of a human CD68 promoter (28). We found that monocyte/macrophage-targeted NADPH oxidase conferred resistance to pulmonary aspergillosis. Consistent with these results, NADPH oxidase in isolated alveolar macrophages was required to limit the growth of phagocytosed spores. In addition, globally NADPH oxidase-deficient mice developed exuberant neutrophilic lung inflammation and pro-inflammatory cytokine responses to zymosan (fungal cell wall-derived particulate beta-glucans) whereas inflammation in transgenic and wildtype mice was minimal. These results support a previously unappreciated role for macrophage NADPH oxidase both in mediating antifungal host defense and in controlling acute neutrophilic inflammation.
Methods
Mice
Mice with a targeted disruption of either the p47phox gene or gp91phox gene have a defective NADPH oxidase, rendering phagocytes incapable of generating measurable superoxide. NADPH oxidase-deficient mice have increased susceptibility to pathogens that afflict CGD patients, including Aspergillus species (29). We used p47phox−/− mice (30) backcrossed to N14 in C57BL/6 and age-and sex-matched wildtype C57BL/6 controls.
Mice with a naturally occurring Ncf1 (which encodes the p47phox protein) mutation (the mutation is Ncf1m1J/m1J and is here abbreviated as Ncf1*/*) have a global NADPH oxidase deficiency (28, 31). The mutation was originally in a B6db/db substrain and was extensively backcrossed to a C57Bl/10.Q strain and ascertained to differ by only one mutation as checked with 10k SNP typing and selected typing around the Ncf1 gene (31). This strain is designated BQ.Ncf1*/*. The p47phox protein variant expressed in mice Ncf1*/* mice results in defective assembly of the NADPH oxidase complex (32). To generate transgenic mice with monocyte/macrophage-restricted NADPH oxidase activity, the wildtype Ncf1 coding sequence was ligated downstream of the promoter and splice site in a vector containing the human CD68 promoter as previously described (28). This construct containing Ncf1 was named MN. The transgene was injected into the BQ.Ncf1*/* strain and maintained by backcrossing. Screening for Ncf1-mutant and the MN transgene was performed by PCR, as described (28). Mice with mutant Ncf1 without the transgene (Ncf1*/*MN−) are globally NADPH oxidase-deficient, whereas mutant mice that harbor the transgene (Ncf1*/*MN+) have monocyte/macrophage-targeted expression of wildtype Ncf1 and NADPH oxidase activity (28, 33). Wildtype B10.Q mice are used as controls.
Mice were bred and maintained under specific pathogen free conditions at the animal care facility at Roswell Park Cancer Institute, Buffalo, NY. All procedures performed on animals in this study were approved by the Animal Care and Use Committee Roswell Park Cancer Institute, and complied with all state, federal, and NIH regulations.
Administration of A. fumigatus
A clinical isolate of A. fumigatus was used in all experiments. Conidial suspensions were prepared as previously described (29), and diluted to desired concentrations. Conidia were administered intratracheally as we described (29) or by oropharyngeal instillation. We found that oropharyngeal instillation leads to similar degrees of fungal pneumonia and mortality in p47phox−/− mice compared to intratracheal administration, but avoids surgery. Oropharyngeal instillation therefore became our preferred approach for intrapulmonary instillations. Mice were anesthetized by isofluorane inhalation using an approved chamber. Following anesthesia, mice were suspended by their upper incisors from a suture thread on a 90° incline board. The tongue was gently extended, and a liquid volume (maximum 50 μl) was delivered into the distal part of the oropharynx. With the tongue extended, the animal was unable to swallow, and the liquid volume was aspirated into the lower respiratory tract. Just prior to liquid delivery, the chest was gently compressed and then released just after deposition of liquid into the oropharynx to enhance aspiration of the liquid into the lung. Mice recovered within 5 minutes of the procedure, and were observed until they resumed normal activity.
Cyclophosphamide administration
The alkylating agent, cyclophosphamide, was used to induce leukopenia. Cyclophosphamide (i.p. 250 mg/kg of body weight) was administered on days −3 and +1 in relation to A. fumigatus challenge. Peripheral blood was collected in blood diluent (Delta Scientific; Ivyland, PA) in K2 EDTA coated tubes (Becton Dickinson, Franklin Lakes, NJ) and white cell counts and differentials were determined by analysis on the Advia 120, Multi-Species Software v. 3.1.8-MS (Bayer HealthCare, Tarrytown, NY). This regimen leads to a total leukocyte count of < 500/ul and absolute neutrophil count of < 100/ul from day 0 until at least day 5 in relation to fungal challenge in wildtype and p47phox−/− mice.
Survival
Following A. fumigatus challenge, mice were monitored twice daily for death and morbidity until at least day 25. Mice with pre-specified criteria for distress that included inability to feed or drink, labored breathing, or general moribund appearance were euthanized by CO2 asphyxiation. The primary endpoint was time to euthanasia.
Broncholalveolar fluid collection and cytology
After sacrifice, bronchoalveolar lavage fluid (BALF) collection was performed as previously described (19). The trachea was cannulated with a 22-gauge i.v. catheter. Using a tuberculin syringe, 1 mL PBS was injected and withdrawn from the lung and again fresh 1 mL PBS was injected and withdrawn from the lung and both were pooled. Cells were pelleted by centrifugation at 1,500 g for 3 min. Supernatants were aliquoted and stored at −80°C. In the cell pellet, the red blood cells (RBCs) were removed by 1 washin ACK lysis buffer, and the cells were suspended in 1 ml of PBS. The total number of leukocytes/ml was counted using a hemocytometer. Cells were then cytocentrifuged onto clean glass slides and stained with the Hema 3 stain set (Fisher Scientific, Pittsburgh, PA, USA), and cell differential counts were assessed blinded to genotype.
Histopathology
After sacrifice and bronchoalveolar lavage, mouse lungs were infused with 10% neutral buffered formalin via the trachea. Paraffin-embedded blocks were prepared and sections were stained with Hemotoxylin and Eosin. In mice administered A. fumigatus, Grocott-Gomori methenamine-silver stain was used to visualize fungi. The percentage of lung involved by granulomatous or consolidative inflammation was scored in each mouse as follows: 0%, 5%, 10%, and then by 10% increments (e.g., 20%, 30%, 40%, etc.). The predominant inflammatory cell type was scored as neutrophilic, monocytic, or lymphocytic. In mice administered A. fumigatus, additional endpoints included necrosis (coagulative and inflammatory), hemorrhage, invasive parenchymal aspergillosis, and angioinvasive aspergillosis, as previously described (34). Coagulative necrosis was defined as acellular necrosis or necrosis with scant inflammation, whereas, inflammatory necrosis was defined as necrosis occurring within inflammatory lesions (34). All slides were analyzed by one of us (BHS) blinded to genotype and treatment.
Analysis of antifungal activity of isolated alveolar macrophages
Alveolar macrophages from unstimulated wildtype and p47phox−/− mice were collected by BAL. The harvested cells were >95% macrophages, based on cytology. Cells were washed, resuspended in RPMI 1640 with 2mM L-glutamine and 10% FBS, and 5 × 105 cells were seeded into each chamber of an 8-chamber glass slide (Thermo Scientific). Macrophages were allowed to adhere overnight at 37°C with 5% CO2, and then seeded with 1 × 106 conidia of an A. fumigatus strain expressing green fluorescent protein (GFP) (35) (kindly provided by Margo Moore, PhD, Simon Fraser University, British Columbia, Canada). After 3 hours, non-phagocytosed conidia were removed and fresh media was added to each well. Simultaneous imaging of wildtype and p47phox−/− macrophages was performed at 30-minute intervals using the Leica AF6000LX live cell imaging platform. ImagePro 6.2 (Media Cybernetics, Silver Spring, MD) was applied to the photographic images (Figure 2) for contrast enhancement and for adjustments of the luminance and color channel settings.
Figure 2. NADPH oxidase in alveolar macrophages limits growth of phagocytosed A. fumigatus spores.
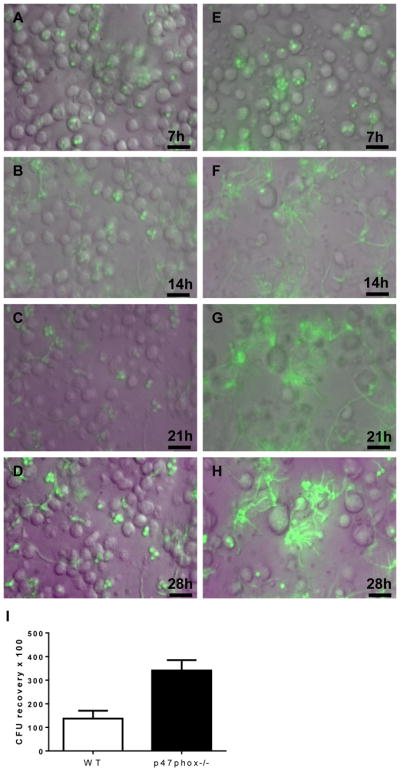
Alveolar macrophages (5 × 105/well) from wildtype (A–D) mice and p47phox−/− mice (E–H) were seeded in parallel wells with GFP-producing A. fumigatus (1 × 106 spores/well). At 3h, non-phagocytosed conidia were removed by gentle washing, and conidial germination was analyzed by simultaneous time lapse imaging at 30-min intervals. The full video clips are shown in Video/Movie 1 and 2. Time-lapse photos at 7-h intervals show that wildtype macrophages, to an extent, limit growth of phagocytosed spores. In contrast, fungal growth led to the destruction of p47phox−/− macrophages with phagocytosed fungi. Note the sparse numbers of p47phox−/− macrophages present at 14h, most of which have ruptured. The green hue that was evident by 14h in wells with p47phox−/− macrophages is probably related to the intensity of the GFP signal within Aspergillus hyphae, and likely spill-over to adjacent areas. Bar, 20 μM. I) NADPH oxidase increases the conidiocidal activity of alveolar macrophages. Alveolar macrophages from WT and p47phox−/− mice (n=6 per genotype) were harvested by BAL and seeded with A. fumigatus spores. Non-phagocytosed spores were removed by washing, followed by lysis of macrophages and assessment of viable fungal recovery by quantitative cultures. CFU, colony forming units. Mann-Whitney, p =0.004.
The conidiocidal activity of wildtype and p47phox−/− alveolar macrophages was assessed, as previously described with modifications (36). Briefly, 2.5 × 105 alveolar macrophages were seeded in 24-well plates (Costar) in 2 ml of complete media (CM; RPMI 1640 with 2 mM L-glutamine, and 10% FBS) and incubated at 37°C with 5% CO2 overnight to allow macrophage to adhere. The following morning, 5 × 105 A. fumigatus conidia were added and the mixtures were incubated for 3h. Wells were then gently washed, and dilutions of supernatants were plated for non-phagocytosed conidia. At 6h, sterile dH2O was added to lyse macrophages, cells were removed with vigorous pipetting, and dilutions were plated on Corn Meal agar plates (Becton, Dickinson and Co). All plates were incubated at 37°C for 24h. Colonies were counted for both the non-phagocytosed conidia collected after 3h, and the number of phagocytosed conidia recovered at 6h.
Confocal laser-scanning microscopy
We evaluate the ability of isolated alveolar macrophages from WT, Ncf1*/*MN+ and Ncf1*/*MN− mice to control growth of phagocytosed Aspergillus spores by confocal microscopy. Mice (n = 3 per genotype) were administered IV 20 μM PKH26 (PKH26 Red Fluorescent Cell Linker Kit, Sigma, St. Louis, MO), which stably accumulates in the membranes of alveolar macrophages, but not in peripheral monocytes or marrow precursors (37–39). Ten days later, alveolar macrophages were harvested by BAL, and cells from the same genotype were pooled and processed as described above for video imaging studies. 8.8 × 105 macrophages were seeded on 22×22 mm cover slips in 6-well plates. Macrophages were allowed to adhere overnight at 37°C with 5% CO2, and then seeded with 8.8 × 106 conidia of GFP-expressing A. fumigatus. Cells were fixed in 1% formaldehyde at 3, 7, and 14h after addition of conidia. After fixation, cover slips were removed, gently washed in PBS, and mounted onto glass slides using Vectashield mounting media with DAPI (Vector Labs, Burlingame, CA). Confocal images were acquired using a Leica TCS SP2 system with a laser base point scanner mounted on DMIRE2 fluorescence microscope. For all images a 63X oil immersion lens was used with a pinhole of 1 airy. Z stacks were acquired with an electronic zoom factor of 2.5 in bright field, DAPI (excitation 405nm; emission range collected 410–499nm), FITC (excitation 488nm; emission range collected 500–537nm), and PKH26 (excitation 543nm; emission range collected 559–613nm). The thickness for a Z stack was determined using DAPI and bright field images. Z stacks were in the range of 14 – 24 μm with individual slices approximately 1 μm thick. For quantification of macrophages, single scans of 1X zoom were performed on at least 6 fields per cover slip. Intact macrophages were identified by confocal microcopy based on PKH26 and DAPI staining of the membrane and nucleus, respectively. Conidia were identified based on FITC staining and bright field image.
Oropharyngeal zymosan administration
Zymosan (Sigma) was diluted to a concentration of 2.5 mg/ml in saline, sonicated until the particles were suspended homogenously and frozen at −20°C. Prior to use, the zymosan stock was diluted to 0.4 mg/mL and autoclaved to ensure sterility. Zymosan (50 ul) was administered by oropharyngeal instillation using the same technique as A. fumigatus administration.
Statistics
Graph Pad Prism 4.0 software was used for statistical analysis and to display graphical data. Kaplan-Meier curves were used to display time to euthanasia and analyzed using the log-rank method. Comparisons between two groups were made using the non-parametric Mann-Whitney method or student t-test. ANOVA with Tukey post-test was used to compare the 3 genotypes when normal distribution was confirmed, and Kruskal-Wallis with Dunn’s multiple comparison test was used when normal distribution was not demonstrated. A p-value of <0.05 was considered statistically significant.
Results
NADPH oxidase-deficient mice are more susceptible to pulmonary aspergillosis than leukopenic wildtype mice
We compared susceptibility to Aspergillus challenge in two well-defined immunocompromised mouse models: wildtype mice rendered leukopenic by cyclophosphamide administration (modeling antineoplastic chemotherapy-induced neutropenia) and non-leukopenic p47phox−/− mice. In prior studies, we observed that immune intact wildtype mice rapidly clear respiratory Aspergillus challenge, and were therefore not used in the current experiments (22). The rationale for cyclophosphamide is that it enables evaluation of antifungal host defense in the absence of recruited inflammatory cells. While repeated cyclophosphamide administration gradually reduces the number of alveolar macrophages in mice (40), the most immediate effect is depletion of circulating neutrophils. Cyclophosphamide treatment led to a total leukocyte count of < 500/ul and absolute neutrophil count of < 100/μl from day 0 until at least day 5 in relation to fungal challenge in wildtype and p47phox−/− mice.
Cyclophosphamide-treated wildtype leukopenic mice were significantly more resistant to Aspergillus challenge than non-leukopenic p47phox−/− mice. Using a range of Aspergillus inocula (1.25 × 103 to 2.5 × 104 spores per mouse), the LD50 of A. fumigatus was more than 10-fold greater in wildtype leukopenic mice (greater than 2.5 × 104 spores/mouse) compared to non-leukopenic p47phox−/− mice (2.5 × 103 spores/mouse) (Figure 1). In separate experiments, mice were challenged with an inoculum of 1.25 × 104 spores/mouse, an inoculum known to cause invasive aspergillosis in p47phox−/− mice, and lungs were harvested on day 5 for histological analysis. Non-leukopenic p47phox−/− mice developed discrete regions of consolidation principally composed of dense neutrophilic infiltrates surrounding invasive hyphae without evidence of hyphal angioinvasion. In contrast, lungs of cyclophosphamide-treated wildtype mice challenged with the same Aspergillus inoculum showed no evidence of fungal disease (Figure 1). Taken together, both the survival data and lung histological analysis support the notion that NADPH oxidase in lung macrophages can confer protection against Aspergillus challenge in the absence of detectable circulating neutrophils.
Figure 1. Non-leukopenic p47phox−/− mice are significantly more susceptible to Aspergillus challenge than leukopenic wildtype mice.

Non-leukopenic p47phox−/− (CGD) mice and cyclophosphamide-treated wildtype mice were administered intratracheally a range of Aspergillus inocula (1.25 × 103 to 2.5 × 104 spores per mouse), and monitored for survival. A) Kaplan-Meier survival curves show that the LD50 of A. fumigatus in non-leukopenic p47phox−/− mice was 2.5 × 103 spores/mouse (n=27, pooled from 3 independent experiments). In contrast, wildtype (WT) mice (n=9) rendered leukopenic with cyclophosphamide (Cyp) had 100% survival after challenge with a 10-fold higher inoculum (2.5 × 104 spores/mouse) than the LD50 in non-leukopenic CGD mice. Log-rank (Mantel-Cox) test, p =0.0002. B and C) In separate experiments, non-leukopenic p47phox−/− mice and Cyp-treated WT mice were administered A. fumigatus (1.25 × 104 spores/mouse), and sacrificed on day 5 (n = 5 mice per group). B) p47phox−/− mice developed multiple foci of predominantly neutrophilic consolidation (H&E, 40x), surrounding invasive hyphae without hyphal angioinvasion (Inset, GMS, 400x; arrow shows invasive hyphae). C) In contrast, Cyp-treated leukopenic wildtype mice that received the same inoculum had normal lung histology (H&E, 40x).
In separate experiments, we evaluated the effect of cyclophosphamide treatment in susceptibility to aspergillosis in p47phox−/− mice. Pulmonary lesions in cyclophosphamide-treated p47phox−/− mice were characterized by coagulative necrosis and extensive hyphal vascular invasion, while angioinvasion was not observed in non-leukopenic p47phox−/− mice (Supplemental Figure 1). These results suggest that recruited inflammatory cells can prevent hyphal angioinvasion through NADPH oxidase-independent pathways (Supplemental Figure 1).
Macrophage NADPH oxidase limits growth of phagocytosed conidia
To investigate the role of macrophage NADPH oxidase in defense against Aspergillus, we evaluate the ability of alveolar macrophages from wildtype and p47phox−/− mice to restrain conidial germination. Alveolar macrophages constituted >95% of the harvested bronchoalveolar lavage (BAL) fluid cells based on cytology. Macrophages (5 × 105/well) from wildtype and p47phox−/− mice were seeded in parallel wells with GFP-producing A. fumigatus (1 × 106 spores/well). At 3h, non-phagocytosed conidia were removed by gentle washing to enable unobstructed visualization of phagocytosed conidia. Conidial germination was then analyzed by simultaneous time lapse imaging at 30-min intervals using GFP fluorescence to visualize fungi. We observed variable ability of wildtype macrophages to contain conidial growth. Even after 28h following fungal seeding, we observed several intact macrophages with phagocytosed fungi. In contrast, fungal growth within p47phox−/− macrophages led to destruction of virtually all of the macrophages (Figure 2 and Video/Movie 1 and 2).
In separate experiments, we found that NADPH oxidase increases the conidiocidal activity of isolated alveolar macrophages. Alveolar macrophages from wildtype and p47phox−/− mice (n = 6 per genotype) were harvested by BAL and seeded with A. fumigatus spores. Non-phagocytosed spores were removed by washing at 3h, followed by lysis of macrophages at 6h, and plating on agar. While the recovery of non-phagocytosed spores was similar between genotypes (not shown), killing of phagocytosed conidia was increased in wildtype compared with p47phox−/− macrophages (Figure 2). Together, our observations with isolated macrophages support a role for NADPH oxidase in restraining the growth of phagocytosed conidia and are consistent with the notion of NADPH oxidase in macrophages conferring protection in leukopenic mice.
Monocyte/macrophage-targeted NADPH oxidase is protective in pulmonary aspergillosis
To more definitively evaluate the contribution of macrophage NADPH oxidase to antifungal host defense in vivo, we used mice in the B10.Q lineage with a naturally acquired mutation in the Ncf1 gene (which encodes the p47phox protein) and transgenic mice (MN+) that harbor wildtype Ncf1 under the control of human CD68 promoter such that NADPH oxidase is functional in the monocyte/macrophage lineage (28). We previously found that the transgene expression is restricted to monocyte/macrophages and not detected in other cell types including neutrophils (28, 41). Whereas the PMA-stimulated respiratory burst in bone marrow-purified wildtype B10.Q neutrophils was robust, ROI generation in Ncf1*/* MN+ neutrophils was minimal (<1% of wildtype neutrophils), but distinguishable from ROI generation in Ncf1*/*MN− neutrophils (33). There was no detectable Ncf1 expression in Ncf1*/* MN+ neutrophils (33). However, expression of Ncf1 protein and NADPH oxidase activity occurred in DCs from Ncf1*/* MN+ mice (42); since myeloid DCs and monocytes have a shared lineage, it is not unexpected that CD68 promoter activity would be present in both cells (43).
Wildtype, Ncf1*/*MN+, and Ncf1*/*MN− mice were administered A. fumigatus by oropharyngeal instillation and monitored for morbidity requiring euthanasia. Based on prior studies, an inoculum of 1.25 × 104 spores/mouse results in lethal pulmonary aspergillosis in p47phox−/− mice within 7 to 15 days of fungal challenge (29). We therefore used this fungal inoculum in globally NADPH oxidase-deficient mice, and challenged transgenic mice and wildtype mice with fungal inocula ranging between 1.25 × 104 to 1.25 × 107 spores/mouse. All transgene-negative mice (Ncf1*/*MN−) administered 1.25 × 104 spores/mouse died by 13 days (Figure 3A). In contrast, there was uniform survival in transgenic mice (Ncf1*/*MN+) administered up to a 100-fold greater inoculum (1.25 × 106 spores/mouse). An inoculum of 1.25 × 107 spores/mouse was lethal in both Ncf1*/*MN+ mice and wildtype B10.Q mice (data not shown). These results point to monocyte/macrophage-targeted NADPH oxidase being protective in pulmonary aspergillosis.
Figure 3. Transgenic mice with monocyte/macrophage-targeted NADPH oxidase are protected from lethal Aspergillus challenge.

A) Wildtype (B10.Q), Ncf1*/*MN+, and Ncf1*/*MN− were administered a range of A. fumigatus inocula by oropharyngeal instillation (1.25 × 104 to 1.25 × 107 spores/mouse). All globally NADPH oxidase-deficient mice (Ncf1*/*MN−; n = 4) administered 1.25 × 104 spores/mouse died by 13 days, whereas there was uniform survival in transgenic mice (Ncf1*/*MN+; n = 5) administered a 100-fold greater inoculum (1.25 × 106 spores/mouse); Log-rank, p < 0.0001. All wildtype mice administered 1.25 × 106 spores/mouse also survived, while an inoculum of 1.25 × 107 spores/mouse was fatal in both wildtype and Ncf1*/*MN+ mice (data not shown). B–D) In separate experiments, wildtype, Ncf1*/*MN+ and Ncf1*/*MN− mice (n = 5 per genotype) were administered A. fumigatus (1.25 × 104 spores/mouse), sacrificed on day 5, and lungs and BAL exudate cells were analyzed. Lungs of wildtype (B) and Ncf1*/*MN+ (C) mice were normal (H&E, 40x), with GMS showing no signs of fungal disease (not shown). D) In contrast, all similarly treated Ncf1*/*MN− mice developed multifocal consolidative lesions (H&E, 40x) and invasive hyphae (inset, GMS 400x; arrow showing hyphae). E–G) Ncf1*/*MN− mice had significantly greater airway neutrophilic leukocytosis and lung consolidative inflammation compared to wildtype and Ncf1*/*MN+ mice at day 5 after A. fumigatus (1.25 × 104 spores/mouse) administration. Although BAL neutrophil (PMN) recovery was greater in wildtype versus Ncf1*/*MN+ mice, this difference was not statistically significant with post-test analysis. H–J) Confocal microscopy of wildtype (H), Ncf1*/*MN+ (I), and Ncf1*/*MN− (J) alveolar macrophages at 14h after seeding with GFP+ A. fumigatus conidia. Intact macrophages are identified based on PKH26 (red) and DAPI (blue) staining of membranes and nuclei, respectively. Solid arrows show intact macrophages with at least 1 phagocytosed spore and arrow heads show hyphae. The GFP signal in hyphae is variable based on the plane of focus of the Z-stacked images. Intact cells with phagocytosed conidia were observed with wildtype and Ncf1*/*MN+ macrophages, but not with Ncf1*/*MN− macrophages. Note ruptured Ncf1*/*MN− macrophage (dashed arrow) and adjacent hypha (arrow head). Bar, 19 μM. K) With serial imaging, macrophages with and without phagocytosed conidia were quantitated. At 3 and 7h, there was no difference among the genotypes, indicating that NADPH oxidase doesn’t influence phagocytosis efficiency. L) At 14h, the number of intact macrophages with and without phagocytosed conidia was similar between wildtype and Ncf1*/*MN+ macrophages. In contrast, Ncf1*/*MN− macrophages were depleted, and we did not observe any intact Ncf1*/*MN− macrophages with phagocytosed spores (ND, not detected). ANOVA with Tukey post-test was used to compare the 3 genotypes when data were normally distributed, and Kruskal-Wallis with Dunn’s multiple comparison test was used when normal distribution was not demonstrated. *, p< 0.05; **, p < 0.01; ***, p < 0.001.
Since survival can be influenced by both fungal burden and inflammation, we compared airway inflammation and lung histology in wildtype, Ncf1*/*MN− and Ncf1*/*MN+ mice following Aspergillus challenge (Figure 3B–G). Mice were administered A. fumigatus (1.25 × 104 spores/mouse) and sacrificed on day 5, a time that preceded morbidity in Ncf1*/*MN− mice. Ncf1*/*MN− mice developed exuberant pulmonary consolidative lesions comprised of neutrophilic and monocytic cells. Hyphae invading the lung parenchyma, but not blood vessels, were observed. In contrast, lung inflammation in similarly treated wildtype and Ncf1*/*MN+ mice was close to nil, and fungi were not observed. Ncf1*/*MN− had significantly greater neutrophilic airway inflammation compared to wildtype and Ncf1*/*MN+ mice. Thus, consistent with survival data, NADPH oxidase in the monocyte/macrophage lineage was protective against pulmonary aspergillosis in the context of NADPH oxidase deficiency in neutrophils.
We next evaluated whether isolated alveolar macrophages from Ncf1*/*MN+ phenocopy wildtype macrophages in limiting growth of phagocytosed conidia. While video imaging described in Figure 2 has the advantage of sequential imaging of the same primary cell culture, confocal microscopy enables more detailed imaging of cells. Wildtype, Ncf1*/*MN+ and Ncf1*/*MN− mice (n = 3 per genotype) were administered IV PKH26, which stably accumulates in the membranes of alveolar macrophages (37–39). Alveolar macrophages were harvested by BAL and seeded with conidia of GFP-expressing A. fumigatus. Cells were fixed at 3, 7, and 14h after addition of conidia. The 3h and 7h time points precede transition to the hyphal stage, and therefore enable comparison of phagocytosis of conidia among the genotypes. In contrast, the 14h time point enables comparison of the genotypes with regard to inhibiting transition of phagocytosed conidia to the tissue-invasive hyphal stage. Intact macrophages were identified by confocal microcopy based on PKH26 and DAPI staining of the membrane and nucleus, respectively, and phagocytosed conidia were identified based on GFP expression. We found that the number of total macrophages and macrophages with ≥1 phagocytosed conidia was similar among the three genotypes at 3h and 7h, reflecting similar phagocytosis efficiency. At 14h, the number of intact macrophages with and without phagocytosed conidia was similar between the wildtype and Ncf1*/*MN+ genotypes. In contrast, we did not observe any intact Ncf1*/*MN− macrophage with a phagocytosed conidium at 14h; rather, remnants of ruptured Ncf1*/*MN− macrophages were observed in association with hyphae (Figure 3H–L). Taken together, our in vivo and in vitro studies demonstrate that monocyte/macrophage NADPH oxidase defends against pulmonary Aspergillus infection.
Monocyte/macrophage-targeted NADPH oxidase can limit zymosan-induced lung inflammation
We next asked whether macrophage NADPH oxidase regulates inflammation independent of its antimicrobial activity. We used oropharyngeal instillation of zymosan as a sterile model of lung inflammation. Zymosan is a fungal cell wall-derived product principally composed of beta-glucan that activates the phagocyte NADPH oxidase through activation of dectin-1 (9). In prior studies using bone marrow chimeras, we showed that NADPH oxidase in the hematopoietic component restrained zymosan-induced lung inflammation (19). We therefore used Ncf1*/*MN+ mice to evaluate whether monocyte/macrophage NADPH oxidase regulates neutrophilic inflammation in the lungs.
Wildtype (B10.Q), Ncf1*/*MN+, and Ncf1*/*MN− mice were administered oropharyngeal zymosan and BALF leukocytosis and lung histology were evaluated on days 1 and 5. On day 1 after zymosan administration, BALF neutrophilic leukocytosis was significantly greater in Ncf1*/*MN− mice compared to similarly treated Ncf1*/*MN+ and wildtype mice (Figure 4A). On day 5, BALF neutrophilic leukocytosis persisted in Ncf1*/*MN− mice, whereas inflammatory cell recovery from BALF from Ncf1*/*MN+ and wildtype mice was close to unstimulated levels, and composed principally of monocytes (Figure 4B). In addition, zymosan resulted in extensive lung consolidation composed of neutrophilic and monocytic cells in Ncf1*/*MN− mice, whereas lung inflammation in similarly treated Ncf1*/*MN+ mice and wildtype mice was close to nil (Figure 4C–E). BALF from zymosan-stimulated Ncf1*/*MN− mice had significantly increased pro-inflammatory cytokine and chemokine levels compared to BALF from similarly treated Ncf1*/*MN+ and wildtype mice (Figure 5). Taken together, these results point to NADPH oxidase in the monocyte/macrophage lineage limiting inflammation independent of its antimicrobial function.
Figure 4. Transgenic mice with monocyte/macrophage-targeted NADPH oxidase limit zymosan-induced lung inflammation.

Wildtype (B10.Q), Ncf1*/*MN+, and Ncf1*/*MN− mice were administered zymosan by oropharyngeal instillation and sacrificed on day 1 or day 5. A) BALF neutrophilic leukocytosis was increased on day 1 after zymosan in Ncf1*/*MN− mice compared to similarly treated Ncf1*/*MN+ and WT mice. B) On day 5, persistent neutrophilic airway inflammation occurred in Ncf1*/*MN− mice, whereas BALF leukocyte recovery returned to unstimulated levels in Ncf1*/*MN+ and WT mice. C and D) Lung inflammation was significantly increased in Ncf1*/*MN− mice as compared to Ncf1*/*MN+ mice and WT mice on day 1 (C) and on day 5 (D) after zymosan administration. E–G) Representative lung sections of WT (E) and Ncf1*/*MN+ (F) mice on day 5 after zymosan administration showing minimal lung inflammation, whereas extensive lung consolidation occurred in Ncf1*/*MN− mice (G) (H&E, 40x). n = 5 mice per genotype per time point. *, p< 0.05; **, p < 0.01; ***, p < 0.001.
Figure 5. Monocyte/macrophage-targeted NADPH oxidase limits zymosan-induced cytokine and chemokine levels in BALF.
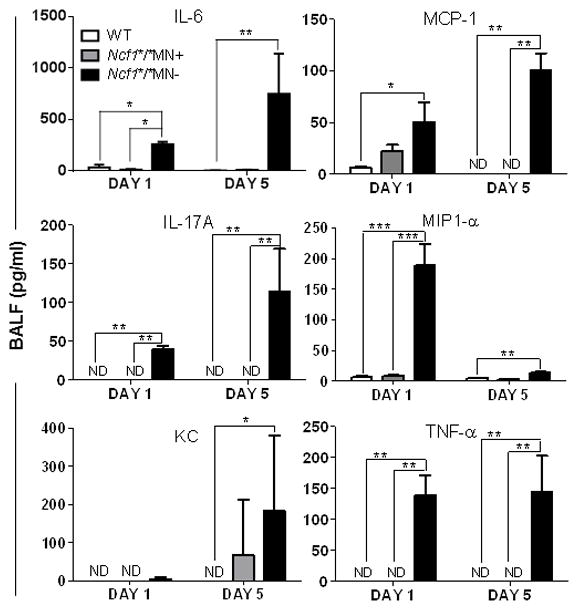
Wildtype (B10.Q), Ncf1*/*MN+, and Ncf1*/*MN− mice were administered zymosan by oropharyngeal instillation, and BALF cytokines and chemokines on days 1 and 5 were measured by ELISA. N = 5 mice per genotype per time point. Groups were compared by Kruskal-Wallis with Dunn’s multiple comparison test. Non-detectable (ND) values were scored as 0 pg/ml for statistical comparisons. * p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Our results show an important role of monocyte/macrophage NADPH oxidase in mediating antifungal host defense and in restraining acute neutrophilic inflammation. In the context of global NADPH oxidase deficiency, transgenic monocyte/macrophage-targeted NADPH oxidase was protective against pulmonary aspergillosis. The lethal inoculum in Ncf1*/*MN+ mice was more than 100-fold greater compared to globally NADPH oxidase-deficient Ncf1*/*MN− mice. Transgenic mice developed no signs of histological fungal disease following a fungal inoculum that led to lethal fungal pneumonia in Ncf1*/*MN− mice. Consistent with the in vivo results, NADPH oxidase in isolated alveolar macrophages limited conidial growth. In addition, monocyte/macrophage-targeted NADPH oxidase attenuated zymosan-induced neutrophilic lung inflammation. Together, these results support a model in which NADPH oxidase in the monocyte/macrophage lineage inhibits fungal growth and restrains inflammation induced by pro-inflammatory fungal products.
Lung macrophages ingest inhaled Aspergillus conidia and limit fungal growth (24). Early neutrophilic accumulation in the lungs is required to prevent fungal germination in mice following challenge with large inocula of A. fumigatus (44, 45). Depletion of alveolar macrophages with clodronate prior to pulmonary A. fumigatus infection did not adversely affect neutrophil recruitment or control of fungal growth, whereas neutrophil depletion increased mortality in mice (45). Studies in isolated human neutrophils show that NADPH oxidase is important in controlling A. fumigatus hyphal growth but is dispensable for conidiocidal activity (46). However, using a reporter system to evaluate fungal viability following pulmonary Aspergillus challenge, Jhingran et al. (47) showed that NADPH oxidase in neutrophils augments killing of phagocytosed conidia. Transgenic Ncf1*/*MN+ mice were able to defend against high A. fumigatus inocula, and demonstrated no obvious impairment in antifungal host defense compared to wildtype mice. These results are consistent with our recent findings that monocyte/macrophage-targeted NADPH oxidase was protective against bacterial infections in mice (33).
While isolated wildtype alveolar macrophages could limit fungal growth of phagocytosed conidia, NADPH oxidase-deficient macrophages failed to do so. These results are consistent with those of Philippe et al. (25), and differ from those of Cornish et al. (26). The approach used by Cornish et al. involved evaluation of fungal germination in isolated alveolar macrophages at 8h after fungal seeding (26). However, we observed that the most dramatic effects of NADPH oxidase in isolated alveolar macrophages in suppressing fungal growth were observed at later time points after fungal seeding (Figure 2 and 3 and Video/Movie 1 and 2). These results support a model in which NADPH oxidase in alveolar macrophages, either through the direct effect of ROIs or via downstream signaling, is sufficiently injurious to spores to limit their germination into tissue-invasive hyphae. Aspergillus species and other molds have a broad repertoire of antioxidative pathways that defend against phagocyte-derived ROIs, and inhibition of these antioxidative pathways can reduce fungal survival during in vivo infection (48). Further studies are required to evaluate the responses of phagocytosed conidia at the genomic and proteomic level to reactive oxygen species produced by macrophage NADPH oxidase. Such studies may identify important fungal stress response pathways that could be targets for drug development.
Although transgenic Ncf1*/*MN+ mice are a valuable tool to delineate the role of monocyte/macrophage NADPH oxidase in antimicrobial host defense and regulation of inflammation, we acknowledge a number of limitations. First, there is the potential for low-level CD68 promoter-independent Ncf1 expression in vivo that can lead to augmented neutrophil NADPH oxidase activity in transgenic mice. Second, Ncf1 expression in myeloid DCs may influence the phenotype. Although NADPH oxidase in DCs can mediate cross-presentation of fungal antigens and vaccine-induced immunity (49), it is unlikely that this effect would be relevant in our experiments that focus on early acute inflammatory responses to a single Aspergillus or zymosan challenge. Third, in transgenic mice, NADPH oxidase-derived oxidants from macrophages may diffuse to neighboring neutrophils; diffused H2O2 could potentially be a substrate for antimicrobial myeloperoxidase-dependent halide generation in NADPH oxidase-deficient neutrophils (50). In addition, although NADPH oxidase is the major source of reactive oxidants in stimulated phagocytes, other oxidant-generating systems, such as xanthine oxidase (51) and mitochondrial respiration (52), can contribute to host defense.
Pathogen recognition receptors on macrophages sense fungal motifs displayed at different stage of fungal growth. Aspergillus cell wall beta-glucans that are unmasked during germination activate macrophages (3–6). Dectin-1 signaling is only activated by particulate beta-glucans that in nature would correspond to direct contact with microbe cell walls rather than soluble products (7). In mice, activation of dectin-1 by fungal beta-glucans leads to NADPH oxidase activation and enhanced fungal clearance in experimental pulmonary aspergillosis (2). Dectin-1 induces pro-inflammatory cytokines and chemokines, including IL-17A (2, 11, 53, 54). In contrast, our current results and prior studies show that NADPH oxidase limits pro-inflammatory cytokine production in response to Aspergillus challenge (17) and to beta-glucan preparations (18, 19). Consistent with our results, Deffert et al. (42) recently reported that intradermal administration of beta-glucan resulted in prolonged neutrophilic inflammation in Ncf1*/*MN− mice, but self-limited inflammation in wildtype and Ncf1*/* MN+ mice. Seen in this light, our results point to macrophage NADPH oxidase both limiting fungal growth and counter-regulating the pro-inflammatory responses induced by fungal cell wall products.
Neutrophils and macrophages co-mingle in the inflammatory milieu, where cross-signaling occurs whereby macrophages recognize and remove dying neutrophils (55, 56). Depending on the nature of the stimulus, neutrophil NADPH oxidase can stimulate neutrophil apoptosis (57, 58). In addition, ROIs produced by NADPH oxidase may coordinately mediate phosphatidylserine externalization in neutrophils and recognition and removal of apoptotic neutrophils (efferocytosis) by macrophages (59, 60). Fernandez-Boyanapalli et al. (61) made the intriguing observation that impaired efferocytosis in CGD macrophages was associated with defective expression and activation of peroxisome proliferator-activated receptor (PPAR)γ, and that a PPARγ agonist limited zymosan-induced peritonitis. We previously showed that activation of Nrf2, a redox-sensitive transcriptional factor that protects against oxidative stress, was impaired in p47phox−/− mice in vivo and in isolated macrophages, and that pharmacological Nrf2 activation reduced zymosan-induced lung inflammation in p47phox−/− mice (19, 62). Since Nrf2 induces PPARγ expression (63), these results suggest that NADPH oxidase-mediated activation of Nrf2 in macrophages may limit inflammation partly through promoting clearance of neutrophils. Macrophage NADPH oxidase and Nrf2 may also modulate inflammation through other mechanisms including regulation of inflammasome activation (64), autophagy (65, 66) and T cell downregulation (28). Further studies to define the interplay between NADPH oxidase and redox sensitive pathways in macrophages could lead to improved understanding of innate immune responses that mediate host defense and regulate inflammation.
Supplementary Material
Acknowledgments
The authors thank Hans Minderman, PhD, Paul Wallace, PhD, and Ree Dolnick, Department of Flow and Image Cytometry, Roswell Park Cancer Institute for their expert assistance with imaging.
Financial support: Chronic Granulomatous Disorder Research Trust (BHS), NIAID R01AI079253 (BHS). This research was supported, in part, by the NCI Cancer Center Support Grant to Roswell Park Cancer Institute (CA016056) and the Academy of Finland and the European Union grant Masterswitch (HEALTH-F2-2008-223404)
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Segal BH. Aspergillosis. N Engl J Med. 2009;360:1870–1884. doi: 10.1056/NEJMra0808853. [DOI] [PubMed] [Google Scholar]
- 2.Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, Faro-Trindade I, Brown GD, Steele C. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol. 2009;182:4938–4946. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aimanianda V, Bayry J, Bozza S, Kniemeyer O, Perruccio K, Elluru SR, Clavaud C, Paris S, Brakhage AA, Kaveri SV, Romani L, Latge JP. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 2009;460:1117–1121. doi: 10.1038/nature08264. [DOI] [PubMed] [Google Scholar]
- 4.Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, Pamer EG. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 2005;1:e30. doi: 10.1371/journal.ppat.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gersuk GM, Underhill DM, Zhu L, Marr KA. Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J Immunol. 2006;176:3717–3724. doi: 10.4049/jimmunol.176.6.3717. [DOI] [PubMed] [Google Scholar]
- 6.Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 2005;1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, Bose N, Chan AS, Magee AS, Danielson ME, Weiss A, Vasilakos JP, Underhill DM. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature. 2011;472:471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle KB, Gyori D, Sindrilaru A, Scharffetter-Kochanek K, Taylor PR, Mocsai A, Stephens LR, Hawkins PT. Class IA phosphoinositide 3-kinase beta and delta regulate neutrophil oxidase activation in response to Aspergillus fumigatus hyphae. J Immunol. 2011;186:2978–2989. doi: 10.4049/jimmunol.1002268. [DOI] [PubMed] [Google Scholar]
- 9.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 11.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 12.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, Espanol T, Fischer A, Kurenko-Deptuch M, Mouy R, Petropoulou T, Roesler J, Seger R, Stasia MJ, Valerius NH, Weening RS, Wolach B, Roos D, Kuijpers TW. Chronic granulomatous disease: the European experience. PLoS ONE. 2009;4:e5234. doi: 10.1371/journal.pone.0005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine (Baltimore) 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O’Brien S, Hilligoss DM, Malech HL, Gallin JI, Holland SM. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114:462–468. doi: 10.1542/peds.114.2.462. [DOI] [PubMed] [Google Scholar]
- 16.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med. 1997;185:207–218. doi: 10.1084/jem.185.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti MC, Grohmann U, Segal BH, Puccetti P. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 18.Schappi M, Deffert C, Fiette L, Gavazzi G, Herrmann F, Belli D, Krause KH. Branched fungal beta-glucan causes hyperinflammation and necrosis in phagocyte NADPH oxidase-deficient mice. J Pathol. 2008;214:434–444. doi: 10.1002/path.2298. [DOI] [PubMed] [Google Scholar]
- 19.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, Blackwell TS. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS ONE. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 21.Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–2622. doi: 10.1182/blood-2009-05-221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vethanayagam RR, Almyroudis NG, Grimm MJ, Lewandowski DC, Pham CT, Blackwell TS, Petraitiene R, Petraitis V, Walsh TJ, Urban CF, Segal BH. Role of NADPH Oxidase versus Neutrophil Proteases in Antimicrobial Host Defense. PLoS ONE. 2011;6:e28149. doi: 10.1371/journal.pone.0028149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, Casanova JL. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12:213–221. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaffner A, Douglas H, Braude A. Selective protection against conidia by mononuclear and against mycelia by polymorphonuclear phagocytes in resistance to Aspergillus. Observations on these two lines of defense in vivo and in vitro with human and mouse phagocytes. J Clin Invest. 1982;69:617–631. doi: 10.1172/JCI110489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philippe B, Ibrahim-Granet O, Prevost MC, Gougerot-Pocidalo MA, Sanchez Perez M, Van der Meeren A, Latge JP. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect Immun. 2003;71:3034–3042. doi: 10.1128/IAI.71.6.3034-3042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cornish EJ, Hurtgen BJ, McInnerney K, Burritt NL, Taylor RM, Jarvis JN, Wang SY, Burritt JB. Reduced nicotinamide adenine dinucleotide phosphate oxidase-independent resistance to Aspergillus fumigatus in alveolar macrophages. J Immunol. 2008;180:6854–6867. doi: 10.4049/jimmunol.180.10.6854. [DOI] [PubMed] [Google Scholar]
- 27.Shi C, Sakuma M, Mooroka T, Liscoe A, Gao H, Croce KJ, Sharma A, Kaplan D, Greaves DR, Wang Y, Simon DI. Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008;112:4699–4711. doi: 10.1182/blood-2008-01-137018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, Holmdahl R. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest. 2007;117:3020–3028. doi: 10.1172/JCI31935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dennis CG, Greco WR, Brun Y, Youn R, Slocum HK, Bernacki RJ, Lewis R, Wiederhold N, Holland SM, Petraitiene R, Walsh TJ, Segal BH. Effect of amphotericin B and micafungin combination on survival, histopathology, and fungal burden in experimental aspergillosis in the p47phox−/− mouse model of chronic granulomatous disease. Antimicrob Agents Chemother. 2006;50:422–427. doi: 10.1128/AAC.50.2.422-427.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knockout model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hultqvist M, Olofsson P, Holmberg J, Backstrom BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A. 2004;101:12646–12651. doi: 10.1073/pnas.0403831101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sareila O, Jaakkola N, Olofsson P, Kelkka T, Holmdahl R. Identification of a region in p47phox/NCF1 crucial for phagocytic NADPH oxidase (NOX2) activation. J Leukoc Biol. 2012 doi: 10.1189/jlb.1211588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J Immunol. 2012;188:5003–5011. doi: 10.4049/jimmunol.1103430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berenguer J, Allende MC, Lee JW, Garrett K, Lyman C, Ali NM, Bacher J, Pizzo PA, Walsh TJ. Pathogenesis of pulmonary aspergillosis. Granulocytopenia versus cyclosporine and methylprednisolone-induced immunosuppression. Am J Respir Crit Care Med. 1995;152:1079–1086. doi: 10.1164/ajrccm.152.3.7663787. [DOI] [PubMed] [Google Scholar]
- 35.Wasylnka JA, Moore MM. Uptake of Aspergillus fumigatus conidia by phagocytic and nonphagocytic cells in vitro: quantitation using strains expressing green fluorescent protein. Infect Immun. 2002;70:3156–3163. doi: 10.1128/IAI.70.6.3156-3163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roilides E, Dimitriadou A, Kadiltsoglou I, Sein T, Karpouzas J, Pizzo PA, Walsh TJ. IL-10 exerts suppressive and enhancing effects on antifungal activity of mononuclear phagocytes against Aspergillus fumigatus. J Immunol. 1997;158:322–329. [PubMed] [Google Scholar]
- 37.Jennings JH, Linderman DJ, Hu B, Sonstein J, Curtis JL. Monocytes recruited to the lungs of mice during immune inflammation ingest apoptotic cells poorly. Am J Respir Cell Mol Biol. 2005;32:108–117. doi: 10.1165/rcmb.2004-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davidson BA, Stewart CC, Russo TA, Chess PR, Knight PR., 3rd Discrimination of resident and infiltrated alveolar macrophages by flow cytometry in influenza A virus-infected mice. Exp Lung Res. 2005;31:323–339. doi: 10.1080/01902140590918524. [DOI] [PubMed] [Google Scholar]
- 39.Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38:380–385. doi: 10.1165/rcmb.2007-0224RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santosuosso M, Divangahi M, Zganiacz A, Xing Z. Reduced tissue macrophage population in the lung by anticancer agent cyclophosphamide: restoration by local granulocyte macrophage-colony-stimulating factor gene transfer. Blood. 2002;99:1246–1252. doi: 10.1182/blood.v99.4.1246. [DOI] [PubMed] [Google Scholar]
- 41.Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R. Reactive Oxygen Species Produced by the NADPH Oxidase 2 Complex in Monocytes Protect Mice from Bacterial Infections. J Immunol. 2012 doi: 10.4049/jimmunol.1103430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deffert C, Carnesecchi S, Yuan H, Rougemont AL, Kelkka T, Holmdahl R, Krause KH, Schappi MG. Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells. J Pathol. 2012 doi: 10.1002/path.4061. [DOI] [PubMed] [Google Scholar]
- 43.Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, Aliberti J, Flick MJ, Jordan MB. Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol. 2010;184:877–885. doi: 10.4049/jimmunol.0902346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonnett CR, Cornish EJ, Harmsen AG, Burritt JB. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus Conidia. Infect Immun. 2006;74:6528–6539. doi: 10.1128/IAI.00909-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mircescu MM, Lipuma L, van Rooijen N, Pamer EG, Hohl TM. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J Infect Dis. 2009;200:647–656. doi: 10.1086/600380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J Immunol. 2007;178:6367–6373. doi: 10.4049/jimmunol.178.10.6367. [DOI] [PubMed] [Google Scholar]
- 47.Jhingran A, Mar KB, Kumasaka DK, Knoblaugh SE, Ngo LY, Segal BH, Iwakura Y, Lowell CA, Hamerman JA, Lin X, Hohl TM. Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell reports. 2012;2:1762–1773. doi: 10.1016/j.celrep.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leal SM, Jr, Vareechon C, Cowden S, Cobb BA, Latge JP, Momany M, Pearlman E. Fungal antioxidant pathways promote survival against neutrophils during infection. J Clin Invest. 2012;122:2482–2498. doi: 10.1172/JCI63239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luca AD, Iannitti RG, Bozza S, Beau R, Casagrande A, D’Angelo C, Moretti S, Cunha C, Giovannini G, Massi-Benedetti C, Carvalho A, Boon L, Latge JP, Romani L. CD4+ T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest. 2012;122:1816–1831. doi: 10.1172/JCI60862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rex JH, Bennett JE, Gallin JI, Malech HL, Melnick DA. Normal and deficient neutrophils can cooperate to damage Aspergillus fumigatus hyphae. J Infect Dis. 1990;162:523–528. doi: 10.1093/infdis/162.2.523. [DOI] [PubMed] [Google Scholar]
- 51.Segal BH, Sakamoto N, Patel M, Maemura K, Klein AS, Holland SM, Bulkley GB. Xanthine oxidase contributes to host defense against Burkholderia cepacia in the p47(phox−/−) mouse model of chronic granulomatous disease. Infect Immun. 2000;68:2374–2378. doi: 10.1128/iai.68.4.2374-2378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.West AP, I, Brodsky E, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, Jamal S, Manguiat A, Rezaei N, Amirzargar AA, Plebani A, Hannesschlager N, Gross O, Ruland J, Grimbacher B. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, Jacobs L, Jansen T, Verheijen K, Masthoff L, Morre SA, Vriend G, Williams DL, Perfect JR, Joosten LA, Wijmenga C, van der Meer JW, Adema GJ, Kullberg BJ, Brown GD, Netea MG. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361:1760–1767. doi: 10.1056/NEJMoa0901053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savill J, Dransfield I, Hogg N, Haslett C. Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature. 1990;343:170–173. doi: 10.1038/343170a0. [DOI] [PubMed] [Google Scholar]
- 57.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 58.Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood. 2011;117:5953–5962. doi: 10.1182/blood-2010-11-322206. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, Henson PM, Bratton DL. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood. 2009;113:2047–2055. doi: 10.1182/blood-2008-05-160564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frasch SC, Fernandez-Boyanapalli RF, Berry KZ, Leslie CC, Bonventre JV, Murphy RC, Henson PM, Bratton DL. Signaling via Macrophage G2A Enhances Efferocytosis of Dying Neutrophils by Augmentation of Rac Activity. J Biol Chem. 2011;286:12108–12122. doi: 10.1074/jbc.M110.181800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fernandez-Boyanapalli R, Frasch SC, Riches DW, Vandivier RW, Henson PM, Bratton DL. PPARgamma activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood. 2010;116:4512–4522. doi: 10.1182/blood-2010-02-272005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davidson BA, Vethanayagam RR, Grimm MJ, Mullan BA, Raghavendran K, Blackwell TS, Freeman ML, Ayyasamy V, Singh KK, Sporn MB, Itagaki K, Hauser CJ, Knight PR, Segal BH. NADPH Oxidase and Nrf2 Regulate Gastric Aspiration-Induced Inflammation and Acute Lung Injury. J Immunol. 2013 doi: 10.4049/jimmunol.1202410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, Kleeberger SR. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. 2010;182:170–182. doi: 10.1164/rccm.200907-1047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 66.Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427–1432. doi: 10.1073/pnas.1014156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.